

Abstract
Clinical trials treating inherited retinal dystrophy caused by RPE65 mutations had put retinal gene therapy at the forefront of gene therapy. Both successes and limitations in these clinical trials have fueled developments in gene vectors, which continue to further advance the field. These novel gene vectors aim to more safely and efficiently transduce retinal cells, expand the gene packaging capacity of AAV, and utilize new strategies to correct the varying mechanisms of dysfunction found with inherited retinal dystrophies. With recent clinical trials and numerous pre-clinical studies utilizing these novel vectors, the future of ocular gene therapy continues to hold vast potential.
Keywords: gene therapy, inherited retinal dystrophies, RPE65, adeno-associated virus, equine infectious anemia virus
Inherited retinal dystrophies (IRD) encompass a diverse group of devastating blinding disorders affecting approximately 1 in 3000 people [1]. Of these rare disorders, retinitis pigmentosa (RP) is the most common and associated with defects in over 60 genes. Altogether, RP and other IRDs involve mutations in over 200 genes, with greatly varying mechanisms of visual dysfunction (RetNet, http://www.sph.uth.tmc.edu). To add to the complexity of these disorders, the aforementioned mutations can result in autosomal recessive, autosomal dominant, x-linked, and even digenic inheritance. These diverse mechanisms of disease pose a unique challenge for gene therapy. For example, autosomal recessive mutations necessitate gene replacement. Autosomal dominant mutations, which can result in dominant negative effects or toxic gain of function, might require gene suppression with or without subsequent gene replacement. Some genes are too large for current vectors. For these diseases where direct gene replacement is not practical, expression of neuroprotective factors that act in a mutation independent fashion might be needed. Finally, conventional gene replacement therapy would not be beneficial in patients with severe or complete loss of photoreceptors. The application of gene therapy to express light-sensitive proteins in inner retinal cells has given rise to the field of optogenetics. Gene therapy techniques continue to advance and evolve to overcome these challenges in new and innovative approaches.
Leber congenital amaurosis (LCA), a more severe dystrophy associated with defects in at least 21 genes was the first ophthalmological disease targeted with gene therapy. Clinical trials of gene replacement therapy for LCA due to RPE65 (retinal pigmented epithelium-specific protein 65kDa) mutations (RPE65-LCA) [2–4] have highlighted the immense potential for treating IRDs, especially those caused by loss of function mutations. Among early viral vector prototypes, such as adenovirus, lentivirus, and adeno-associated virus (AAV); AAV showed the best combination of safety and retinal cell transduction [5]. Following the success of RPE65-LCA trials, AAV has emerged as the leading delivery vector for ocular gene therapy. However, many challenges and questions remain as to how to broaden the spectrum of treatable diseases as well as how to better regulate and sustain gene expression. Although AAV2 has been shown to efficiently transduce RPE cells, newer vectors are needed to selectively and efficiently target other retinal cells, particularly photoreceptors. Second, the 4.7 kb gene capacity of AAV has hindered treatment of mutations in larger genes. Developments in AAV dual vectors and non-AAV viral vectors have opened up gene targets such as CEP290, ABCA4, and MYO7A, associated with LCA, Stargardt, and Type 1B Usher syndromes. However, some genes, such as USH2A, still exceed the capacity of these newer techniques and will likely require alternative techniques, such as compacted DNA nanoparticles [6].
We will briefly review the seminal RPE65-LCA studies with a focus on how findings and limitations of these studies have shaped the future direction of ocular gene therapy. RPE65-LCA studies have highlighted potential adverse effects from a subretinal delivery, which have spurred advancements in viral gene vectors and attempts to achieve efficient photoreceptor cell transduction through a safer intravitreal delivery route [7]. This review will cover advancements in gene therapy that have since emerged and broadened treatment possibilities, and the new frontiers in treatment of ocular and inherited retinal diseases being explored.
RPE65-LCA clinical trials: how we got there and what have we learned
The RPE65 story provides an ideal example of how translational research can move from the clinic to the lab bench top and ultimately back to the clinic. While genetic testing might be able to identify the causative mutation, much preclinical work was necessary to produce a treatment. Elucidation of the function of RPE65 at the cellular level permitted an understanding of the mechanism of the disease. The identification and creation of both small and large animal models furthered this understanding and provided a platform for the development of treatments. Finally, an understanding of the disease phenotype provided the evidence that human clinical trials would be feasible.
In LCA, the most severe of retinal dystrophies, patients show nystagmus, unrecordable electroretinograms (ERGs), and a profound visual deficiency at birth or within the first few years of life that progresses to total blindness by mid- to late-adulthood. Cross sectional genotype-phenotype studies have suggested that RPE65-LCA patients show a slower and less severe photoreceptor degeneration relative to other LCA genes. For example, patients at the earliest ages tested have been found to have severely reduced but detectable cone electroretinograms [8,9], greater visual acuity within the first decade of life [10–12], recordable visual fields with peripheral concentric constriction [10–12], and residual islands of retained vision in adulthood [9,12]. Another characteristic phenotype of RPE65-LCA patients is the relative preservation of the outer nuclear layer (ONL) demonstrated through in vivo structural optical coherence tomography (OCT) studies, which was in agreement with ONL preservation in animal models. An advanced modeling study further demonstrated that ONL preservation in patients was greater than expected for the level of vision loss in the fovea and in rod-rich regions temporally and superiorly to the fovea [13]. This greatly encouraged advancements into gene therapy since successful visual gains depended on the presence of surviving photoreceptors.
The initial step in understanding RPE65 was the identification and cloning of the RPE65 gene in humans and mice [14], followed closely by the generation of the RPE65 knockout mouse [15]. Comprehensive pre-clinical characterization of multiple mouse (rd12, Rpe65R91W-knockin, and Rpe65-knockout mice) and canine (RPE65-mutant Briard dog) models of RPE65-LCA had elucidated important groundwork information on the pathophysiology of disease. RPE65 is an isomerase in the retinoid cycle involved in regeneration of the essential visual pigment, 11-cis-retinal, specifically in conversion of all-trans-retinyl esters to 11-cis-retinol [15]. With loss of function of RPE65, there is progressive retinal degeneration [15], a decrease in lipofuscin granules, and accumulation of retinyl esters in lipid droplets in RPE cells [15–17]. A lack of 11-cis-retinal regeneration leads to a severe decrease in visual function with abnormal rod- and cone-mediated electroretinograms (ERGs) at approximately 10% of normal levels by 2–4 months of age in Rpe65-KO mice [18]. Despite visual function deficits, histologic studies showed relatively slow rod photoreceptor loss in all animal models [18]. Rpe65-KO mice show a particularly severe cone cell loss following early cone opsin misclocalization, in contrast to the foveal sparing in RPE65 patients. Foveal preservation may stem from an inherent resistance to degeneration, residual enzymatic activity with specific RPE65 mutations, and an alternative isomerase in Muller glial cells for chromophore regeneration [19]. While gene replacement would not restore vision loss secondary to photoreceptor degeneration, it could increase the 11-cis-retinal chromophore to increase photon capture and subsequent light sensitivity.
In addition to the promising characteristics of the RPE65 phenotype, simultaneous developments in AAV vectors were essential to the road towards treatment of RPE65-LCA. Adeno-associated virus (AAV) is a linear single-stranded DNA virus belonging to the Parvoviridae family and the Dependovirus genus, first found as a contaminant amongst adenovirus (Ad) [20]. The high safety profile, transduction efficiency and longevity of AAV over other viruses have established it as a leading vector [20]. AAV vectors largely exist as an episomal, non-integrating virus [21], which ensures stable transgene expression in postmitotic cells. Wildtype AAV shows non-pathogenicity in humans with 40–80% of adults seropositive without associated symptoms or disease [22]. Despite its safety, neutralizing antibodies have been reported to decrease vector efficiency, particularly with systemic administration [23]. Although less likely to be problematic in ocular tissues, a potential reduction in transduction efficiency is concerning in administration of rAAV treatment to the contralateral eye.
The prototypic AAV2 used in RPE65-LCA clinical trials, was first cloned into a bacterial plasmid in 1982 [24,25]. Currently, production of recombinant AAV vectors involves co-transfection of two engineered plasmids. The first plasmid is the transgene cassette that has removed all viral genes except for two palindromic inverted terminal repeats (ITRs) flanking the transgene of interest [26,27]. This replication-deficient plasmid carrying the transgene of interest requires co-transfection with plasmids expressing viral replication (rep) and packaging capsid (cap) genes (Figure 1).
Figure 1.
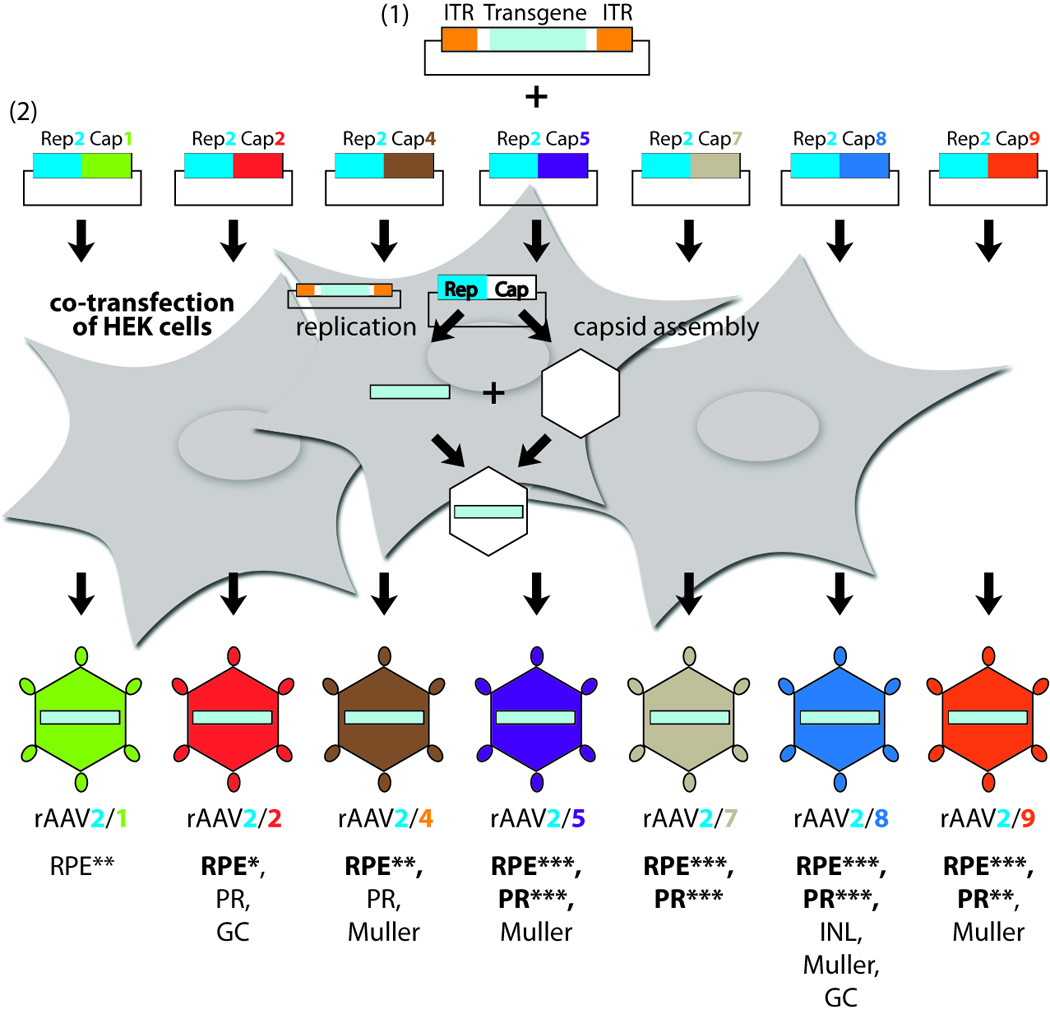
AAV capsid pseudotyping and retinal cell tropism. Production of rAAV involves co-transfection of two plasmids: (1) the transgene cassette with two ITR regions flanking the transgene of choice, and (2) a helper plasmid encoding for the necessary replication (Rep) genes from AAV2 and capsid (Cap) genes from the desired AAV serotype. The retinal cell tropism of each AAV serotype listed below show the most effective retinal cell type transduced in bold. Relative comparisons of transduction efficiency of the RPE and PRs are shown from lowest (*) to highest (***) for particularly effective serotypes [38,42]. Note that information pertaining to transduction efficiencies is representative of murine retina [38,42], but variances between species have been reported [150]. Abbreviations: ITR, inverted terminal repeats; rAAV, recombinant adeno-associated virus; RPE, retinal pigmented epithelium; PR, photoreceptors; GC, ganglion cells; INL, inner nuclear layer. Adapted from [39].
