

 PRMT inhibitors can compete with cofactor, substrate, or bind at allosteric sites found in the active or inactive states.
PRMT inhibitors can compete with cofactor, substrate, or bind at allosteric sites found in the active or inactive states.
Abstract
Protein arginine methyltransferases (PRMTs), an emerging target class in drug discovery, can methylate histones and other substrates, and can be divided into three subgroups, based on the methylation pattern of the reaction product (monomethylation, symmetrical or asymmetrical dimethylation). Here, we review the growing body of structural information characterizing this protein family, including structures in complex with substrate-competitive and allosteric inhibitors. We outline structural differences between type I, II and III enzymes and propose a model underlying class-specificity. We analyze the structural plasticity and diversity of the substrate, cofactor and allosteric binding sites, and propose that the conformational dynamics of PRMTs can be exploited towards the discovery of allosteric inhibitors that would antagonize conformationally active states.
Introduction
PRMTs catalyze the transfer of a methyl group from the cofactor S-adenosylmethionine (SAM) to the guanidinium group of arginine side-chains on nuclear and cytoplasmic proteins including histones. There are nine known PRMTs in humans, but the large number of putative methyltransferases suggests that this number may increase.1,2 PRMTs can be divided into three classes: Type I (PRMT1, the major Type I PRMT, PRMT2, PRMT3, CARM1/PRMT4, PRMT6, and PRMT8) can mono- and asymmetrically di-methylate arginines; Type II (PRMT5) mono- and symmetrically di-methylate their substrates, and Type III (PRMT7) only mono-methylate the guanidinium group.3 PRMT substrates are involved in multiple and diverse cellular mechanisms such as chromatin mediated signaling (PRMT1, PRMT2, CARM1, PRMT5, PRMT6 and PRMT7 methylate histone tails4,5), the ribosomal machinery (PRMT3 methylates the 40S ribosomal protein S2 (ref. 6)), and DNA repair or mRNA splicing7; PRMTs affect cell growth, proliferation and differentiation,5 can be deregulated in cancer and other disease conditions, and are emerging as potential therapeutic targets.8–10
Domain architecture
The nine human PRMTs are organized around a conserved catalytic core composed of a Rossman fold, where the cofactor binds, and a β-barrel that contributes to substrate binding11 (Fig. 1A). Protein–protein interaction modules are sometimes observed at the N-terminus of the PRMT core. These include an SH3 domain in PRMT2, a zinc finger in PRMT3, a pleckstrin homology (PH) domain in CARM1,12 and a TIM barrel in PRMT5.13 It is expected that these structural domains participate in substrate binding, recruit other proteins that are necessary for the formation of a catalytically active complex, or contribute to the homo-oligomerization of PRMT subunits. For instance, the TIM barrel of PRMT5 interacts with MEP50, a binding partner that is necessary for full PRMT5 catalytic activity, and largely contributes to the homo-tetramerization of the enzyme (discussed below).13
Fig. 1. Structural overview of Type I PRMTs. (A) Domain architecture of human PRMTs. The C-terminal Rossman fold and β-barrel of PRMT7, shown in lighter colors, have low sequence similarity with template sequences, and are catalytically inactive. Dashed lines indicate putative structural elements for which no structural data is available. (B) Canonical dimeric arrangement of Type I PRMTs, illustrated by the structure of CARM1 (PDB code 3B3F). In CePRMT7, the C-terminal catalytic core replaces the second subunit. (C) Connolly surface representation of CARM1 colored by electrostatic potential highlighting the cofactor and substrate binding sites. (D) Schematic representation illustrating the structural mechanism of Type I PRMTs: folding of the α-helix on the cofactor completes the formation of the substrate binding site.

Structural overview of Type I and III PRMTs
Class I PRMT structures solved to date adopt a head-to-tail dimeric arrangement, where a dimerization arm projecting off of the β-barrel interacts with the Rossman fold of another subunit (Fig. 1B).12,14–18 A dynamic α-helix at the N-terminus of the Rossman fold also contributes to inter-subunit contacts in the active state of rCARM1.12 This important and conserved structural element is composed of 2 segments, α-X and α-Y that fold onto the bound cofactor, and participates in recruitment and proper positioning of the substrate peptide. As a result, the cofactor becomes buried in the core of the Rossman fold, and a substrate-binding site at the boundary of the Rossman fold, the β-barrel and the α-helix becomes clearly delineated (Fig. 1C). This suggests a mechanism where cofactor binding stabilizes a conformationally active state of the α-helix (Fig. 1D). Cofactor-binding alone may however not be sufficient as the α-helix was captured in a great diversity of conformational states in holo-structures of PRMT1, PRMT3, CARM1, and PRMT6.12,18,19
While no structure of mammalian PRMT7 was reported, the CePRMT7 structure reveals a variation on the head-to-tail dimerization theme observed in Type I PRMTs:14 the N-terminal catalytic core composed of a Rossman fold and a β-barrel adopts a canonical PRMT structure, but a quaternary arrangement places a C-terminal pseudo-catalytic core that is unique to PRMT7 in a position that mimics the second subunit in Type I PRMTs and in TbPRMT7.14,16 An important pair of glutamic acids that are conserved at the catalytic sites of active PRMTs (detailed later) is missing from the C-terminal CePRMT7 core, and a loop partially occludes the cofactor pocket.
