

Abstract
A 46-year-old female with interstitial lung disease presented with proximal muscle weakness, worsening hypertension, microangiopathic hemolysis, thrombocytopenia and deteriorating renal function. She had no sclerodactyly, but had abnormal capillaroscopy. She tested positive for PM-Scl antibodies, and a renal biopsy showed an acute thrombotic microangiopathy consistent with scleroderma renal crisis (SRC). She failed to respond to corticosteroids, plasmapheresis and renin–angiotensin pathway inhibitors. She recovered quickly with the anti-C5 antibody, eculizumab. She had no genetic abnormalities associated with atypical hemolytic uremic syndrome except a DNA variant of unknown significance in C3. This case suggests that eculizumab may be effective for SRC.
Keywords: AKI, autoantibodies, complement, plasmapheresis, scleroderma renal crisis
Introduction
Systemic sclerosis (SSc) is characterized by chronic inflammation and fibrosis in various organ systems. Scleroderma renal crisis (SRC) is a major complication of SSc and is recognized by malignant hypertension, acute kidney injury (AKI), thrombocytopenia and microangiopathic hemolytic anemia (MAHA) [1, 2]. Despite ACE inhibition, patients with SRC have a high incidence of end-stage renal disease [2]. Herein, we report a case of AKI with thrombotic microangiopathy (TMA), MAHA and hypocomplementemia in a patient with PM-Scl overlap syndrome without evidence of skin involvement who had prompt resolution of her MAHA and AKI with the complement C5 inhibitor, eculizumab. She had not responded earlier to treatment with enalapril, aliskiren, plasmapheresis and glucocorticoids.
Case report
A 46-year-old Caucasian female who was diagnosed with nonspecific interstitial pneumonitis (NSIP) on a lung biopsy 9 months previously was admitted with worsening dyspnea, progressive over the past year. Her prior treatment included mycophenolate mofetil (MMF) and courses of prednisone and trimethoprim-sulfamethoxazole (TMP-SMX) for ‘pneumocystis’ prophylaxis. Her platelet count and serum creatinine checked 2 months before admission were 241 × 109/L and 61.9 µmol/L (0.7 mg/dL), respectively. One month before admission, she developed swelling of her hands and feet followed by proximal muscle weakness. She also complained of nausea and intermittent non-bloody diarrhea that appeared to have begun after she started MMF. At admission, she denied fevers, rash or Raynaud's phenomenon.
Physical examination showed a blood pressure (BP) of 152/89 (ranged from 129/67 to 188/78) and a body mass index of 39.1 kg/m2. Her oxygen saturation was 96% on 6 L of oxygen. She had bibasilar crackles with diminished breath sounds. Her muscle strength was 3 (on a 5-point scale) in her neck flexors along with her proximal upper and lower extremity muscles. There was no rash, sclerodactyly, digital cyanosis, pedal edema or synovitis. Capillaroscopy exam showed dilated capillary loops.
Pertinent studies at admission included an ESR of 8 and a CRP of 1.4, WBC of 21.6 × 109/L, hemoglobin of 124 g/L (12.4 mg/dL), platelets of 51 × 109/L and creatinine of 141.4 µmol/L (1.6 mg/dL). Peripheral smear revealed 3+ schistocytes. She had a haptoglobin of <2 µmol/L (20 mg/dL), LDH of 1427 U/L, total and direct bilirubin of 58.1 and 13.7 µmol/L (3.4 and 0.8 mg/dL), AST of 99 U/L and ALT of 95 U/L. Her CK was 877 U/L, and aldolase was 44.7 U/L (normal 1.5–8.1 U/L). Urinalysis demonstrated 2+ blood, 2+ protein, 4 WBC and 1 RBC per high-power field. Her urine protein-to-creatinine ratio was 0.9 g/g.
Her ANA was positive at >1:2560 with a speckled pattern. She had a PM-Scl-100 antibody of 165 U (normal <11 U) and an RNA polymerase III antibody of 21 U (normal <20 U). Rheumatoid factor, anti-CCP, anti-SSA, anti-SSB, anti-Scl-70, anti-Jo-1, anti-RNP, anti-Smith, anti-double-stranded DNA, anti-cardiolipin and anti-beta-2 glycoprotein I antibodies were negative. Complement C3 was 0.97 g/L (97 mg/dL) and C4 was 0.15 g/L (15 mg/dL). ADAMTS13 activity level was 49% (normal >66%). Initial stool studies were negative but later tested positive for Clostridium difficile toxin. CT chest showed changes consistent with NSIP. Renal ultrasound with Doppler showed elevated resistive indices within the kidneys averaging 0.80.