With well-defined animal models and advancements in AAV gene delivery, the next progression towards treatment for RPE65-LCA patients was proof-of-concept vision rescue in animal models. Subretinal administration of AAV2 packaging the Rpe65 gene under a hybrid cytomegalovirus (CMV) immediate early enhancer/chicken β-actin (CBA) promoter provided vision rescue as measured through ERGs in 1–2 month old Rpe65-KO and rd12 mice [28]. The same AAV2 construct successfully provided long-term improvements up to 3 years in the canine RPE65-mutant model in rod- and cone-ERGs, and 11-cis-retinal regeneration [29]. A variety of AAV pseudotypes and gene promoters in studies from other groups also showed visual improvements in the RPE65-canine model [30–32], as well as long-term improvements in photoreceptor outer segment morphology and lipid inclusions [31]. Although pre-clinical canine studies showed high safety profiles, a potential adverse effect of dose-dependent retinal thinning with AAV2-CBA-hRPE65 has been observed [33].
Initiation of three independent RPE65-LCA clinical trials in 2007 occurred following the mounting evidence of safe and successful proof-of-concept visual improvements across multiple Rpe65 animal models and the promising phenotype of RPE65-LCA patients. All studies demonstrated a relatively high safety profile of AAV2-RPE65 gene replacement without toxicity, adverse surgical events, or serious immunologic events [2–4]. Slight differences in visual outcomes have been reported between studies, particularly as long-term follow-ups are released. Changes in visual acuity (VA), which is often the gold standard outcome for clinical trials, have been variable. This is not surprising considering the low baseline VA in many of the treated patients. In initial patient cohorts, one of three trials reported VA improvements in three out of three patients [4], while insignificant changes in VA were found in the other two trials. However, the patients with improved VA showed the lowest baseline VA, with improved VA generally not surpassing the baseline VA of the other patients from the initial cohorts. In addition to individual baseline visual function, differences in visual outcomes may be attributed to the differences between the three independent clinical trials in AAV viral vector production, gene promoter use, and dosage concentration and volume of viral solution administered (Table 1). No direct comparisons of efficiency have been conducted on these three specific gene promoters used. In addition to varying volume, subtle differences in administration methods may alter the amount of solution forming the subretinal bleb. Despite these differences, a larger number of patients from each trial showed improved visual sensitivity as assessed through microperimetry [2], full-field sensitivity testing [3], and transient pupillary light response (TPLR) [4] (Table 2).
Table 1. RPE65-LCA Clinical Trials.
Details of RPE65-LCA clinical trials in gene promoter, viral titer, and volume of the viral solution used. *Results not yet published. **All clinical trial information was obtained from the Clinical Trials online registry, https://clinicaltrials.gov [165].
| NCT# | Sponsor | Phase | Vector | Promoter | Viral titer (by cohort) |
Volume (by cohort) |
Subjects | Ref **[165] |
|---|---|---|---|---|---|---|---|---|
| 00643747 | UCL | I/II | rAAV2/2 | hRPE65 | 1.00×1011 vp/ml | 1.0 ml | N=12 | [2] |
| 00481546 | U Penn - Scheie | I | rAAV2/2 | CBSB | 1. 5.96×1010 vp/ml 2. 11.92×1010 vp/ml 3. 8. 94×1010 vp/ml 4. 17.88×1010 vp/ml 5. 17.88×1010 vp/ml (7.95×1010 vp/ml) |
0.15 ml 0.30 ml 0.225 ml 2×0.225 ml 2×0.225 ml (0.20 ml) |
N=3 N=3 N=3 N=2 N=4 (N=1) |
[3,7,34,35] |
| 00516477 | U Penn - CHOP | I | rAAV2/2 | CBA | 1. 1.50×1010 vp/ml 2. 4.8×1010 vp/ml 3. 1.50×1011 vp/ml |
0.15 ml 0.15 ml 0.30 ml |
N=3 N=6 N=3 |
[4,151–155] |
| 00749957 | AGTC | I/II | rAAV2/2 | CBA | 1. 4.00×1011 vp/ml 2. 1.33×1012 vp/ml |
0.45 ml 0.45 ml |
N=6 N=6 |
[156] |
| 00821340 | Hadassah | I | AAV2/2 | CBSB | 1.19×1011 vp/ml | 0.30 ml | N=10 | [157] |
| 01496040 | Nantes | I/II | rAAV2/4 | hRPE65 | 6.0×1010 vp/ml | 0.4–0.8 ml | N=9 | [158] |
| 00999609 | Sparks Therapeutics |
III | AAV2/2 | CBA | 1.50×1011 vp/mL | 0.30 ml? | N=24 | * |
Abbreviations: NCT#, ClinicalTrials.gov identifier number; UCL, University College of London; U Penn-Scheie, University of Pennsylvania – Scheie Scheie Eye Institute; U Penn-CHOP, University of Pennsylvania – The Children’s Hospital of Philadelphia. hRPE65p, human RPE65 promoter; hRPE65, human RPE65; CBSB, hybrid modified short cytomegalovirus (CMV) enhancer and chicken β-actin promoter (CBA); vp, viral particles; ml, milliliter. Ref, references.
Table 2. Results from RPE65-LCA Clinical Trials.
Visual outcomes of RPE65-LCA patients following treatment. *NCT#01208389 is a Phase I follow-up study treating the contralateral eye to patients enrolled in NCT#00516477. **All clinical trial information was obtained from the Clinical Trials online registry, https://clinicaltrials.gov [165].
| NCT# | Sponsor | Cohort | Subjects | Reported improvements in visual outcomes | Ref **[165] |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| VA | MP | FST | TPLR | Motility Studies | New or additional pseudofovea formation |
|||||
| 00643747 | UCL | 1 | N=3 | 0/3 patients |
1/3 patients |
n/a | n/a | 1/3 patients |
n/a | [2] |
| 00481546 | U Penn - Scheie | 1 | N=3 | 0/3 patients |
n/a | 3/3 patients |
2/3 patients |
2/3 patients |
1/3 patients |
[3,7,34,35] |
| 2 | N=3 | 1/3 patients |
n/a | 3/3 patients |
3/3 patients |
0/3 patients |
2/3 patients |
[7,35] | ||
| 3 | N=3 | 1/3 patients |
n/a | 3/3 patients |
3/3 patients |
1/3 patients |
0/3 patients |
[7] | ||
| 4 | N=2 | 1/2 patients |
n/a | 2/2 patients |
1/2 patients |
1/2 patients |
0/2 patients |
[7] | ||
| 5 | N=4 | 0/4 patients |
n/a | 4/4 patients |
2/4 patients |
1/4 patients |
1/4 patients |
[7,35] | ||
| 00516477 | U Penn - CHOP | 1 | N=3 | 3/3 patients |
n/a | 3/3 patients |
3/3 patients |
3/3 patients |
n/a | [4,151] |
| 2 | N=6 | 3/6 patients |
n/a | 4/5 patients (1 n/a) |
4/5 patients (1 n/a) |
3/5 patients |
n/a | [151] | ||
| 3 | N=3 | 1/3 patients |
n/a | 1/2 patients (1 n/a) |
3/3 patients |
1/3 patients |
n/a | [151] | ||
| *01208389 | Spark Therapeutics |
1 | N=3 (N=12) |
3/3 patients |
n/a | 2/3 patients |
3/3 patients |
2/3 patients |
n/a | [154] |
Abbreviations: NCT#, ClinicalTrials.gov identifier number; UCL, University College London; U Penn – Scheie, University of Pennsylvania, Scheie Eye Institute; U Penn – CHOP, University of Pennsylvania – The Children’s Hospital of Philadelphia. VA, visual acuity; MP, microperimetry; FST, full-field sensitivity testing; TPLR, transient pupillary light response sensitivity; n/a, non-applicable or not-tested. Ref, references.
A particularly interesting finding from one group of investigators has been the development of “pseudo-foveas” initially observed in one patient at one year post-treatment [34], with similar findings in an additional three patients in further follow-up studies [7,35]. These patients demonstrated the worst baseline VAs with non-foveal fixation [7]. They gained the greatest improvements in VA post-treatment, which was found to stem from improved vision at their extrafoveal fixation locus or “pseudo-fovea”, an area of best visual function.
Cautionary tales from early trials using first-generation AAV vectors
Despite encouraging visual gains in the Phase I studies, three out of five RPE65-LCA patients in one trial had significant foveal thinning at 1 or 3 year post-operative follow-up [7]. While one patients’ thinning was attributed to the natural course of the disease, the cause of thinning in the other two patients was less apparent. Unfortunately, foveal thinning was not measured in other clinical trials, limiting a full assessment of the risks and benefits of this treatment. It remains unclear whether foveal thinning in patients was a complication secondary to vector toxicity, foveal detachment following subretinal injection, or a combination of these factors. Elucidation of factors that might contribute to potential photoreceptor loss will be important in developing the next generation of vectors. Erring on the side of caution, it has been therefore suggested to include the fovea only when there is already significant foveal atrophy and when fixation is parafoveal [7]. However, treatment of the fovea will be essential to prevent significant vision loss as younger patients are treated and in other diseases such as Stargardt dystrophy.
Overcoming obstacles to improve efficacy
Since development of the first-generation rAAV2 vector, vast innovations and improvements in viral gene vectors have emerged largely driven by inefficient transduction of immune-competent organ systems, such as the liver, muscle, and heart [36]. These advancements address the potential causes of foveal thinning in RPE65 patients in vector toxicity, transgene overexpression, and adverse events from subfoveal detachment.
Optimizing viral transduction with AAV vector advancements
In canine models, cone cell loss occurred in a dose-dependent manner indicating the potential for cellular toxicity due to high viral titers [33]. Therefore, one strategy to prevent potential vector toxicity is to lower the number of viral particles used. Viral transduction has been enhanced through various approaches, such as providing improved cell-specific tropism to decrease off-target transduction, increased transduction efficiency, and through the evasion of immune responses that would otherwise decrease efficacy. Although distinctly enumerated below, alterations to the viral capsid may achieve more than one of these advantageous properties simultaneously or synergistically.
Improvements in cell-specific tropism
One innovation in AAV capsid engineering has been the development of pseudotyping. Following the characterization of AAV2, AAV serotypes 1 through 9 were subsequently isolated (reviewed in [37]) and it became evident that differences in capsid proteins directed cell tropism. Taking advantage of variations in tropism, “pseudotyped” rAAV vectors express AAV2 replication proteins with capsid proteins of other serotypes (Figure 1). In the retina, all serotypes thus far have shown RPE cell transduction, with AAV1, 2, 4, and 6 transducing RPE cells either specifically or more efficiently than neural retina [38–41]. Conversely, retinal photoreceptor transduction is best achieved with AAV5, 7, 8, and 9 [40–42]. Selecting the appropriate AAV pseudotype would effectively reduce the viral titer required for transduction of the desired retinal cell type.
Improved transduction efficiency through directed mutagenesis
Beyond pseudotyping, AAV capsid engineering through directed capsid peptide mutagenesis has improved transduction efficiency by minimizing viral vector degradation. In directed mutagenesis, specific surface-exposed tyrosine residues are altered to phenylalanine (Y-F mutants) to evade tyrosine phosphorylation, which would normally target the vector for ubiquitination and proteasome-mediated degradation [43] [44]. Therefore, these capsid mutants escape degradation and show improved intracellular trafficking and transduction efficiency [44,45]. In addition to improved efficacy, Y-F mutants showed novel tropism, with some Y-F mutants achieving photoreceptor- and RPE-specific cell targeting, whereas others provided widespread pan-retinal transduction [45,46].
Surface-exposed tyrosine residue mutants also mitigate binding of neutralizing antibodies to the neutralizing epitope regions in the AAV capsid [46], that would otherwise prevent efficient viral binding and uptake into desired cells [47]. This potential adverse immune response may prohibit effective viral re-administration and/or treatment of the contralateral eye.