In depth structural analysis of rCARM1 by Troffer-Charlier demonstrated a profound structural rearrangement of the conserved Type I α-helix, which flips by 180° and partially converts into a β-strand in the apo conformation12 (Fig. 2A and B). This dramatic motion should re-position the PH domain located immediately at the N-terminus of the α-helix to the other side of the CARM1 dimer, which probably necessitates dissociation of the dimer, and suggests a regulatory mechanism whereby cofactor binding and enzymatic activity correlate with positioning of the PH domain and its binding partners relative to the CARM1 catalytic core (Fig. 2B).
Fig. 2. Structural dynamics of the Type I α-helix. The α-helix observed in Type I PRMTs is structurally dynamic and can adopt strikingly divergent conformations in the active (A – PDB: 3B3F) and inactive (B – PDB: ; 3B3J) states, correlating with distinct positioning of distant structural modules, such as the PH domain of CARM1 (shown), the SH3 domain of PRMT2, or the ZnF domain of PRMT3, relative to the catalytic domain. (C) Substrate specificity of Rossman-fold methyltransferases may rely on the nature of a regulatory element (red; inactive state: blue) N-terminal to the conserved Rossman fold (yellow). PDB codes: active CARM1: ; 3B3F; inactive CARM1: ; 3B3J; PRMT5: ; 4GQB; active DOT1L: ; 1NW3; inactive DOT1L: ; 4ER5; DNMT1: ; 3PTA; NSUN4: ; 4FP9; COMT: ; 1VID. (C is inspired from ref. 40).

A catalytic Rossman fold is also observed in non-SET domain lysine methyltransferases,20 DNA, RNA and small-molecule methyltransferases21–23 (Fig. 2C). In all these structures, a secondary element located at the N- or C-terminus of the Rossman fold and positioned in the 3D space at the site of PRMTs' α-helix contributes to cofactor binding. This element is an α-helix in Type I and III PRMTs, as well as in DNMTs, and a loop in small-molecule, lysine, RNA methyltransferases, and PRMT5 (Fig. 2C). The structural plasticity of the corresponding loop was demonstrated in the case of lysine methyltransferase DOT1L: the size and hydrophobicity of the cofactor binding pocket are significantly increased when the loop adopts a conformationally inactive state, which is captured in complex with pico-molar small molecule inhibitors that compete with the cofactor.24,25 The fact that the structure of this dynamic element varies with the nature of the substrate (Fig. 2C), and its proximity to the site of methyl transfer, supports the idea that it plays an important role in substrate recognition.
Structural features unique to PRMT5
A recent crystal structure of PRMT5 revealed that the core Rossman fold and β-barrel were conserved in this Type II methyltransferase (Fig. 3A and B) but, in contrast with the α-X and α-Y helices observed in Type I enzymes, PRMT5 harbors a short α-Y helix preceded by a large loop that links the catalytic Rossman fold to an N-terminal, PRMT5-specific TIM-barrel.13 The TIM-barrel in turn binds MEP50, a WD-repeat protein that is a core component of PRMT5-containing chromatin complexes and acts as a structural scaffold for the recruitment of substrates13 (Fig. 3B). The loose secondary structural arrangement of the linker domain suggests that it corresponds to a structurally variable region, as observed in the corresponding α-helix of Type I PRMTs.
Fig. 3. Structural overview of PRMT5. (A) The structure core of CARM1 is shown as a reference [PDB: 3B3F]. (B) PRMT5 monomers have a conserved catalytic core composed of a Rossman fold and a β-barrel; specific structural features include a TIM barrel that recruits MEP50, and a linker region that contributes to cofactor and substrate binding. Interactions between the TIM barrel and Rossman fold + linker mediate dimerization; orthogonal stacking of dimers produces the PRMT5 tetramer [; 4GQB].