She was treated with intravenous methylprednisolone for her NSIP. She had eight sessions of plasmapheresis with fresh frozen plasma replacement as thrombotic thrombocytopenic purpura (TTP) was in the initial differential diagnosis. Despite this, her renal function worsened with her creatinine rising to 256.3 µmol/L (2.9 mg/dL) and hemoglobin dropping to 89 g/L (8.9 mg/dL) with evidence of ongoing hemolysis. Her mean arterial pressure continued to remain elevated at 100–120 mmHg. She underwent a kidney biopsy, which showed capillary endothelial cell swelling in most glomeruli. Several glomeruli showed segmental fibrin thrombi and ischemic collapse. Most hilar arterioles showed luminal occlusion with fibrin admixed with fragmented erythrocyte fragments, consistent with an acute TMA (Figure 1).
Fig. 1.
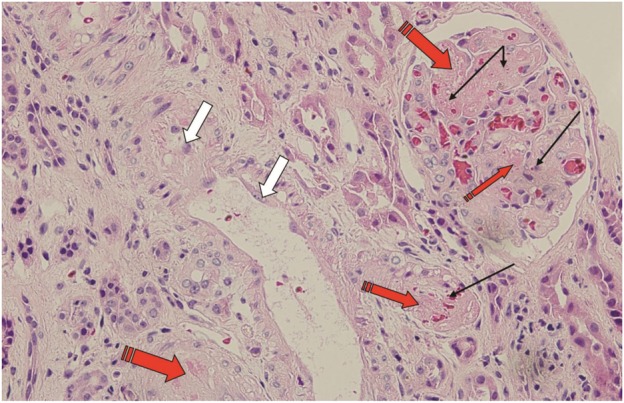
Renal biopsy stained with hematoxylin and eosin demonstrating acute TMA with occlusive endothelial swelling of the capillary loops (thick white arrows), fragmented red blood cells (thin black arrows) and fibrin thrombi in glomerular capillary loops and arterioles (red tail-traced arrows).
Given the NSIP, positive capillaroscopy, muscle weakness, positive ANA and PM-Scl autoantibodies, we suspected a scleroderma/polymyositis overlap syndrome (scleromyositis) with limited cutaneous systemic sclerosis (lcSSc) as a differential. The rising BP, worsening AKI with TMA and MAHA made us consider SRC and atypical hemolytic-uremic syndrome (aHUS) [3]. As high-dose steroids can precipitate SRC, her steroids were immediately tapered. Enalapril was initiated and 5 days later, aliskiren was added as her creatinine continued to rise to 353.6 µmol/L (4.0 mg/dL). Serum complement levels were rechecked and were low (CH50, C3 and C4 were <35%, 0.64 and 0.08 g/L, respectively) consistent with consumption by the disease process or modulation by plasmapheresis. She received a dose of meningococcal conjugate vaccine and then was started on ciprofloxacin 250 mg daily and given 900 mg of eculizumab. Within 24 h, her platelet count rose with a corresponding fall in her LDH and within a week of treatment, her renal function, BP and dyspnea began to improve (Figure 2). The improvement in her renal function and blood pressure was likely due to resolution of her TMA, and the improvement in her dyspnea may have been from better BPs, improvement in her interstitial pneumonitis or from increase in respiratory muscle strength. She was discharged after receiving the second dose of eculizumab 7 days after the first dose. After 5 weekly induction doses were given, the patient was continued on 1200 mg of eculizumab every 2 weeks.
Fig. 2.

Lab value trends in our patient following plasmapheresis (PP) and rescue treatment with eculizumab (E). A total of eight PPs were given over a period indicated by the horizontal bar. Eculizumab was commenced on Day 0 and continued for a total of 8 months. Methylprednisolone was started on Day 12 and continued till Day 1. Enalapril was started on Day 6 and the patient remains on it till today. Aliskiren was started on Day 1 and continued until Day 5. (A) Hemoglobin, creatinine and functional C5 are plotted. (B) Platelets, LDH and haptoglobin are plotted. Note brisk improvement in renal function and markers of hemolysis after introduction of eculizumab therapy.