Improved transduction efficiency through directed evolution
In contrast to site-directed mutagenesis, AAV capsid engineering via directed evolution introduces random large-scale alterations to the capsid envelop. While various strategies of directed evolution have been thoroughly reviewed [48–50], the general methodology first involves generation of a large library of chimeric AAV capsid mutants through combinatorial techniques such as error prone PCR that generate random mutagenesis, site specific mutagenesis at key binding sites, and DNA shuffling of multiple cap genes (Figure 2). AAV capsid libraries are subsequently applied to selection conditions in in vitro or in vivo systems to generate desired phenotypes, such as the aforementioned retinal properties. AAV variants that are able to effectively transduce specific cells, bind to receptors, or overcome barriers or antibodies are identified and selected for amongst the large library of AAV capsid mutants.
Figure 2.
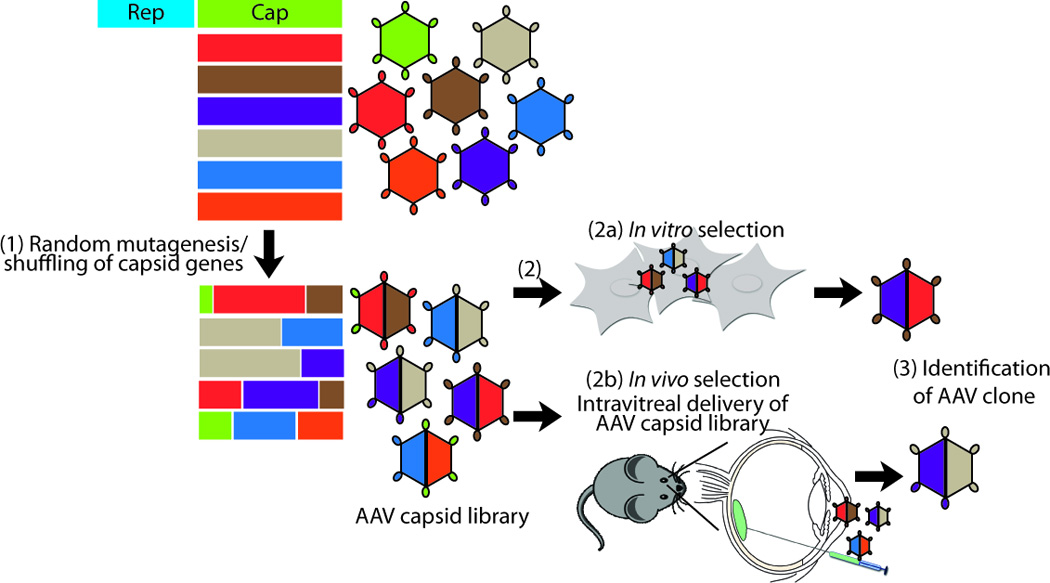
AAV capsid engineering through directed evolution. Directed evolution involves (1) random mutagenesis, recombination, and/or DNA shuffling of AAV capsid genes from all AAV serotypes generate a large AAV capsid library with chimeric AAV capsids. (2) This capsid library is applied to (2a) in vitro or (2b) in vivo selection systems, and (3) AAV capsids that successfully overcome the selection criterion are identified.
Improved transgene expression through gene promoters
Strategic use of gene promoters can also impart cell-specific tropism and improved transduction efficiency, providing another means of viral transduction optimization thus reducing the number of viral particles needed. While ubiquitous promoters, such as CMV and CBA have classically been used to obtain maximal expression across multiple cell types, there is a movement toward achieving cell-specific physiological levels of expression. Compared to the wide therapeutic range of RPE65 replacement, gene replacement of other RPE genes such as BEST1 [51] and MERTK [52] have utilized the retinal pigmented epithelium-specific vitelliform macular dystrophy 2 (VMD2) promoter.
Photoreceptor-specific genes are particularly fastidious and might need to be well-titrated to achieve sufficient but non-toxic levels of expression, such as with RHO [53–55], MYO7A [56], or BBS1 [57]. Cone cell damage with overexpression of the reporter GFP gene has also been reported [58]. In these cases, photoreceptor-specific opsin and human G-protein-coupled receptor protein kinase 1 (hGRK1) promoters have been utilized [59]. A number of promoters have also been developed for cone-specific targeting, including the human red opsin pR2.1 promoter [60], a 569 bp S-opsin promoter known as HB569 [61], and an chimeric IRBPe-GNAT2 (inter-retinoid binding protein enhancer element – transducin alpha-subunit) promoter [62], largely useful for treatment of achromatopsia (ACHM).
Minimizing cell loss through better surgical methods
In addition to vector toxicity, foveal thinning in RPE65 patients may have occurred secondary to subfoveal detachment. To avoid this potential adverse outcome, novel methods of creating a subretinal bleb have been developed [63] and alternate intravitreal routes of photoreceptor transduction have been highly sought after [64].
The delicate nature of the degenerating retina poses inherent surgical challenges and requires extreme care with subretinal administration of gene vectors. A recent two-step injection technique attempts to minimize trauma from generation of the subretinal bleb. An initial subretinal detachment is formed over the targeted area through injecting a balanced salt solution applied through a continuous footswitch-operated pressure device. The viral vector is subsequently deposited through the same retinotomy site, extending the existing detachment site [63].
Directed evolution of viral vectors has opened up the possibility of using intravitreal rather than subretinal injections [64]. While Y-F capsid mutants provided initial proof-of-concept for intravitreal transduction of outer retinal cells in mice [46,65], transduction was highly limited or absent in canine [66] and non-human primates (NHPs) [64]. This discrepancy has been partly attributed to the biophysical barrier imparted by the significantly thicker inner limiting membrane (ILM) in large animals [67]. Additionally, a complex balance occurs between a vector’s ability to bind to ILM proteins such as heparan sulfate and laminin. For example, AAV2, AAV8 and AAV9 bind heparan sulfate, while AAV1 and AAV5 primarily bind sialic acid, a monosaccharide that is absent in the ILM [68]. The “7m8” capsid mutant, produced through directed evolution, contains a seven-amino acid loop insertion in the heparan sulfate binding domain [64]. It has demonstrated the most effective effective pan-retinal transduction via intravitreal delivery in NHPs to date, improving upon the results from Y-F mutants. Greater retinal penetration of the 7m8 clone in NHPs was likely attributed to reduced heparan sulfate affinity which decreased sequestration at the ILM.
Expansion of gene therapy: large genes and new disease frontiers
Following the success of RPE65 clinical trials, a multitude of gene targets are currently or will soon be entering clinical trials (Table 3 and Table 4). However, the endogenous 4.7 kb packaging capacity of AAV has limited utility in treating larger genes. Particularly desirable large gene targets are ABCA4 and MYO7A, mutations which cause Stargardt (STGD1) and Usher (USH1B) syndromes. Expanding the packaging capacity is therefore an important second aim in AAV vector development. Multiple strategies are currently being explored including: oversized AAV vectors, dual AAV vector systems, non-AAV viral vectors such as equine infectious anemia virus (EIAV), and compacted DNA nanoparticles.
Table 3. Other Gene Therapy Trials under progress.
Gene therapy clinical trials recently initiated. **All clinical trial information was obtained from the Clinical Trials online registry, https://clinicaltrials.gov [165]. Additional references in parentheses indicate pre-clinical data, and references without parenthesis indicate clinical trial publications.
| NCT# | Trial status |
Sponsor | Phase | Disease | Gene | Vector | Titer | Volume | Subjects | Ref **[165] |
|---|---|---|---|---|---|---|---|---|---|---|
| 01367444 | Recruiting | Sanofi | I/II | Stargardt | ABCA4 | EIAV | n.p. | n.p. | N=8 N=4 N=4 N=12 |
([88]) |
| 01505062 | Recruiting | Sanofi | I/II | Usher 1B |
MYO7A | EIAV | n.p. | n.p. | N=6 N=3 N=3 N= |
([56,87]) |
| 01461213 | Recruiting | U Oxford | I II |
CHM | CHM | AAV2 | 1×1010 vp/ml 1×1011 vp/ml |
0.1 ml | N=6 N=6 |
[63] |
| 02341807 | Recruiting | Spark Therapeutics |
I II |
CHM | CHM | AAV2 | n.p. n.p. |
n.p. n.p. |
N=5 N=5 |
([159,160]) |
| 02077361 | Not yet recruiting |
Ian M. MacDonald |
I | CHM | CHM | AAV2 | 1×1010 vp/ml | 0.1 ml | N=12 | ([159,160]) |
| 01482195 | Recruiting | King Khaled Eye Hospital |
I | LCA/RP | MERTK | AAV2 | n.p. | n.p. | N=6 | ([52]) |
| 01301443 | Active, not recruiting |
Oxford Biomedica |
I | AMD | EIAV | n.p. n.p. n.p. MTD |
n.p. | N=3 N=3 N=3 N=9 |
([105]) | |
| 01024998 | Active, not recruiting |
Genzyme/ Sanofi |
I | AMD | sFLT01 | AAV2 | 2×108 vp/ml 2×109 vp/ml 6×109 vp/ml 2×1010 vp/ml MTD |
0.1 ml 0.1 ml 0.1 ml 0.1 ml |
N=6 N=6 N=6 N=6 N=10 |
([112]) |
| 01494805 | Active, not recruiting |
Lions Eye Institute/ Avalanche |
I II |
AMD | sFLT01 | AAV | 1×1010 vp/ml 1×1011 vp/ml |
n.p. | N=40 | ([112]) |
| 02317887 | Recruiting | NEI | I/IIa | XLRS | RS1 | AAV8 | 1×109 vp/ml 1×1010 vp/ml 1×1011 vp/ml |
n.p. | N=3 N=3 N=3 |
([116,117]) |
Abbreviations: NCT#, ClinicalTrials.gov identifier number; U Oxford, University of Oxford; NEI, National Eye Institute. CHM, choroideremia; LCA, Leber congenital amaurosis; RP, retinitis pigmentosa; AMD, age-related macular degeneration; XLRS, X-linked retinoschisis. EIAV, equine infectious anemia virus; AAV, adeno-associated virus. vp, viral particles; ml, milliliter; n.p., not published; MTD, most tolerated dose. Ref, references.
Table 4. Proposed Future Clinical Trials.
Proposed or potential clinical trials following evidence of pre-clinical treatment efficacy.
| Sponsor | Disease | Gene | Proposed Vector |
Preclinical Evidence/ Animal Models |
Ref | Sponser Ref |
|---|---|---|---|---|---|---|
| AGTC | Achromatopsia | CNGB3 | AAV | Mouse (Cngb3-KO) Canine (CNGB3-KO, CNGB3m/m) |
[136,161] | [166] |
| AGTC | Achromatopsia | CNGA3 | AAV | Mice (Cpfl5Cnga3-KO) | [137,138] | [166] |
| Genable | adRP | RHO | AAV | Mouse (P347SRHO) | [142] | [167] |
| University of Alberta | Choroideremia | CHM | AAV2 | Mouse (Chmnull/WT) | [159,160] | [168] |
| Spark Therapeutics | Choroideremia | CHM | AAV2 | Mouse (Chmnull/WT) | [159,160] | [169] |
| Genzyme | LCA Type 1 | GUCY2D | AAV | Mice (Gucy2e–KO, Gucy2e/Gucy2f dKO) |
[126,127,162] | [170] |
| LCA Type 10 | CEP290 | AAV? | Mouse (rd16; Nrl−/−) Feline (rdAc Abyssinian cat) |
[130,131] | ||
| AGTC | XLRP | RPGR | AAV5 | Canine (XLPRA1, XLPRA2) | [141] | [171] |
| AGTC | XLRS | RS1 | AAV5 | Mouse (Rs1-KO) | [118,163,164] | [172] |
| RdCVF | Mouse (rd10) | [145] |
Abbreviations: AGTC, Applied Genetic Technologies Corp. adRP, autosomal dominant retinitis pigmentosa; LCA, Leber congenital amaurosis; XLRP, X-linked retinitis pigmentosa; XLRS, X-linked retinoschisis. CNGB3, cyclic nucleotide-gated channel beta 3; CNGA3, cyclic nucleotide-gated channel alpha 3; RHO, rhodopsin; CHM, choroideremia (Rab escort protein-1); GUCY2D, guanylate cyclase 2D, membrane (retina-specific), CEP290, centrosomal protein 290 kDa; RPGR, retinitis pigmentosa GTPase regulator; RS1, retinoschisin-1; RdCVF, rod-derived cone viability factor. AAV, adeno-associated virus; hIRBP, human interstitial retinol binding protein 3. KO, knockout; dKO, double knockout. Ref, references.