It is unclear whether the α-helix of Type I PRMTs interacts with residues of the target peptide other than the substrate arginine,16 but the corresponding linker segment of PRMT5 makes extensive interactions with the backbone and side-chains of at least 8 histone residues centered on the substrate arginine (Fig. 3B).13 Three tyrosine side-chains from the PRMT5 linker (Y297, Y304 and Y307) are phosphorylated by an oncogenic mutant of the kinase JAK2, resulting in impaired methyltransferase activity.26 Two of these (Y304 and Y307) are making direct interactions with the substrate histone, which would be disrupted by phosphorylation (Fig. 3B), and phosphorylation of the third would also dramatically affect the electrostatics and geometry of the substrate binding site. In a related mechanism, phosphorylation of Ser217 in CARM1 abrogates its interaction with Tyr154 of the α-X helix, and prevents locking of the α-helix in a conformationally active state.27 Both Tyr154 and Ser217 are conserved in Type I PRMTs, suggesting that phosphorylation affecting structural elements at the catalytic site may be a common mechanism to regulate the activity of PRMTs.
In addition to the presence of a TIM barrel and a linker region, another PRMT5-specific feature is its tetrameric form. Dimerization is not mediated by a dimerization arm, as observed in Type I PRMTs, and in CePRMT5,28 but is stabilized by extensive interactions between the TIM barrel of one subunit and the Rossman fold and linker of another. Orthogonal stacking of two dimers generates a tetrameric arrangement at the heart of which lies the substrate binding site, while MEP50 is recruited at the periphery (Fig. 3B). Electron microscopy analysis conducted on XlPRMT5 indicates that the bulk of the interaction with nucleoplasmin, a PRMT5 substrate, is taking place with MEP50 rather than PRMT5 itself, and it was proposed that a channel linking MEP50 to the substrate arginine binding pocket may guide the peptide substrate to the site of methyl transfer.29 The PRMT5–MEP50 complex has about fifty times stronger affinity than PRMT5 for a substrate H4 peptide,13 supporting the idea that MEP50 contributes to the recruitment of diverse PRMT5 substrates.
The fact that MEP50 – a core component of PRMT5 complexes necessary for full PRMT5 activity – interacts mainly with the TIM barrel of PRMT5, the position of which is tightly correlated with the conformation of the linker segment, suggests a model where interactions at the TIM barrel would dictate the activation state of the linker domain (α-helix in CARM1). This would echo a structural mechanism proposed for CARM1 where protein–protein interactions at the PH domain would dictate the conformational state of the α-helix and control enzymatic activity. Further structural analysis will be necessary to support or disprove this model.
Structural determinants of product specificity
Structural determinants underlying asymmetrical dimethylation by Type I PRMTs, symmetrical dimethylation by Type II PRMTs and monomethylation by Type III enzymes have not been clearly established, but recent structures of PRMT5 and CePRMT7 in complex with a substrate arginine as well as CARM1 in complex with potent inhibitors occupying the substrate arginine site are providing important insights.12,13,16 A first observation is that mutating F327 to methionine turns PRMT5 into a dual Type I and Type II PRMT,28 suggesting that the catalytic mechanism is conserved between the two PRMT classes, and relies on the only two catalytic site residues that are absolutely conserved across all PRMTs: a pair of glutamates from the Rossman fold “double-E loop” that is probably involved in deprotonating and activating the methyl-accepting nitrogen.13,18 If Type I, II and III enzymes share the same catalytic mechanism, steric factors are probably responsible for product specificity. While F327 stacks against the substrate guanidinium group in the PRMT5 structure and holds the substrate arginine tightly in place, a more flexible methionine, conserved at this position in Type I enzymes (M163 in CARM1), probably allows sufficient spatial adjustment of the substrate arginine to accommodate two methyl groups on the same nitrogen, though the efficiency of asymmetrical di-methylation seems limited28 (Fig. 4).
Fig. 4. Structural determinants for product specificity of Type I, II and III PRMTs. Type I and II PRMTs asymmetrically and symmetrically dimethylate arginine side-chains respectively, while Type III PRMTs mono-methylate only. The conserved “double-E loop” glutamates are shown, along with residues dictating class specificity in the crystal structures of CARM1 [PDB: 2Y1X], PRMT5 [; 4GQB] and TbPRMT7 [; 4M38]. The substrate arginine (positioned manually in the case of CARM1 by superimposing the guanidinium group on the alanine moiety of a co-crystallized inhibitor [; 2Y1X]) is shown in magenta. Orange: residues from the α-helix of CARM1 and TbPRMT7, and from the linker region of PRMT5.

In Type I PRMTs, a conserved YFxxY motif (Y150, F151 and Y154 in CARM1) adopts a particular conformation where the 3 aromatic rings stack in an orthogonal fashion with each other and with the adenine ring of the cofactor; this stabilizes a conformational state where the two hydroxyl groups of the tyrosine side-chains form hydrogen-bonds with one of the conserved glutamates of the double-E loop (E267 in CARM1). The other carboxylic oxygen of the glutamate forms a hydrogen bond with the substrate guanidinium. This structural arrangement has two consequences: (1) it locks in place the substrate arginine, and (2) it generates a large Type I-specific volume surrounding the methyl-accepting nitrogen of the arginine substrate, compatible with binding of unmethylated arginine for monomethylation, or monomethylated arginine for asymmetrical dimethylation. This is in contrast with the tight environment of the other ω-nitrogen of the arginine substrate, which is buttressed against the side-chain of M163 and forms hydrogen-bonds with the side-chains of H415 and the second “double E-loop” glutamate (E258), leaving no room for deposition of a methyl group. Importantly, all these residues are conserved in Type I PRMTs. In summary, the substrate arginine of CARM1 is locked in place by three hydrogen-bonds with the double-E loop glutamates (E258, E267), as well as H415, and by steric constraints from M16317. This arrangement is further stabilized by the YFxxY motif (Y150, F151 and Y154) that stacks against the cofactor and denies any rotational freedom at E267. Importantly, these residues are conserved exclusively in Type I PRMTs, and four of them are part of the dynamic α-helix that adopts distinct conformations in the active and inactive states (Fig. 4).
The situation is very different in PRMT5: while a large volume is observed at the methyl-accepting nitrogen in CARM1, this volume is closed in PRMT5 due to a difference in the relative orientation of the substrate guanidinium group and E444 (E267 in CARM1).28 Methylation of this nitrogen would preclude its repositioning at the same site for asymmetrical dimethylation. Instead, extra volume is present on the other side of the guanidinium group, due to the replacement of a histidine group in Type I enzymes (H415 in CARM1) with a serine group (S578 in PRMT5). This volume can accommodate a monomethylated N-ω-nitrogen, accompanied by placement of the unmethylated N′-ω-nitrogen at the site of methyl transfer, an arrangement that is in agreement with symmetrical dimethylation (Fig. 4).
Finally, in TbPRMT7, the orientation of the guanidinium group relative to E181 is similar to that observed in PRMT5 (relative to E444), which antagonizes asymmetrical dimethylation. Hydrogen bonding of the N′-ω-nitrogen with Q329 recapitulates the effect of H415 in CARM1 and prevents symmetrical dimethylation16 (Fig. 4). Further supporting this model, the Q329H TbPRMT7 mutant retains Type III specificity.16 The fact that the Q329S mutant largely loses catalytic activity suggests that the substrate arginine is no longer stabilized in a conformation where it aligns with the departing methyl, maybe due to the replacement of F327, which is tightly stacked against the substrate guanidinium group in PRMT5, with a more permissive methionine (M75) in PRMT7.
In summary, we propose a model where variations between Type I, Type II and Type III PRMTs in spatial constraints and in hydrogen-bonds at the substrate N- and N′-ω-nitrogens are controlled by class-specific combinations of residues (M163, H415, Y150, F151, and Y154 in CARM1, S578 and F327 in PRMT5, Q329 and M75 in TbPRMT7) that dictate product specificity. An open question is whether these class-specific features could be exploited to design class-specific PRMT inhibitors.
Structural diversity of cofactor and substrate binding sites
The α-helix of Type I PRMTs and the linker domain of PRMT5 fold on the cofactor and participate in the formation of the substrate binding site (Fig. 5A and B). The cofactor site is entirely buried and the substrate site is more open, but sufficiently enclosed to be targeted by potent inhibitors. For instance, the substrate-binding pocket of CARM1 has been co-crystallized with 30 nM inhibitors (Fig. 5A).30 Bisubstrate inhibitors composed of cofactor analogs that extend into the methyl-arginine binding channel have also been described,31 but these extremely polar compounds are not drug-like and reflect the challenge of designing cofactor competitors that are sufficiently polar to have high affinity for the hydrophilic cofactor binding site, but sufficiently hydrophobic to cross cell membranes. An important question is whether the structural diversity of these sites is sufficient to develop highly selective chemical probes that could be used as tools for target validation and may serve as the starting point for drug discovery.32,33
Fig. 5. Structural plasticity of the cofactor and substrate binding sites. The conformationally dynamic α-helix of CARM1 [PDB code 2Y1W] (A) and the corresponding linker domain of PRMT5 [; 4GQB] (B) contribute to the formation of both cofactor and substrate binding pockets. The cofactor binding pocket is absent in the inactive state of CARM1 [; 3B3J] (C), and a novel pocket is formed at the interface of the α-Y helix and the structure core.