Preclinical evidence/animal models: CNGB3m/m canine model, misssense mutation in exon 6; cpfl5 mouse, naturally occurring missense mutation in Cnga3; P347S–RHO mouse, missense mutation in RHO and a model of RHO-adRP; Chmnull/WT mouse, female heterozygous-null carriers with the choroideremia phenotype; Gucy2e, guanylate cyclase 2E, membrane (retina-specific), the mouse homolog of GUCY2D or RetGC1, Gucy2f, guanylate cyclase 2F, membrane (retina-specific), the mouse homolog of GUCY2F or RetGC2; Gucy2e/Gucy2f dKO mice are missing both RetGC1 and RetGC2. Rd16 mice, naturally occurring deletion of exons 35–39 of Cep290; Nrl/- mice, mouse lacking the neural retina leucine zipper gene involved in rod photoreceptor differentiation leading to an all-cone phenotype; rdAC, retinal degeneration Abyssinian cat with truncation mutation in CEP290; XLPRA1, X-linked progressive retinal atrophy-1, canine with ORF15 microdeletion (del 1028–1032) in RPGR; XLPRA2, X-linked progressive retinal atrophy-2, canine with ORF15 microdeletion (del 1084–1085) in RPGR; rd10, retinal degeneration 10 mouse with missense point mutation in Pde6b.
“Overstuffed” AAV vectors
With oversized or “overstuffed” AAV vectors, transgenes exceeding the conventional 4.7 kb capacity are packaged as usual. A sizeable extension to a packaging capacity of 8.9 kb was reported specifically with AAV5, with other overstuffed AAV serotypes showing a significant decrease in viral titer production [69]. Although this study showed that oversized AAV5 viral particles held intact, full-length genomes that mediated in vitro and in vivo full-length expression [69], further studies have had more limited success. Although additional studies produced oversized AAV1–5 and AAV8 vectors packaging 5.2 kb [70] to 6.0 kb [71], it was noted that viral titer yields may be decreased ten-fold compared to AAV < 4.7 kb [70] and oversized vectors may undergo intracellular proteasomal degradation post-entry [71]. In contrast to successful encapsulation of full-length genomes [69,71], other studies demonstrated fragmentation of genomes greater than 5.3 kb that subsequently recombine intracellularly to produce full-length expression [70,72]. These discrepancies between studies in the ability to encapsulate oversized genomes without fragmentation, size capacity, and transduction efficiency are controversial and remains to be further examined.
Dual AAV vectors
In contrast to a single oversized vector, three strategies to accommodate larger genes utilize a dual vector approach where the transgene of interest is divided into 5’ and 3’ halves (Figure 3). The first strategy, known as overlapping vectors, overtly takes advantage of recombination between two halves of the transgene that contain a homologous overlapping sequence [73]. The second strategy termed, trans-splicing, utilizes vectors containing a 3’ splice donor signal on one cassette and a 5’ splice acceptor signal on a second cassette, with head-to-tail concatemerization of the two vectors producing full-length transgene expression [74]. A third hybrid strategy adds a highly recombinant region from an exogenous gene, such as alkaline phosphatase (AP) and the F1 phage (AK), and places it adjacent to the splice sites, thus utilizing both recombination and splicing [75,76].
Figure 3.
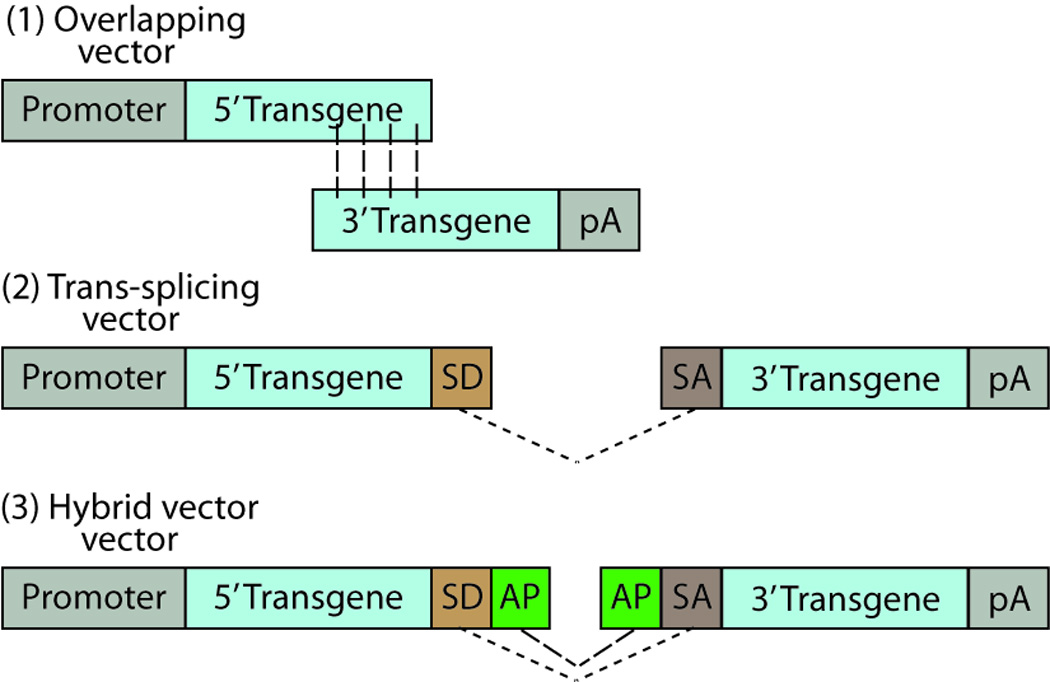
Three AAV dual vector types. (1) Overlapping vectors have a homologous overlapping region between the two halves of the transgene of choice, which undergo homologous recombination for reconstitution of a full-length transcript of the transgene. (2) Trans-splicing vectors undergo splicing through their splice donor (SD) and splice acceptor (SA) sites at the ends of the transgene halves. (3) Hybrid vectors undergo splicing through the SD and SA sites, as well as recombination through a highly recombinogenic site from a exogenous gene, such as alkaline phosphatase (AP). Dashed lines show homologous recombination, dotted lines show splicing between SD and SA sites. Abbreviations: pA, polyadenylation tail; SD, splice donor; SA, splice acceptor; AP, alkaline phosphatase. Adapted from [76].
Use of dual vector AAV approaches in retinal applications has been hindered by several factors. Dual vector systems require co-transfection of two vectors in a single cell with subsequent occurrence of homologous recombination or splicing of gene products. The low levels of homologous recombination that occurs in post-mitotic neurons, such as photoreceptors, decreases the efficiency of overlapping vectors significantly [76]. Additionally, in vivo retinal transduction efficiencies of trans-splicing and hybrid dual vectors have been highly variable, ranging from ∼5% to 100% of the transduction efficiency of single AAV vectors [76,77], although the reason for this variability remains unknown. Lastly, while no aberrantly truncated proteins were detected in vivo, thorough evaluation of this would be required moving forward into clinical trials.
Non-AAV viral vectors for retinal gene therapy
With these limitations in extended capacity of AAV vectors, alternative non-AAV vectors with large packaging capacities have been examined. One particular viral vector that has shown promise is the equine infectious anemia virus (EIAV). EIAV is a non-HIV1-based, non-primate lentivirus in the Retroviridae family able to package transgenes up to 8 kb [78]. Although it is associated with a self-limiting equine infectious anemia in horses, donkeys, and mules; humans show no pre-existing immunity or pathogenicity to EIAV [79]. It has shown a high safety profile in CNS, muscle, and hematopoietic pre-clinical studies [80–82]. In fact, EIAV was the first lentivirus used in clinical trials for the treatment of Parkinson’s disease [83], following its demonstrated safety and efficacy in simultaneously delivering three enzymes involved in dopamine synthesis to rodent [84] and NHP [85] disease models. In ocular studies, EIAV has shown high safety and transduction efficiency in neonatal rodent [56,86,87] and adult NHP photoreceptors [79,88].
The use of HIV1-based lentiviral vectors has been more limited than non-HIV1-based lentiviruses in retinal gene therapy due to inefficient photoreceptor transduction in adult retina [41,89]. No HIV1 pseudotypes have been found to significantly improve photoreceptor transduction in adult rodents to date [78,90]. While similar findings in rodents were also reported with EIAV, EIAV efficiently transduces photoreceptors in adult NHPs, whereas photoreceptor transduction with an HIV1-lentiviral vector in ex vivo adult human retinal explants remained highly inefficient [91]. Definitive in vivo NHP studies with HIV1 vectors are required. The utility of HIV1-based lentivirus, however, may be in RPE transduction with HIV-1-Mokola [41] and HIV1-venezuelan equine encephalitis virus-derived glycoprotein (VEEV-G) [91] pseudotypes showing improved RPE transduction. Another limitation in HIV1-lentiviral vectors stems from its property of random integration into the chromosome. Recent developments in integration-deficient vectors, however, may address concerns of insertional mutagenesis [92].
Although adenoviral vectors offer a large packaging capacity of up to 37 kb [93], the transient transgene expression following adenoviral vector gene delivery had limited its use. This decrease in transgene expression is attributed to immunogenic responses that lead to clearance of the virus and/or of transduced cells [94]. The development of helper-dependent adenoviral vectors, which are devoid of all viral genes except for encapsidation, has improved long-term transgene expression following subretinal delivery [90]. Additionally, improved photoreceptor targeting through alterations to the penton base in combination with use of photoreceptor-specific gene promoters [95,96] has recently increased the utility of adenoviral vectors in the treatment of IRDs.
Nanoparticles as an alternative vector to deliver large genes
A last technology developed to overcome size limitations with AAV vectors has been DNA-compacted nanoparticles (NPs), which are single DNA molecules compacted polyethylene glycol-substituted polylysine 30-mers (CK30PEG). Its 20 kb packaging capacity [97] provides immense possibilities in gene targets with particular utility in treatment of Type 2A Usher syndrome associated with mutations in the Usherin gene, where its 15.6 kb size prohibits packaging into any AAV or non-AAV viral vectors. Its safety and utility has been demonstrated in the lung [98], with introduction of the cystic fibrosis transmembrane conductance regulator (CFTR) through an intranasal route resulting in Phase I/II clinical trials for cystic fibrosis patients [99]. In retinal gene therapy, nanoparticles have also demonstrated safety and transduction efficiency in Abca4-KO mice [6]. However, nanoparticles have shown some limitations hindering its widespread use in ocular gene therapy. It showed lower transduction efficiency per vector genome as compared to AAV [100], and silencing of transgene expression potentially related to the gene promoter [100] or the DNA plasmid backbone used [101], thus higher vector titers and repeated dosing might be required. Although not problematic in intranasal treatment of lung diseases, repeated subretinal injections would be less than ideal.
Other retinal gene therapies underway or in preparation
Ocular gene therapy has extended to a whole host of eye conditions, including expansion of the gene target size, treatment of autosomal dominant and X-linked IRDs, and expression of anti-angiogenic genes to treat neovascular age-related macular degeneration (Table 3 and Table 4). Treatment has also gone beyond simple gene replacement into gene-independent strategies, such as expression of neuroprotective agents and light-sensitive proteins with optogenetics.
Gene therapy for Stargardt dystrophy
Mutations in the 6.8 kb ABCA4 gene are associated with STGD1, which is the most common form of juvenile-onset macular degeneration. ABCA4 encodes for ABCR, a flippase for N-retinylidene phosphatidylethanolamine (N-retinylidene-PE) to the cytoplasmic side of photoreceptor outer segment disk membranes. A deficiency of the protein leads to accumulation of all-trans-retinal, N-retinylidene-PE and the subsequent toxic by-product, N-retinylidene-N-retinylethanolamine, commonly known as A2E [102]. Successful treatment of the commonly used Abca4-STGD1 mouse model has been demonstrated with a variety of these large gene capacity vectors, including the oversized AAV5 vector [69], the trans-splicing and hybrid AAV2 dual vector systems [76], and the EIAV vector [86]. However, only EIAV has thus far has been translated into clinical trials with recent initiation of escalating dose Phase I/IIa EIAV-ABCA4 StarGentm clinical trials (Sanofi), following its demonstrated safety in NHPs [88].