When clustering methyltransferases based on the structural diversity of their cofactor site using the atomic property fields method,34 SET domain protein methyltransferases form a cluster of their own, while PRMTs are grouped with small molecule-, DNA- and RNA-methyltransferases (Fig. 6A). This reflects a risk of off-target activity when targeting the cofactor site of PRMTs. This is in agreement with the fact that, unlike SET domain protein methyltransferases, PRMTs share a cofactor-binding Rossman fold with non-protein methyltransferases (Fig. 2C). Previous work indicated that the structural diversity of the cofactor binding pocket of protein methyltransferases is similar to that of the ATP binding pocket of kinases.35 Since it is generally possible to develop selective kinase inhibitors, the same should apply to protein methyltransferases in general, and to PRMTs in particular. Alignment of residues lining the cofactor site of Type I PRMTs shows that the pocket is generally conserved, but highlights a few variable positions, such as F138, M164 or A216 (CARM1 nomenclature) that may be exploited to develop isoform-selective inhibitors (Fig. 6B). Nevertheless, the relatively high sequence conservation at the cofactor site should represent a challenge for drug design.
Fig. 6. Structural diversity of the cofactor and substrate binding pockets. (A) Clustering of human methyltransferases in the protein databank based on the structural similarity of their cofactor site (Molsoft's APF method was used34). (B) Side-chains within 5 Å of the cofactor in the CARM1 structure (PDB code ; 3B3F) were extracted from a multiple sequence alignment of Type I PRMTs. Sequence conservation at these positions was used for color-coding. Conserved residues forming direct hydrogen-bonds with the cofactor are marked with an “*”. Positions with significant sequence variability are highlighted in magenta (CARM1 numbering). (C) The same procedure as in (B) was applied to the substrate binding pocket, defined by side-chains within 5 Å of 2 substrate competitors co-crystallized to CARM1 (PDB codes ; 2Y1W and ; 2Y1X). (PNMT: phenylethanolamine N-methyltransferase).

The situation is a little more favorable at the substrate-binding site. A multiple sequence alignment of residues lining potent CARM1 inhibitors that occupy the substrate binding pocket30 shows high sequence conservation at the substrate arginine channel, but significant diversity at the other end of the pocket, at positions occupied by S146, Q149, N266, R446 or S448 in CARM1 (Fig. 6C). The fact that the two 30 nM CARM1 inhibitors are inactive against PRMT1 and PRMT3 (ref. 36 and 37) supports the idea that selective inhibition can be achieved at the substrate binding site, though profiling these inhibitors against other PRMTs would be informative. Interestingly, different warheads in each inhibitor are anchored in the methyl-arginine binding channel of CARM1, but in both cases, hydrogen bonds are formed with the side-chains of H415 and the double E-loop glutamate E258, two residues that are absolutely conserved in Type I PRMTs, as well as the backbone carbonyl of M260 (Fig. 6C). This suggests that potent Type I PRMT inhibitors binding at the substrate-binding pocket should efficiently exploit these positions. Considering the high sequence conservation at the methyl-arginine channel, combinatorial libraries focused on these two warheads should be enriched in Type I PRMT inhibitors.
Allosteric inhibition of PRMT3
Compounds from a third chemical series were co-crystallized at an allosteric pocket of PRMT3, located at the base of the dimerization arm (Fig. 7A).15,38 In spite of their binding mode, the compounds do not prevent homodimerization of PRMT3. This unexpected binding site is 15 Å away from the site of methyl transfer, and it is surprising that binding at such a distant pocket could have an inhibitory effect, but the mechanistic relevance of the crystal structure was confirmed by mutagenesis studies showing that mutating residues at the allosteric site (ex K392A) preserved the catalytic activity of the enzyme, but reduced the effect of the inhibitor (Fig. 7B).15 The structure clearly shows that the benzothiadiazole moiety of the compound is inserted in an enclosed cavity of the β-barrel of a PRMT3 subunit, while the 2-oxo-phenyl end of the inhibitor extends out of the cavity and is buttressed against the α-helix of the other PRMT3 subunit (Fig. 7B). Interestingly, the α-helix is in an inactive state, suggesting that the inhibitor induces some conformational strain that antagonizes proper folding of the α-X helix on the cofactor. While the reported inhibitors only have IC50s in the low micromolar range, it is expected that potent compounds could be targeting this site.
Fig. 7. Allosteric inhibition of PRMT3. (A and B) An allosteric PRMT3 inhibitor binds in a cavity of the β-barrel, at the base of the dimerization arm, and is buttressed against the α-helix (orange) of the other PRMT3 subunit. The cofactor-binding pocket is highlighted by a cofactor molecule (blue), and the substrate binding pocket by a substrate competitor (magenta) co-crystallized to CARM1 (superimposed CARM1 structure not shown). Both the cofactor and CARM1 inhibitor are shown as references, but are absent from the PRMT3 structure. (C) Entrance to the allosteric pocket is occluded by R396 in the absence of the inhibitor. (D) Sequence diversity of residues lining the allosteric pocket (constructed as in Fig. 6).

The entrance of the pocket is occluded in the structure of PRMT3 in complex with the cofactor, due to an alternate rotameric state of R396 (PDB code 2FYT) (Fig. 7C). The fact that the pocket is not seen in other PRMT structures does therefore not necessarily mean that it is unique to PRMT3. A multiple sequence alignment of Type I PRMTs based on PRMT3 residues lining the co-crystallized inhibitor reveals significant sequence variability, indicating that this pocket can be exploited towards the development of selective inhibitors (Fig. 7D).
Unexplored allosteric binding pockets
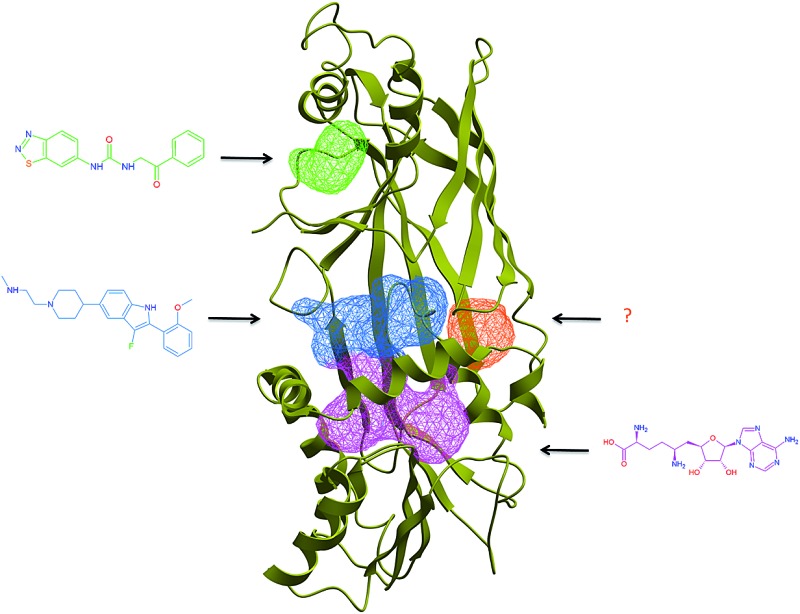
The dynamic nature of the α-helix of Type I PRMTs translates into significant structural plasticity at both cofactor and substrate binding sites. In the structure of apo CARM1, the cofactor binding pocket is gone, and a profound remodeling takes place at what used to be the substrate binding site, where an entirely new cavity emerges from the relocation of the α-helix (Fig. 5C and 8). Compounds that occupy this site would presumably lock the enzyme in an inactive conformation.
Fig. 8. The PRMT pocketome. ICMPocketFinder (Molsoft, San Diego) was used to map pockets present in PRMT structures. Blue: merged cofactor and substrate binding pockets. Green: experimentally validated PRMT3 allosteric pocket. Orange: unexplored allosteric pocket observed in PRMT1, PRMT3 and PRMT6. Red: novel CARM1 pocket formed by the inactive α-helix. Light blue: pocket formed at the interface of the C-terminal β-barrel and pseudo-dimerization arm of CePRMT7 (C-terminal pseudo-PRMT core shown in white). PDB codes: PRMT1: 1OR8; PRMT3: ; 3SMQ; CARM1 (active): ; 2Y1W; CARM1 (inactive): ; 3B3J; PRMT5: ; 4GQB; PRMT6: ; 4HC4; CePRMT7: ; 3WSS. Red: α-helix of Type I/III PRMTs and linker domain of PRMT5.

In an attempt to identify other potential allosteric binding sites, we used Molsoft's ICMPocketFinder39 to systematically search for binding pockets in PRMT structures available in the PDB (Fig. 8). In addition to the substrate and cofactor binding sites, and the PRMT3 allosteric site, this exercise reveals the existence of unexplored binding pockets. First, a cavity present in PRMT1, PRMT3, PRMT6 and apo-CARM1 is located at the interface of the Rossman fold and the β-barrel, immediately next to the second double-E loop glutamate (E355 in PRMT3). The entrance to this cavity is occupied by the α-helix in the active conformation of CARM1, suggesting a model where inhibitors occupying this site would destabilize the catalytically competent conformation of the α-helix. Another pocket is identified at the interface of the linker domain and the TIM-barrel of PRMT5. It is located in the vicinity of the substrate-binding site, and compounds binding at this site may affect recruitment of PRMT5 substrates. A large cavity is also present at the junction of the C-terminal β-barrel and the pseudo-dimerization arm of CePRMT7, immediately next to the N-terminal catalytic site (Fig. 8, light blue). However, CePRMT7 shares only 32% sequence identity with hPRMT7, and the corresponding pocket may not be present in the latter. Finally, a pocket is found at the back of the β-barrel of inactive CARM1 (Fig. 8, purple). However, unlike the pocket targeted by PRMT3 allosteric inhibitors, this cavity is facing away from the other subunits, and the putative effect of ligands binding at this site is unclear.