Gene therapy for Usher Type 1B
The second important large gene target was the 6.6 kb MYO7A gene associated with Type 1B Usher syndrome, the most common inherited combined deaf-blind condition [103]. A spectrum in the severity of deafness and retinal degeneration defines the phenotypes of Usher 1, 2, and 3. Usher 1 patients show the greatest severity in hearing and visual impairment, with progression of retinitis pigmentosa beginning in childhood and profound deafness from birth to within a year of life. Shaker1 (sh1) mice, lacking the Myo7a gene, are deaf and exhibit a characteristic circling and head shaking behavior for which it is named after due to cochlear and vestibular dysfunction. In the retina, myosin VIIa is an actin-based motor protein involved in rhodopsin transport in photoreceptor cilia, melanosome localization at RPE microvilli, and phagosome motility within RPE cells. Sh1 mice therefore display ultrastructural opsin and melanosome mislocalization defects. Gene replacement in sh1 mice with the oversized AAV5 vector [69], dual vector AAV2 [76], and the EIAV vector [56,87] successfully rescued ultrastructural defects. Similar to ABCA4, safety studies in NHPs [87] allowed for the EIAV-MYO7A vector, known as UshStattm, to enter Phase I/IIa dose escalation clinical trials (Sanofi).
Gene therapy for treatment of age-related macular degeneration
While neovascular age-related macular degeneration (AMD) has been successfully treated with routine repeated intravitreal injections of anti-vascular endothelial growth factor (anti-VEGF) compounds, gene therapy alternatively could provide stable expression of anti-angiogenic molecules for a prolonged period. The large packaging capacity of EIAV has been used to treat neovascular AMD with RetinoStattm, an EIAV vector expressing two anti-angiogenic genes, endostatin and angiostatin. Subretinal delivery of RetinoStattm provided safe and efficient transgene expression in rodents [104] and NHPs [105], and significantly suppressed choroidal neovascular lesions [104]. These proof-of-concept studies progressed RetinoStattm into Phase I clinical trials (Oxford Biomedica) (Table 3), with potential to also improve ocular neovascularization in multiple conditions, such as diabetic retinopathy [106,107], retinopathy of prematurity (ROP) [108,109], and post-transplant neovascularization [110].
Successful treatment for neovascular AMD through gene therapy has also been achieved through intravitreal delivery of AAV2 expressing soluble Flt1 receptor (sFlt01), a chimeric VEGF binding agent [111]. Following the demonstrated safety, long-term expression up to 18 months post-treatment, and efficacy in inhibiting choroidal neovascularization in NHPs [112,113], a dose-escalation Phase 1 study was initiated (Genzyme) (Table 3). NHP studies, however, pointed to several potential obstacles and precautions with intravitreal AAV2-sFlt01. The first was variable expression levels, attributed to unpredictable release and diffusion of the AAV vector in the vitreous. Additionally, mild to moderate inflammatory reactions against the AAV2 capsid were demonstrated in 4 out of 6 macaques receiving the highest viral titer. Although transient mild to moderate inflammation was found with EIAV-RetinoStattm, inflammation was found to be relatively persistent lasting 5 to 15 months post-treatment with intravitreal AAV2-sFlt01, which was also attributed to the slow diffusion and trapping of the vector in the vitreous [112].
Gene therapy treatment for X-linked retinoschisis
The only gene currently associated with XLRS is RS1, which encodes for retinoschisin (Rs1). Rs1 is a 224 amino acid secreted protein that binds anionic phospholipids on photoreceptor and bipolar cell membranes, potentially mediating cell-matrix and cell-cell interactions, and playing a role in maintaining photoreceptor inner segment stability, synaptic structure, and overall retinal architecture [114,115]. Defects in RS1 lead to characteristic splitting or schisis of retinal layers and loss of ERG b-waves due to the disruption of synaptic processes. In X-linked retinoschisis (XLRS), retinal detachments are not uncommon and schisis cavities render the retina highly susceptible to further retinal damage with subretinal gene delivery. Therefore intravitreal delivery is desirable. Intravitreal gene replacement of RS1 with AAV8 is currently in Phase I/IIa dose-escalation clinical trials (National Eye Institute), with high safety and efficacy demonstrated in rs1-deficient mice and in rabbits [116,117]. However, use of intravitreally injected AAV2 is also being explored (AGTC), following the long-term safety and efficacy of AAV2 demonstrated in rs1-knockout mice [118].
Gene therapy treatment for choroideremia
Subretinally injected AAV2 is being used in clinical trials for choroideremia (CHM). CHM is an X-linked recessive disease with characteristic fundus findings of peripheral retinal, choroidal, and RPE degeneration with macular-sparing until late in the disease progression. CHM is associated with functionally null mutations in the 1.9 kb CHM gene, encoding for Rab escort protein-1 (REP-1). REP-1 is involved in the post-translational prenylation lipid-modification of Rab small GTpases (Rabs) needed for proper intracellular vesicular transport [119]. Initial results have been promising from Phase I/II clinical trials from six patients treated with the initial dose of AAV2-REP1 [63]. Two of the six patients showed improvements in visual acuity, and five out of the six patients showed improved retinal sensitivity as assessed through microperimetry [63].
Gene therapy for retinitis pigmentosa
Mutations in MERTK, which encodes for a transmembrane receptor of tyrosine kinases involved in RPE cell phagocytosis of photoreceptor outer segments, are associated with autosomal recessive retinitis pigmentosa and LCA with characteristic findings of discrete autofluorescent dots and subretinal debris [120,121]. A subset of patients display preservation of peripheral fields [120,121] showing potential for rescue. The well-characterized Mertk-RCS (Royal College of Surgeon’s) rat model was previously successfully treated with AAV2 [122,123], with further optimization through use of the RPE-specific promoter, VMD2 [52]. This improved AAV2-hVMD2-MERTK vector, is currently being used in Phase I clinical trials targeting MERTK-RP patients (King Khaled Eye Hospital) (Table 3).
Gene therapy for GUC2YD-LCA
Many more pre-clinical trials are currently underway and show great promise for future entry into clinical trials (Table 4). One of the most common causes of LCA are mutations in GUCY2D (LCA1), accounting for about 20% of cases. Human GUCY2D and the mouse Gucy2e isoform encode for the retinal guanylate cyclase-1 (GC1), an essential photoreceptor outer segment protein required for synthesis of the cGMP visual transduction signaling molecule. Similar to RPE65 patients, GUCY2D–LCA1 patients were found to have relatively preserved macular ONL and retinal laminar architecture [124,125] despite severe vision loss. Combined with successful pre-clinical rescue of GC1-knockout mice [126] and double GC1- and GC2-knockout mice [127], GUCY2D–LCA therefore shows high potential for entry into clinical trials.
Gene therapy for CEP290-LCA
A last genotype that has been associated with advantageous macular ONL preservation is CEP290-LCA10 patients [124,128]. The focus on rescue of the preserved foveal cones in patients has been recapitulated through generation of an all-cone mouse model lacking Cep290 [129,130], allowing for future testing of treatment efficacy. With recent characterization of the feline Cep290 model [131] that also allows for pre-clinical large animal model testing and recent developments in large gene capacity vectors, the potential to treat mutations in the 6.4 kb CEP290 gene is high.
Gene therapy for achromatopsia
In line with attempts to treat conditions with macular preservation and advancements in cone-cell targeting [61,62,132], the treatment potential of ACHM patients has been closely examined. This has been facilitated through structural imaging of the macula and cone cells through high-resolution spectral-domain OCT (sdOCT) [133,134] and adaptive optics in ACHM patients [135]. Although cross-sectional studies have shown variability and a lack of clear age-related disease progression [133,134], the pre-clinical successes of treatment of the canine CNGB3-ACHM model [136], the rodent CNGA3-ACHM model [137,138], and recent characterization of a large-animal sheep model [139] provide encouragement for potential future clinical trials.
Gene therapy for X-linked retinitis pigmentosa
In addition to autosomal recessive IRDs, X-linked and autosomal dominant IRDs are being targeted for treatment. RPGR, a ciliary protein that is associated with over 70% of cases of X-linked retinitis pigmentosa (XLRP) is an important gene target. Multiple rodent models of RPGR-XLRP exist with the Rpgr-knockout mouse, transgenic mutant Rpgr mouse models, as well as the naturally occurring rd9 mouse model [140]. Rescue of two naturally occurring canine RPGR models [141] with AAV2/5-hIRBP-hRPGR has provided evidence for safety and efficacy.
Gene therapy for autosomal dominant retinitis pigmentosa
Autosomal dominant IRDs, such as autosomal dominant RP (adRP), provide more of a treatment challenge than gene replacement for autosomal recessive IRDs. The most common cause of adRP is mutations in RHO, encoding for the essential visual transduction rhodopsin protein. With the high heterogeneity of RHO-adRP mutations of over 150 different mutations (RetNet, http://www.sph.uth.tmc.edu), an efficient rescue strategy has been through RNA-interference (RNAi) with simultaneous introduction of a modified rhodopsin gene replacement resistant to RNAi suppression. This two-vector strategy has demonstrated efficient rescue of the RHO-adRP P347S RHO-mutant mouse model [142].
Gene-independent strategies: neuroprotection and optogenetics
Distinct from gene replacement, gene therapy has extended to mutation-independent strategies that provide a generalized means of vision rescue without the need to identify genetic defects causing disease. The first strategy has been in expression of neuroprotective factors, which provide gene-independent rescue effects for retinal dystrophies. A particularly promising neuroprotective factor is rod-derived cone viability factor (RdCVF), a thioredoxin-like protein secreted by rods that promotes cone photoreceptor survival [143]. While previous subretinal delivery of the RdCVF provided some retinal protection in the P23H–RHO adRP mouse [144], gene expression of RdCVF provides an improved strategy. Intravitreal expression of RdCVF with the novel 7m8 AAV2 capsid mutant developed through directed evolutionary methods was found to improve degeneration in the rd10 RP mouse model [145]. Administration of neuroprotective agents would primarily be useful in slowing down retinal degeneration while awaiting genetic characterization.
A second mutation-independent strategy is in the field of optogenetics. In addition to being gene-independent, the advantage of this strategy is to restore visual function following loss of photoreceptor cells. While thoroughly reviewed elsewhere [146,147], optogenetics briefly works through expression of light-sensitive proteins in residual inner retinal cells, such as retinal ganglion cells or bipolar cells. Expression of channelrhodopsin-2 (ChR2) and halorhodopsin, ionotropic rhodopsins found in green algae and halobacteria, respond to light stimulation through conformational changes that cause opening of the channel and cell depolarization [146]. Although visual-evoked potentials were demonstrated following expression of ChR2 in ganglion cells of the rd1 mouse model of retinal degeneration [148,149], limitations include requirements for a high level of light stimulus and/or transgene expression [149]. Although a highly promising tool in gene therapy, much work remains in improvement of methodology, such as increasing sensitivity and generating ON and OFF light responses, and determining whether the inner retinal circuitry in various retinal degenerations would benefit from optogenetic conversion.
Expert commentary
Generally considered as the most compelling success story in gene therapy, the big picture achievements of RPE65 clinical trials have been pivotal to the larger field of gene therapy. The seminal RPE65-LCA clinical trials have provided the foundation for translational success and demonstrated safety of AAV in ocular gene therapy. Equally astounding have been the rapid advancements in response to the issues that have surfaced from and since RPE65 studies. These recent advances in viral vectors have diversely expanded ocular gene therapy to provide viable treatments for a vast array of inherited retinal degenerations and ocular diseases. With further developments on the horizon, the future prospects of ocular gene therapy are tremendous.
Despite these large-scale achievements, limitations in vision rescue exist with treatment of a severe RPE65-LCA phenotype. Identification of diseases and genotype-phenotype patient subsets that may be more amenable to vision rescue is a key aspect in moving forward with ocular gene therapy. Continuing advancements in imaging, such as with high-resolution optical coherence tomography and adaptive optics, are needed to provide a more detailed genotype-phenotype characterization and identify individual patient candidates. Also needed are continuing technologies in measurements of visual function to standardize baseline and post-treatment visual outcomes in patients with inherited retinal degenerations.
With multiple divergent avenues currently being explored in further advancements to AAV, alternate use of EIAV, and use of non-viral nanoparticles, much work remains to demonstrate the same safety and efficacy of AAV2 in a clinical setting. However, AAV2-RPE65 studies had provided an exemplary groundwork to base future pre-clinical and clinical trials on.
Five-year review
In the coming years, additional follow-up results from Phase I/IIa and initial results from Phase III RPE65-LCA clinical trials will provide important information on dosing, and the treatment efficacy and visual gains that can be realistically provided by gene replacement therapy for a severe LCA retinal degeneration phenotype. Preliminary results of the safety and efficacy of the EIAV vector in treatment of Stargardt and Type 1B Usher syndrome will also emerge, which may potentially shape the use of AAV or non-AAV viral vectors. However, rapid developments in AAV vectors continue to address and improve upon limitations with the first-generation AAV2 vector. The great diversity in AAV capsid modifications as well as the potential to expand the AAV packaging capacity continues to make AAV an adaptable vector with vast potential in treating multiple IRDs. Ongoing pre-clinical treatment of IRDs with these novel AAVs will soon elucidate the successful modifications that best improve transduction efficiency, retinal-cell specificity, and intravitreal transduction of outer retinal cells across the new diseases and genes being targeted, which will further influence movement into clinical trials.