Conclusion
PRMTs share a catalytic Rossman fold with DNA-, RNA-, and small molecule-methyltransferases, as well as some lysine protein methyltransferases, and novel enzymes with PRMT activity may emerge from the human methyltransferasome.1 We propose a model where steric constraints dictate class (i.e. product) specificity of PRMTs. Structural determinants for substrate specificity, another dimension to the signaling versatility of PRMTs, remain largely unexplored, and more structures such as the recent PRMT5- or TbPRMT7-histone 4 complexes will be important to address this question. Another area for future research is the influence of protein–protein interaction modules such as the PH domain of CARM1, or the Zn finger of PRMT3 on the activation state of PRMTs. This may necessitate the introduction of additional components of chromatin complexes, as was done in the case of PRMT5.13 The discovery and co-crystal structure of potent CARM1 inhibitors demonstrated that the substrate-binding pocket of PRMTs is chemically tractable. The cofactor-binding site is also very enclosed, but low diversity and high hydrophilicity make it a challenging target. More promising is the opportunity to develop allosteric inhibitors that exploit the structural plasticity of PRMTs, as was demonstrated in the case of PRMT3.15,38
Acknowledgments
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, Genome Canada through the Ontario Genomics Institute [OGI-055], GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z].
References
- Petrossian T. C., Clarke S. G. Mol. Cell. Proteomics. 2011;10:M110000976. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon V. M., Johnston D., Sneeringer C. J., Jin L., Majer C. R., Elliston K., Jerva L. F., Scott M. P., Copeland R. A. Chem. Biol. Drug Des. 2011;78:199–210. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- Zurita-Lopez C. I., Sandberg T., Kelly R., Clarke S. G. J. Biol. Chem. 2012;287:7859–7870. doi: 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A., Bedford M. T. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Bedford M. T. Nat. Rev. Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Swiercz R., Cheng D., Kim D., Bedford M. T. J. Biol. Chem. 2007;282:16917–16923. doi: 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- Bedford M. T., Clarke S. G. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B., Jho E. H. Expert Opin. Ther. Targets. 2012;16:651–664. doi: 10.1517/14728222.2012.688030. [DOI] [PubMed] [Google Scholar]
- Copeland R. A., Solomon M. E., Richon V. M. Nat. Rev. Drug Discovery. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- Krause C. D., Yang Z. H., Kim Y. S., Lee J. H., Cook J. R., Pestka S. Pharmacol. Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Cheng X., Collins R. E., Zhang X. Annu. Rev. Biophys. Biomol. Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troffer-Charlier N., Cura V., Hassenboehler P., Moras D., Cavarelli J. EMBO J. 2007;26:4391–4401. doi: 10.1038/sj.emboj.7601855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S., Bonday Z., Campbell R. M., Doyle B., Druzina Z., Gheyi T., Han B., Jungheim L. N., Qian Y., Rauch C., Russell M., Sauder J. M., Wasserman S. R., Weichert K., Willard F. S., Zhang A., Emtage S. Proc. Natl. Acad. Sci. U. S. A. 2012;109:17960–17965. doi: 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Toma-Fukai S., Kim J. D., Fukamizu A., Shimizu T. FEBS Lett. 2014;588:1942–1948. doi: 10.1016/j.febslet.2014.03.053. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A., Senisterra G., Allali-Hassani A., Dong A., Dobrovetsky E., Wasney G. A., Chau I., Marcellus R., Hajian T., Liu F., Korboukh I., Smil D., Bolshan Y., Min J., Wu H., Zeng H., Loppnau P., Poda G., Griffin C., Aman A., Brown P. J., Jin J., Al-Awar R., Arrowsmith C. H., Schapira M., Vedadi M. Structure. 2012;20:1425–1435. doi: 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Wang C., Zhu Y., Caceres T. B., Liu L., Peng J., Wang J., Chen J., Chen X., Zhang Z., Zuo X., Gong Q., Teng M., Hevel J. M., Wu J., Shi Y. Structure. 2014;22:756–768. doi: 10.1016/j.str.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Yue W. W., Hassler M., Roe S. M., Thompson-Vale V., Pearl L. H. EMBO J. 2007;26:4402–4412. doi: 10.1038/sj.emboj.7601856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Cheng X. Structure. 2003;11:509–520. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhou L., Cheng X. EMBO J. 2000;19:3509–3519. doi: 10.1093/emboj/19.14.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J., Feng Q., Li Z., Zhang Y., Xu R. M. Cell. 2003;112:711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Song J., Rechkoblit O., Bestor T. H., Patel D. J. Science. 2011;331:1036–1040. doi: 10.1126/science.1195380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahr H., Habermann B., Gustafsson C. M., Larsson N. G., Hallberg B. M. Proc. Natl. Acad. Sci. U. S. A. 2012;109:15253–15258. doi: 10.1073/pnas.1210688109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidgren J., Svensson L. A., Liljas A. Nature. 1994;368:354–358. doi: 10.1038/368354a0. [DOI] [PubMed] [Google Scholar]
- Basavapathruni A., Jin L., Daigle S. R., Majer C. R., Therkelsen C. A., Wigle T. J., Kuntz K. W., Chesworth R., Pollock R. M., Scott M. P., Moyer M. P., Richon V. M., Copeland R. A., Olhava E. J. Chem. Biol. Drug Des. 2012;80:971–980. doi: 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- Yu W., Chory E. J., Wernimont A. K., Tempel W., Scopton A., Federation A., Marineau J. J., Qi J., Barsyte-Lovejoy D., Yi J., Marcellus R., Iacob R. E., Engen J. R., Griffin C., Aman A., Wienholds E., Li F., Pineda J., Estiu G., Shatseva T., Hajian T., Al-Awar R., Dick J. E., Vedadi M., Brown P. J., Arrowsmith C. H., Bradner J. E., Schapira M. Nat. Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Liu F., Zhao X., Perna F., Wang L., Koppikar P., Abdel-Wahab O., Harr M. W., Levine R. L., Xu H., Tefferi A., Deblasio A., Hatlen M., Menendez S., Nimer S. D. Cancer Cell. 2011;19:283–294. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q., He B., Jung S. Y., Song Y., Qin J., Tsai S. Y., Tsai M. J., O'Malley B. W. J. Biol. Chem. 2009;284:36167–36174. doi: 10.1074/jbc.M109.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Wang M., Lv Z., Yang N., Liu Y., Bao S., Gong W., Xu R. M. Proc. Natl. Acad. Sci. U. S. A. 2011;108:20538–20543. doi: 10.1073/pnas.1106946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. C., Wilczek C., Bonanno J. B., Xing L., Seznec J., Matsui T., Carter L. G., Onikubo T., Kumar P. R., Chan M. K., Brenowitz M., Cheng R. H., Reimer U., Almo S. C., Shechter D. PLoS One. 2013;8:e57008. doi: 10.1371/journal.pone.0057008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack J. S., Thieffine S., Bandiera T., Fasolini M., Duke G. J., Jayaraman L., Kish K. F., Klei H. E., Purandare A. V., Rosettani P., Troiani S., Xie D., Bertrand J. A. Biochem. J. 2011;436:331–339. doi: 10.1042/BJ20102161. [DOI] [PubMed] [Google Scholar]
- Dowden J., Hong W., Parry R. V., Pike R. A., Ward S. G. Bioorg. Med. Chem. Lett. 2010;20:2103–2105. doi: 10.1016/j.bmcl.2010.02.069. [DOI] [PubMed] [Google Scholar]
- Bunnage M. E., Chekler E. L., Jones L. H. Nat. Chem. Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Frye S. V. Nat. Chem. Biol. 2010;6:159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- Totrov M. BMC Bioinf. 2011;12(1):S35. doi: 10.1186/1471-2105-12-S1-S35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna-Slater V., Mok M. W., Nguyen K. T., Feher M., Najmanovich R., Schapira M. J. Chem. Inf. Model. 2011;51:612–623. doi: 10.1021/ci100479z. [DOI] [PubMed] [Google Scholar]
- Therrien E., Larouche G., Manku S., Allan M., Nguyen N., Styhler S., Robert M. F., Goulet A. C., Besterman J. M., Nguyen H., Wahhab A. Bioorg. Med. Chem. Lett. 2009;19:6725–6732. doi: 10.1016/j.bmcl.2009.09.110. [DOI] [PubMed] [Google Scholar]
- Huynh T., Chen Z., Pang S., Geng J., Bandiera T., Bindi S., Vianello P., Roletto F., Thieffine S., Galvani A., Vaccaro W., Poss M. A., Trainor G. L., Lorenzi M. V., Gottardis M., Jayaraman L., Purandare A. V. Bioorg. Med. Chem. Lett. 2009;19:2924–2927. doi: 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- Liu F., Li F., Ma A., Dobrovetsky E., Dong A., Gao C., Korboukh I., Liu J., Smil D., Brown P. J., Frye S. V., Arrowsmith C. H., Schapira M., Vedadi M., Jin J. J. Med. Chem. 2013;56:2110–2124. doi: 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J., Totrov M., Abagyan R. Mol. Cell. Proteomics. 2005;4:752–761. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- Yu W., Smil D., Li F., Tempel W., Fedorov O., Nguyen K. T., Bolshan Y., Al-Awar R., Knapp S., Arrowsmith C. H., Vedadi M., Brown P. J., Schapira M. Bioorg. Med. Chem. 2013;21:1787–1794. doi: 10.1016/j.bmc.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]