Key issues.
Inherited retinal dystrophies (IRDs) are genetically heterogenous and show diverse mechanisms of disease and inheritance, such as autosomal dominant, X-linked, and digenic inheritance requiring more complex treatment than gene replacement therapy for autosomal recessive diseases. Novel gene therapy strategies are required to address these additional and more complex IRDs.
The translational pathway exemplified by RPE65-LCA studies included a thorough understanding of the mechanism of dysfunction in animal models, pre-clinical treatment in these animal models, and detailed characterization of patient phenotype. This pathway greatly contributed to the success of RPE65 clinical trials and would serve well to be modeled in the treatment of additional IRDs.
- Despite promising improvements in retinal sensitivity, foveal thinning occurred following subfoveal delivery of AAV2 in Phase I/IIa RPE65-LCA clinical trials in three patients. The cause of foveal thinning remains unclear, and may be attributed to the location of subretinal administration or viral vector toxicity.
- Assessment of risks and benefits of foveal inclusion are important in future studies.
- An alternate intravitreal route of foveal transduction is in development.
- Optimizing transduction efficiency may minimize viral vector toxicity.
The limited 4.7 kb packaging capacity of adeno-associated virus requires alternative strategies to target large gene mutations causing IRDs, including expansion of the AAV capacity, use of other viral vectors, and non-viral vectors.
Current clinical trials are using AAV2 to treat autosomal recessive RP associated with MERTK mutations, and X-linked recessive choroideremia associated with CHM mutations.
Current clinical trials are treating large gene targets ABCA4 and MYO7A associated with Stargardt dystrophy and Type 1B Usher syndrome with EIAV, a non-AAV viral vector.
Application of gene therapy forstable expression of anti-VEGF compounds in the treatment of AMD.
Mutation-independent gene therapy strategies may be required to aid vision rescue.
Acknowledgments
M Pennesi received funding from Research to Prevent Blindness, Foundation Fighting Blindness and the National Eye Institute.
Footnotes
Financial and competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125(2):151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. doi: 10.1056/NEJMoa0802268. *Seminal Phase I/IIa RPE65-LCA study from University College, London with initial patient cohort results.
- 3. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19(10):979–990. doi: 10.1089/hum.2008.107. *Seminal Phase I RPE65-LCA study from University of Pennsylvania, Scheie Eye Institute with initial patient cohort results.
- 4. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–2248. doi: 10.1056/NEJMoa0802315. *Seminal Phase I RPE65-LCA study from University of Pennsylvania, Children’s Hospital of Philadelphia with initial patient cohort results.
- 5.Buch PK, Bainbridge JW, Ali RR. AAV-mediated gene therapy for retinal disorders: from mouse to man. Gene Ther. 2008;15(11):849–857. doi: 10.1038/gt.2008.66. [DOI] [PubMed] [Google Scholar]
- 6.Han Z, Conley SM, Makkia RS, Cooper MJ, Naash MI. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J Clin Invest. 2012;122(9):3221–3226. doi: 10.1172/JCI64833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130(1):9–24. doi: 10.1001/archophthalmol.2011.298. *Follow-up study from additional patient cohorts from the Phase I RPE65-LCA study from University of Pennsylvania, Scheie Eye Institute with initial patient cohort results.
- 8.Jacobson SG, Aleman TS, Cideciyan AV, et al. Defining the Residual Vision in Leber Congenital Amaurosis Caused by RPE65 Mutations. Invest Ophthalmol Vis Sci. 2009;50(5):2368–2375. doi: 10.1167/iovs.08-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson DA, Gyurus P, Fleischer LL, et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci. 2000;41(13):4293–4299. [PubMed] [Google Scholar]
- 10.Perrault I, Rozet JM, Ghazi I, et al. Different functional outcome of RetGC1 and RPE65 gene mutations in Leber congenital amaurosis. Am J Hum Genet. 1999;64(4):1225–1228. doi: 10.1086/302335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23(4):306–317. doi: 10.1002/humu.20010. [DOI] [PubMed] [Google Scholar]
- 12.Paunescu K, Wabbels B, Preising MN, Lorenz B. Longitudinal and cross-sectional study of patients with early-onset severe retinal dystrophy associated with RPE65 mutations. Graefes Arch Clin Exp Ophthalmol. 2005;243(5):417–426. doi: 10.1007/s00417-004-1020-x. [DOI] [PubMed] [Google Scholar]
- 13.Jacobson SG, Aleman TS, Cideciyan AV, et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc Natl Acad Sci U S A. 2005;102(17):6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamel CP, Jenkins NA, Gilbert DJ, Copeland NG, Redmond TM. The gene for the retinal pigment epithelium-specific protein RPE65 is localized to human 1p31 and mouse 3. Genomics. 1994;20(3):509–512. doi: 10.1006/geno.1994.1212. [DOI] [PubMed] [Google Scholar]
- 15.Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet. 1998;20(4):344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 16.Katz ML, Redmond TM. Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42(12):3023–3030. [PubMed] [Google Scholar]
- 17.Katz ML, Wendt KD, Sanders DN. RPE65 gene mutation prevents development of autofluorescence in retinal pigment epithelial phagosomes. Mech Ageing Dev. 2005;126(4):513–521. doi: 10.1016/j.mad.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 18. Cideciyan AV. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res. 2010;29(5):398–427. doi: 10.1016/j.preteyeres.2010.04.002. **In-depth review of pre-clinical studies and patient characterization leading up to Phase I RPE65-LCA clinical trials.
- 19.Kaylor JJ, Yuan Q, Cook J, et al. Identification of DES1 as a vitamin A isomerase in Muller glial cells of the retina. Nat Chem Biol. 2013;9(1):30–36. doi: 10.1038/nchembio.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atchison RW, Casto BC, Hammon WM. Adenovirus-associated defective virus particles. Science. 1965;149(3685):754–756. doi: 10.1126/science.149.3685.754. [DOI] [PubMed] [Google Scholar]
- 21.Muzyczka N, Samulski RJ, Hermonat P, Srivastava A, Berns KI. The genetics of adeno-associated virus. Adv Exp Med Biol. 1984;179:151–161. doi: 10.1007/978-1-4684-8730-5_15. [DOI] [PubMed] [Google Scholar]
- 22.Sprecher-Goldberger S, Thiry L, Lefebvre N, Dekegel D, de Halleux F. Complement-fixation antibodies to adenovirus-associated viruses, cytomegaloviruses and herpes simplex viruses in patients with tumors and in control individuals. Am J Epidemiol. 1971;94(4):351–358. doi: 10.1093/oxfordjournals.aje.a121330. [DOI] [PubMed] [Google Scholar]
- 23. Willett K, Bennett J. Immunology of AAV-Mediated Gene Transfer in the Eye. Front Immunol. 2013;4:261. doi: 10.3389/fimmu.2013.00261. *Detailed review of the immunologic issues of ocular gene therapy, covering both mechanistic information and clinical interpretations and precautions.
- 24.Samulski RJ, Berns KI, Tan M, Muzyczka N. Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc Natl Acad Sci U S A. 1982;79(6):2077–2081. doi: 10.1073/pnas.79.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hermonat PL, Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci U S A. 1984;81(20):6466–6470. doi: 10.1073/pnas.81.20.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni TH, Zhou X, McCarty DM, Zolotukhin I, Muzyczka N. In vitro replication of adeno-associated virus DNA. J Virol. 1994;68(2):1128–1138. doi: 10.1128/jvi.68.2.1128-1138.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X, Muzyczka N. In vitro packaging of adeno-associated virus DNA. J Virol. 1998;72(4):3241–3247. doi: 10.1128/jvi.72.4.3241-3247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennicelli J, Wright JF, Komaromy A, et al. Reversal of blindness in animal models of leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther. 2008;16(3):458–465. doi: 10.1038/sj.mt.6300389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12(6):1072–1082. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Meur G, Stieger K, Smith AJ, et al. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther. 2007;14(4):292–303. doi: 10.1038/sj.gt.3302861. [DOI] [PubMed] [Google Scholar]
- 31.Vaegan Narfstrom K, Katz M, Bragadottir R, Rakoczy EP, Seeliger M. Assessment of structure and function over a 3-year period after gene transfer in RPE65−/− dogs. Doc Ophthalmol. 2005;111(1):39–48. doi: 10.1007/s10633-005-3159-0. [DOI] [PubMed] [Google Scholar]
- 32.Aguirre GK, Komaromy AM, Cideciyan AV, et al. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med. 2007;4(6):e230. doi: 10.1371/journal.pmed.0040230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobson SG, Acland GM, Aguirre GD, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther. 2006;13(6):1074–1084. doi: 10.1016/j.ymthe.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 34.Cideciyan AV, Hauswirth WW, Aleman TS, et al. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med. 2009;361(7):725–727. doi: 10.1056/NEJMc0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cideciyan AV, Aguirre GK, Jacobson SG, et al. Pseudo-fovea formation after gene therapy for RPE65-LCA. Invest Ophthalmol Vis Sci. 2014 doi: 10.1167/iovs.14-15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12(5):341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 37.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5(3):285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 38.Lebherz C, Maguire A, Tang W, Bennett J, Wilson JM. Novel AAV serotypes for improved ocular gene transfer. J Gene Med. 2008;10(4):375–382. doi: 10.1002/jgm.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beltran WA. The use of canine models of inherited retinal degeneration to test novel therapeutic approaches. Vet Ophthalmol. 2009;12(3):192–204. doi: 10.1111/j.1463-5224.2009.00694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang GS, Schmidt M, Yan Z, et al. Virus-mediated transduction of murine retina with adeno-associated virus: effects of viral capsid and genome size. J Virol. 2002;76(15):7651–7660. doi: 10.1128/JVI.76.15.7651-7660.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet. 2001;10(26):3075–3081. doi: 10.1093/hmg/10.26.3075. [DOI] [PubMed] [Google Scholar]
- 42.Allocca M, Mussolino C, Garcia-Hoyos M, et al. Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J Virol. 2007;81(20):11372–11380. doi: 10.1128/JVI.01327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong L, Zhao W, Wu J, et al. A dual role of EGFR protein tyrosine kinase signaling in ubiquitination of AAV2 capsids and viral second-strand DNA synthesis. Mol Ther. 2007;15(7):1323–1330. doi: 10.1038/sj.mt.6300170. [DOI] [PubMed] [Google Scholar]
- 44.Zhong L, Li B, Mah CS, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A. 2008;105(22):7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrs-Silva H, Dinculescu A, Li Q, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther. 2009;17(3):463–471. doi: 10.1038/mt.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petrs-Silva H, Dinculescu A, Li Q, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther. 2011;19(2):293–301. doi: 10.1038/mt.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moskalenko M, Chen L, van Roey M, et al. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J Virol. 2000;74(4):1761–1766. doi: 10.1128/jvi.74.4.1761-1766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitchell AM, Nicolson SC, Warischalk JK, Samulski RJ. AAV’s anatomy: roadmap for optimizing vectors for translational success. Curr Gene Ther. 2010;10(5):319–340. doi: 10.2174/156652310793180706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, Xiao X. Creation of a cardiotropic adeno-associated virus: the story of viral directed evolution. Virol J. 2013;10:50. doi: 10.1186/1743-422X-10-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hida K, Hanes J, Ostermeier M. Directed evolution for drug and nucleic acid delivery. Adv Drug Deliv Rev. 2007;59(15):1562–1578. doi: 10.1016/j.addr.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 51.Guziewicz KE, Zangerl B, Komaromy AM, et al. Recombinant AAV-mediated BEST1 transfer to the retinal pigment epithelium: analysis of serotype-dependent retinal effects. PLoS One. 2013;8(10):e75666. doi: 10.1371/journal.pone.0075666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conlon TJ, Deng WT, Erger K, et al. Preclinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retinitis pigmentosa. Hum Gene Ther Clin Dev. 2013;24(1):23–28. doi: 10.1089/humc.2013.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9(5):815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 54.Tan E, Wang Q, Quiambao AB, et al. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2001;42(3):589–600. [PubMed] [Google Scholar]
- 55.Mao H, James T, Jr, Schwein A, et al. AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum Gene Ther. 2011;22(5):567–575. doi: 10.1089/hum.2010.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hashimoto T, Gibbs D, Lillo C, et al. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 2007;14(7):584–594. doi: 10.1038/sj.gt.3302897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seo S, Mullins RF, Dumitrescu AV, et al. Subretinal gene therapy of mice with Bardet-Biedl syndrome type 1. Invest Ophthalmol Vis Sci. 2013;54(9):6118–6132. doi: 10.1167/iovs.13-11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beltran WA, Boye SL, Boye SE, et al. rAAV2/5 gene-targeting to rods:dose-dependent efficiency and complications associated with different promoters. Gene Ther. 2010;17(9):1162–1174. doi: 10.1038/gt.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boye SE, Alexander JJ, Boye SL, et al. The human rhodopsin kinase promoter in an AAV5 vector confers rod- and cone-specific expression in the primate retina. Hum Gene Ther. 2012;23(10):1101–1115. doi: 10.1089/hum.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Q, Timmers AM, Guy J, Pang J, Hauswirth WW. Cone-specific expression using a human red opsin promoter in recombinant AAV. Vision Res. 2008;48(3):332–338. doi: 10.1016/j.visres.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 61.Glushakova LG, Timmers AM, Pang J, Teusner JT, Hauswirth WW. Human blue-opsin promoter preferentially targets reporter gene expression to rat s-cone photoreceptors. Invest Ophthalmol Vis Sci. 2006;47(8):3505–3513. doi: 10.1167/iovs.05-1670. [DOI] [PubMed] [Google Scholar]
- 62.Dyka FM, Boye SL, Ryals RC, Chiodo VA, Boye SE, Hauswirth WW. Cone specific promoter for use in gene therapy of retinal degenerative diseases. Adv Exp Med Biol. 2014;801:695–701. doi: 10.1007/978-1-4614-3209-8_87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;383(9923):1129–1137. doi: 10.1016/S0140-6736(13)62117-0. *Seminal Phase I/II study using the AAV vector intraocularly for treatment of choroideremia.
- 64. Dalkara D, Byrne LC, Klimczak RR, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5(189) doi: 10.1126/scitranslmed.3005708. 189ra176. *Directed evolution of AAV mutagenesis leading up to successful intravitreal transduction of foveal cones in NHPs.
- 65.Kay CN, Ryals RC, Aslanidi GV, et al. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS One. 2013;8(4):e62097. doi: 10.1371/journal.pone.0062097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mowat FM, Gornik KR, Dinculescu A, et al. Tyrosine capsid-mutant AAV vectors for gene delivery to the canine retina from a subretinal or intravitreal approach. Gene Ther. 2014;21(1):96–105. doi: 10.1038/gt.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yin L, Greenberg K, Hunter JJ, et al. Intravitreal injection of AAV2 transduces macaque inner retina. Invest Ophthalmol Vis Sci. 2011;52(5):2775–2783. doi: 10.1167/iovs.10-6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dalkara D, Kolstad KD, Caporale N, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17(12):2096–2102. doi: 10.1038/mt.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allocca M, Doria M, Petrillo M, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118(5):1955–1964. doi: 10.1172/JCI34316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18(1):80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grieger JC, Samulski RJ. Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J Virol. 2005;79(15):9933–9944. doi: 10.1128/JVI.79.15.9933-9944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dong B, Nakai H, Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther. 2010;18(1):87–92. doi: 10.1038/mt.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duan D, Yue Y, Engelhardt JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol Ther. 2001;4(4):383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- 74.Yan Z, Zhang Y, Duan D, Engelhardt JF. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc Natl Acad Sci U S A. 2000;97(12):6716–6721. doi: 10.1073/pnas.97.12.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghosh A, Yue Y, Lai Y, Duan D. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol Ther. 2008;16(1):124–130. doi: 10.1038/sj.mt.6300322. [DOI] [PubMed] [Google Scholar]
- 76.Trapani I, Colella P, Sommella A, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. 2014;6(2):194–211. doi: 10.1002/emmm.201302948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Colella P, Trapani I, Cesi G, et al. Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 2014;21(4):450–456. doi: 10.1038/gt.2014.8. [DOI] [PubMed] [Google Scholar]
- 78.Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43C:108–128. doi: 10.1016/j.preteyeres.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Balaggan KS, Binley K, Esapa M, et al. Stable and efficient intraocular gene transfer using pseudotyped EIAV lentiviral vectors. J Gene Med. 2006;8(3):275–285. doi: 10.1002/jgm.845. [DOI] [PubMed] [Google Scholar]
- 80.Mazarakis ND, Azzouz M, Rohll JB, et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum Mol Genet. 2001;10(19):2109–2121. doi: 10.1093/hmg/10.19.2109. [DOI] [PubMed] [Google Scholar]
- 81.O’Rourke JP, Hiraragi H, Urban K, Patel M, Olsen JC, Bunnell BA. Analysis of gene transfer and expression in skeletal muscle using enhanced EIAV lentivirus vectors. Mol Ther. 2003;7(5 Pt 1):632–639. doi: 10.1016/s1525-0016(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 82.O’Rourke JP, Olsen JC, Bunnell BA. Optimization of equine infectious anemia derived vectors for hematopoietic cell lineage gene transfer. Gene Ther. 2005;12(1):22–29. doi: 10.1038/sj.gt.3302350. [DOI] [PubMed] [Google Scholar]
- 83.Palfi S, Gurruchaga JM, Ralph GS, et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet. 2014;383(9923):1138–1146. doi: 10.1016/S0140-6736(13)61939-X. [DOI] [PubMed] [Google Scholar]
- 84.Azzouz M, Martin-Rendon E, Barber RD, et al. Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model of Parkinson’s disease. J Neurosci. 2002;22(23):10302–10312. doi: 10.1523/JNEUROSCI.22-23-10302.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jarraya B, Boulet S, Ralph GS, et al. Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci Transl Med. 2009;1(2) doi: 10.1126/scitranslmed.3000130. 2ra4. [DOI] [PubMed] [Google Scholar]
- 86.Kong J, Kim SR, Binley K, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15(19):1311–1320. doi: 10.1038/gt.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zallocchi M, Binley K, Lad Y, et al. EIAV-based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: development of UshStat. PLoS One. 2014;9(4):e94272. doi: 10.1371/journal.pone.0094272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Binley K, Widdowson P, Loader J, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest Ophthalmol Vis Sci. 2013;54(6):4061–4071. doi: 10.1167/iovs.13-11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bainbridge JW, Stephens C, Parsley K, et al. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene Ther. 2001;8(21):1665–1668. doi: 10.1038/sj.gt.3301574. [DOI] [PubMed] [Google Scholar]
- 90.Puppo A, Cesi G, Marrocco E, et al. Retinal transduction profiles by high-capacity viral vectors. Gene Ther. 2014;21(10):855–865. doi: 10.1038/gt.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lipinski DM, Barnard AR, Charbel Issa P, et al. Vesicular stomatitis virus glycoprotein- and Venezuelan equine encephalitis virus-derived glycoprotein-pseudotyped lentivirus vectors differentially transduce corneal endothelium, trabecular meshwork, and human photoreceptors. Hum Gene Ther. 2014;25(1):50–62. doi: 10.1089/hum.2013.009. [DOI] [PubMed] [Google Scholar]
- 92.Yanez-Munoz RJ, Balaggan KS, MacNeil A, et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. 2006;12(3):348–353. doi: 10.1038/nm1365. [DOI] [PubMed] [Google Scholar]
- 93.Parks RJ, Chen L, Anton M, Sankar U, Rudnicki MA, Graham FL. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci U S A. 1996;93(24):13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bennett J. Immune response following intraocular delivery of recombinant viral vectors. Gene Ther. 2003;10(11):977–982. doi: 10.1038/sj.gt.3302030. [DOI] [PubMed] [Google Scholar]
- 95.Cashman SM, McCullough L, Kumar-Singh R. Improved retinal transduction in vivo and photoreceptor-specific transgene expression using adenovirus vectors with modified penton base. Mol Ther. 2007;15(9):1640–1646. doi: 10.1038/sj.mt.6300203. [DOI] [PubMed] [Google Scholar]
- 96.Sweigard JH, Cashman SM, Kumar-Singh R. Adenovirus vectors targeting distinct cell types in the retina. Invest Ophthalmol Vis Sci. 2010;51(4):2219–2228. doi: 10.1167/iovs.09-4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fink TL, Klepcyk PJ, Oette SM, et al. Plasmid size up to 20 kbp does not limit effective in vivo lung gene transfer using compacted DNA nanoparticles. Gene Ther. 2006;13(13):1048–1051. doi: 10.1038/sj.gt.3302761. [DOI] [PubMed] [Google Scholar]
- 98.Ziady AG, Gedeon CR, Muhammad O, et al. Minimal toxicity of stabilized compacted DNA nanoparticles in the murine lung. Mol Ther. 2003;8(6):948–956. doi: 10.1016/j.ymthe.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 99.Konstan MW, Davis PB, Wagener JS, et al. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther. 2004;15(12):1255–1269. doi: 10.1089/hum.2004.15.1255. [DOI] [PubMed] [Google Scholar]
- 100.Han Z, Conley SM, Makkia R, Guo J, Cooper MJ, Naash MI. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. PLoS One. 2012;7(12):e52189. doi: 10.1371/journal.pone.0052189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yew NS, Zhao H, Wu IH, et al. Reduced inflammatory response to plasmid DNA vectors by elimination and inhibition of immunostimulatory CpG motifs. Mol Ther. 2000;1(3):255–262. doi: 10.1006/mthe.2000.0036. [DOI] [PubMed] [Google Scholar]
- 102.Tsybovsky Y, Molday RS, Palczewski K. The ATP-binding cassette transporter ABCA4: structural and functional properties and role in retinal disease. Adv Exp Med Biol. 2010;703:105–125. doi: 10.1007/978-1-4419-5635-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nagel-Wolfrum K, Baasov T, Wolfrum U. Therapy strategies for Usher syndrome Type 1C in the retina. Adv Exp Med Biol. 2014;801:741–747. doi: 10.1007/978-1-4614-3209-8_93. [DOI] [PubMed] [Google Scholar]
- 104.Kachi S, Binley K, Yokoi K, et al. Equine infectious anemia viral vector-mediated codelivery of endostatin and angiostatin driven by retinal pigmented epithelium-specific VMD2 promoter inhibits choroidal neovascularization. Hum Gene Ther. 2009;20(1):31–39. doi: 10.1089/hum.2008.046. [DOI] [PubMed] [Google Scholar]
- 105.Binley K, Widdowson PS, Kelleher M, et al. Safety and biodistribution of an equine infectious anemia virus-based gene therapy, RetinoStat((R)), for age-related macular degeneration. Hum Gene Ther. 2012;23(9):980–991. doi: 10.1089/hum.2012.008. [DOI] [PubMed] [Google Scholar]
- 106.Shyong MP, Lee FL, Kuo PC, et al. Reduction of experimental diabetic vascular leakage by delivery of angiostatin with a recombinant adeno-associated virus vector. Mol Vis. 2007;13:133–141. [PMC free article] [PubMed] [Google Scholar]
- 107.Raisler BJ, Berns KI, Grant MB, Beliaev D, Hauswirth WW. Adeno-associated virus type-2 expression of pigmented epithelium-derived factor or Kringles 1–3 of angiostatin reduce retinal neovascularization. Proc Natl Acad Sci U S A. 2002;99(13):8909–8914. doi: 10.1073/pnas.122247299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Auricchio A, Behling KC, Maguire AM, et al. Inhibition of retinal neovascularization by intraocular viral-mediated delivery of anti-angiogenic agents. Mol Ther. 2002;6(4):490–494. doi: 10.1006/mthe.2002.0702. [DOI] [PubMed] [Google Scholar]
- 109.May CA, Ohlmann AV, Hammes H, Spandau UH. Proteins with an endostatin-like domain in a mouse model of oxygen-induced retinopathy. Exp Eye Res. 2006;82(2):341–348. doi: 10.1016/j.exer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 110.Parker M, Bellec J, McFarland T, et al. Suppression of neovascularization of donor corneas by transduction with equine infectious anemia virus-based lentiviral vectors expressing endostatin and angiostatin. Hum Gene Ther. 2014;25(5):408–418. doi: 10.1089/hum.2013.079. [DOI] [PubMed] [Google Scholar]
- 111.Pechan P, Rubin H, Lukason M, et al. Novel anti-VEGF chimeric molecules delivered by AAV vectors for inhibition of retinal neovascularization. Gene Ther. 2009;16(1):10–16. doi: 10.1038/gt.2008.115. [DOI] [PubMed] [Google Scholar]
- 112.Maclachlan TK, Lukason M, Collins M, et al. Preclinical safety evaluation of AAV2-sFLT01- a gene therapy for age-related macular degeneration. Mol Ther. 2011;19(2):326–334. doi: 10.1038/mt.2010.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lukason M, DuFresne E, Rubin H, et al. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther. 2011;19(2):260–265. doi: 10.1038/mt.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012;31(3):195–212. doi: 10.1016/j.preteyeres.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vijayasarathy C, Ziccardi L, Sieving PA. Biology of retinoschisin. Adv Exp Med Biol. 2012;723:513–518. doi: 10.1007/978-1-4614-0631-0_64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Park TK, Wu Z, Kjellstrom S, et al. Intravitreal delivery of AAV8 retinoschisin results in cell type-specific gene expression and retinal rescue in the Rs1-KO mouse. Gene Ther. 2009;16(7):916–926. doi: 10.1038/gt.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marangoni D, Wu Z, Wiley HE, et al. Preclinical Safety Evaluation of a Recombinant AAV8 Vector for X-Linked Retinoschisis After Intravitreal Administration in Rabbits. Hum Gene Ther Clin Dev. 2014;25(4):202–211. doi: 10.1089/humc.2014.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kjellstrom S, Bush RA, Zeng Y, Takada Y, Sieving PA. Retinoschisin gene therapy and natural history in the Rs1h–KO mouse: long-term rescue from retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48(8):3837–3845. doi: 10.1167/iovs.07-0203. [DOI] [PubMed] [Google Scholar]
- 119.Larijani B, Hume AN, Tarafder AK, Seabra MC. Multiple factors contribute to inefficient prenylation of Rab27a in Rab prenylation diseases. J Biol Chem. 2003;278(47):46798–46804. doi: 10.1074/jbc.M307799200. [DOI] [PubMed] [Google Scholar]
- 120.Charbel Issa P, Bolz HJ, Ebermann I, Domeier E, Holz FG, Scholl HP. Characterisation of severe rod-cone dystrophy in a consanguineous family with a splice site mutation in the MERTK gene. Br J Ophthalmol. 2009;93(7):920–925. doi: 10.1136/bjo.2008.147397. [DOI] [PubMed] [Google Scholar]
- 121.Mackay DS, Henderson RH, Sergouniotis PI, et al. Novel mutations in MERTK associated with childhood onset rod-cone dystrophy. Mol Vis. 2010;16:369–377. [PMC free article] [PubMed] [Google Scholar]
- 122.Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR. AAV-Mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8(2):188–195. doi: 10.1016/s1525-0016(03)00144-8. [DOI] [PubMed] [Google Scholar]
- 123.Deng WT, Dinculescu A, Li Q, et al. Tyrosine-mutant AAV8 delivery of human MERTK provides long-term retinal preservation in RCS rats. Invest Ophthalmol Vis Sci. 2012;53(4):1895–1904. doi: 10.1167/iovs.11-8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pasadhika S, Fishman GA, Stone EM, et al. Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2010;51(5):2608–2614. doi: 10.1167/iovs.09-3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jacobson SG, Cideciyan AV, Peshenko IV, et al. Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum Mol Genet. 2013;22(1):168–183. doi: 10.1093/hmg/dds421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boye SL, Conlon T, Erger K, et al. Long-term preservation of cone photoreceptors and restoration of cone function by gene therapy in the guanylate cyclase-1 knockout (GC1KO) mouse. Invest Ophthalmol Vis Sci. 2011;52(10):7098–7108. doi: 10.1167/iovs.11-7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Boye SL, Peshenko IV, Huang WC, et al. AAV-mediated gene therapy in the guanylate cyclase (RetGC1/RetGC2) double knockout mouse model of Leber congenital amaurosis. Hum Gene Ther. 2013;24(2):189–202. doi: 10.1089/hum.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cideciyan AV, Aleman TS, Jacobson SG, et al. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum Mutat. 2007;28(11):1074–1083. doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- 129.Cideciyan AV, Rachel RA, Aleman TS, et al. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet. 2011;20(7):1411–1423. doi: 10.1093/hmg/ddr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boye SE, Huang WC, Roman AJ, et al. Natural history of cone disease in the murine model of Leber congenital amaurosis due to CEP290 mutation: determining the timing and expectation of therapy. PLoS One. 2014;9(3):e92928. doi: 10.1371/journal.pone.0092928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Narfstrom K, Holland Deckman K, Menotti-Raymond M. The domestic cat as a large animal model for characterization of disease and therapeutic intervention in hereditary retinal blindness. J Ophthalmol. 2011;2011:906943. doi: 10.1155/2011/906943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mancuso K, Hendrickson AE, Connor TB, Jr, et al. Recombinant adeno-associated virus targets passenger gene expression to cones in primate retina. J Opt Soc Am A Opt Image Sci Vis. 2007;24(5):1411–1416. doi: 10.1364/josaa.24.001411. [DOI] [PubMed] [Google Scholar]
- 133.Yang P, Michaels KV, Courtney RJ, et al. Retinal morphology of patients with achromatopsia during early childhood: implications for gene therapy. JAMA Ophthalmol. 2014;132(7):823–831. doi: 10.1001/jamaophthalmol.2014.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aboshiha J, Dubis AM, Cowing J, et al. A prospective longitudinal study of retinal structure and function in achromatopsia. Invest Ophthalmol Vis Sci. 2014;55(9):5733–5743. doi: 10.1167/iovs.14-14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dubis AM, Cooper RF, Aboshiha J, et al. Genotype-dependent variability in residual cone structure in achromatopsia: toward developing metrics for assessing cone health. Invest Ophthalmol Vis Sci. 2014;55(11):7303–7311. doi: 10.1167/iovs.14-14225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Komaromy AM, Alexander JJ, Rowlan JS, et al. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19(13):2581–2593. doi: 10.1093/hmg/ddq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Michalakis S, Muhlfriedel R, Tanimoto N, et al. Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. 2010;18(12):2057–2063. doi: 10.1038/mt.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pang JJ, Deng WT, Dai X, et al. AAV-mediated cone rescue in a naturally occurring mouse model of CNGA3-achromatopsia. PLoS One. 2012;7(4):e35250. doi: 10.1371/journal.pone.0035250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ezra-Elia R, Banin E, Honig H, et al. Flicker cone function in normal and day blind sheep: a large animal model for human achromatopsia caused by CNGA3 mutation. Doc Ophthalmol. 2014;129(3):141–150. doi: 10.1007/s10633-014-9458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Raghupathy RK, McCulloch DL, Akhtar S, Al-Mubrad TM, Shu X. Pathogenesis of X-linked RP3: insights from animal models. Adv Exp Med Biol. 2014;801:477–485. doi: 10.1007/978-1-4614-3209-8_61. [DOI] [PubMed] [Google Scholar]
- 141.Beltran WA, Cideciyan AV, Lewin AS, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012;109(6):2132–2137. doi: 10.1073/pnas.1118847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Millington-Ward S, Chadderton N, O’Reilly M, et al. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol Ther. 2011;19(4):642–649. doi: 10.1038/mt.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Leveillard T, Mohand-Said S, Lorentz O, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36(7):755–759. doi: 10.1038/ng1386. [DOI] [PubMed] [Google Scholar]
- 144.Yang Y, Mohand-Said S, Danan A, et al. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther. 2009;17(5):787–795. doi: 10.1038/mt.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Byrne LC, Dalkara D, Luna G, et al. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J Clin Invest. 2014 doi: 10.1172/JCI65654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.G N, Tan A, Farhatnia Y, et al. Channelrhodopsins: visual regeneration and neural activation by a light switch. N Biotechnol. 2013;30(5):461–474. doi: 10.1016/j.nbt.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mutter M, Swietek N, Munch TA. Salvaging ruins: reverting blind retinas into functional visual sensors. Methods Mol Biol. 2014;1148:149–160. doi: 10.1007/978-1-4939-0470-9_10. [DOI] [PubMed] [Google Scholar]
- 148.Bi A, Cui J, Ma YP, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron. 2006;50(1):23–33. doi: 10.1016/j.neuron.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thyagarajan S, van Wyk M, Lehmann K, Lowel S, Feng G, Wassle H. Visual function in mice with photoreceptor degeneration and transgenic expression of channelrhodopsin 2 in ganglion cells. J Neurosci. 2010;30(26):8745–8758. doi: 10.1523/JNEUROSCI.4417-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rolling F. Recombinant AAV-mediated gene transfer to the retina: gene therapy perspectives. Gene Ther. 2004;11(Suppl 1):S26–S32. doi: 10.1038/sj.gt.3302366. [DOI] [PubMed] [Google Scholar]
- 151.Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374(9701):1597–1605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18(3):643–650. doi: 10.1038/mt.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ashtari M, Cyckowski LL, Monroe JF, et al. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest. 2011;121(6):2160–2168. doi: 10.1172/JCI57377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4(120) doi: 10.1126/scitranslmed.3002865. 120ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Testa F, Maguire AM, Rossi S, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013;120(6):1283–1291. doi: 10.1016/j.ophtha.2012.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Stout T, Weleber R, McBride M, et al. Treatment of patients with Leber congenital amaurosis type 2 with an AAV vector expressing RPE65. Invest Ophthalmol Vis Sci. 2013;54 E-Abstract 5964. [Google Scholar]
- 157.Banin E, Bandah-Rozenfeld D, Obolensky A, et al. Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum Gene Ther. 2010;21(12):1749–1757. doi: 10.1089/hum.2010.047. [DOI] [PubMed] [Google Scholar]
- 158.Le Meur G, Lebranchu P, Pereon Y, et al. Efficacy and safety of gene therapy with AAV4 in childhood blindness due to rpe65 mutations. Acta Ophthalmologica. 2012;90(Supplement S249):0. [Google Scholar]
- 159.Tolmachova T, Tolmachov OE, Barnard AR, et al. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med (Berl) 2013;91(7):825–837. doi: 10.1007/s00109-013-1006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Black A, Vasireddy V, Chung DC, et al. Adeno-associated virus 8-mediated gene therapy for choroideremia: preclinical studies in in vitro and in vivo models. J Gene Med. 2014;16(5–6):122–130. doi: 10.1002/jgm.2768. [DOI] [PubMed] [Google Scholar]
- 161.Carvalho LS, Xu J, Pearson RA, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. 2011;20(16):3161–3175. doi: 10.1093/hmg/ddr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mihelec M, Pearson RA, Robbie SJ, et al. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum Gene Ther. 2012;22(10):1179–1190. doi: 10.1089/hum.2011.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 Gene Delivery to an Adult Rs1h Knockout Mouse Model Restores ERG b-Wave with Reversal of the Electronegative Waveform of X-Linked Retinoschisis. Invest Ophthalmol Vis Sci. 2004;45(9):3279–3285. doi: 10.1167/iovs.04-0576. [DOI] [PubMed] [Google Scholar]
- 164.Takada Y, Vijayasarathy C, Zeng Y, Kjellstrom S, Bush RA, Sieving PA. Synaptic pathology in retinoschisis knockout (Rs1-/y) mouse retina and modification by rAAV-Rs1 gene delivery. Invest Ophthalmol Vis Sci. 2008;49(8):3677–3686. doi: 10.1167/iovs.07-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 165.ClinicalTrials.gov Home Page. [Accessed 07 March 2015];U.S. National Institutes of Health. https://ClinicalTrials.gov.
- 166.AGTC. [Accessed 08 March 2015]; http://www.agtc.com/products/achromatopsia.
- 167.Genable. [Accessed 08 March 2015]; http://www.genable.net/genable-technology.
- 168.CHM Gene Therapy at University of Alberta. [Accessed 08 March 2015]; http://www.chmgenetherapy.ca/
- 169.Spark Therapeutics. [Accessed 08 March 2015]; http://www.sparktx.com/pipeline/inherited-retinal-dystrophies.
- 170.Genzyme. [Accessed 08 March 2015]; http://news.genzyme.com/press-release/genzyme-collaborates-gene-therapy-rare-disease-causes-childhood-blindness
- 171.AGTC. [Accessed 08 March 2015]; http://www.agtc.com/products/x-linked-retinitis-pigmentosa.
- 172.AGTC. [Accessed 08 March 2015]; http://www.agtc.com/products/x-Linked-retinoschisis.