One month after discharge, her serum creatinine had dropped to 141.4 µmol/L (1.6 mg/dL) and her BP had improved. Her hemoglobin was 117 g/L (11.7 mg/dL), platelets were 222 × 109/L, LDH 249 U/L, haptoglobin 17 µmol/L (170 mg/dL) and C3 was 0.91 g/L (91 mg/dL). Three months after the first dose, a second dose of meningococcal conjugate vaccine was given. Eight months after hospitalization, she continued to do well with a creatinine of 141.4 µmol/L (1.6 mg/dL). She had no evidence of hemolysis or thrombocytopenia and her eculizumab infusions were discontinued 8 months after her presentation with AKI and TMA.
Genetic studies drawn at the time of admission for thrombomodulin, complement factor H, membrane cofactor protein, factor B, factor I and complement factor H-related gene CFHR5 were all negative for mutations known to cause aHUS. No deletion or duplication of the CFHR3-CFHR1 region was detected. She has a single base substitution in one allele of C3 causing a non-synonymous change (c.835G>A, p.Glu279Lys). This has a predicted Conseq (http://conseq.bioinfo.tau.ac.il/) score of 4, a PROVEAN (http://provean.jcvi.org/index.php) score of −1.618 (neutral) and a Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/) score of 0.012 (benign), and this variant is seen in <1 in 1000 genomes and is thus of unknown significance but probably benign.
Three and a half years after her initial presentation, she remains without evidence of MAHA or hypocomplementemia and has a serum creatinine of 132.6 µmol/L (1.5 mg/dL) with no hematuria or proteinuria. She now has evidence of sclerodactyly. Her NSIP is being treated with mycophenolic acid and rituximab.
Discussion
Our patient presented with NSIP, polymyositis and positive PM-Scl antibody without definite scleroderma making us consider early lcSSc or an SSc/PM overlap syndrome. This overlap syndrome has been well-described, particularly in patients with positive PM-Scl and anti-RNP antibodies [4, 5]. The precise clinical features associated with PM-Scl antibody syndrome are not defined, but muscle inflammation, lung disease, calcinosis, Raynaud's and limited skin involvement have been reported [4, 6–8].
Our patient developed hypocomplementemia and rapidly deteriorating renal function. Renal biopsy showed acute TMA consistent with SRC. SRC has been reported in lcSSC and SSc/PM overlap syndrome [9–11]. Recent data suggest that anti-RNA polymerase III antibodies may confer an increased risk of SRC [12]. She was refractory to treatment with the usual treatments for SRC: ACE inhibitor and/or a renin inhibitor [12].
Thrombocytopenia and MAHA are also features of malignant hypertension, TTP, HUS and anti-phospholipid syndrome. A patient with SSc/PM overlap who presented with TTP and responded to plasmapheresis has been described [13]. Our patient had an ADAMTS13 of >10% and was resistant to plasmapheresis, features that make TTP less likely. She responded to eculizumab, suggesting that her MAHA was mediated by complement activation.
Complement activation can occur via the classical, alternative or lectin binding pathways. These pathways converge on C5, which is then cleaved to C5a and C5b and leads to the assembly of C5b-9. aHUS results from dysregulation of the alternative pathway and is frequently manifest by a low C3 but normal C4. Heterozygous mutations in a complement regulator or from autoantibodies to these regulators are the pathogenic mechanism in the large majority [3]. Although the abnormal complement studies and her response to eculizumab supports complement activation, the diagnosis of aHUS is difficult to substantiate. First, the presence of TMA is not sufficient to establish aHUS. Furthermore, the association of the patient's primary diagnosis with TMA historically makes it more likely that her TMA was caused by her rheumatologic disease. Third, mutation screening of known complement regulatory genes was negative except for a likely benign non-synonymous change in C3. Conversely, the lack of a supporting complement mutation does not exclude the possibility that our patient has aHUS. Only 55% of patients with aHUS have an identifiable gene mutation; therefore, finding a mutation is not a prerequisite for making a diagnosis of aHUS [3].
Complement activation has been described in autoimmune and alloimmune disorders including SSC [14, 15]. In these situations, complement is generally activated via the classical pathway and is expected to lead to consumption of both C3 and C4 [16]. Eculizumab has been shown to reverse the hemolysis and thrombocytopenia and to resolve severe tissue injury from TMA in anti-phospholipid antibody syndrome and in antibody-mediated rejection from anti-HLA and anti-ABO antibodies, situations where complement is activated via the classical pathway [17, 18]. Although in our patient we cannot exclude the possibility that aHUS co-exists with SSc/PM overlap syndrome, her entire clinical presentation is compatible with SRC.
In summary, we report a patient presenting with SSc/PM overlap syndrome who developed SRC. Her rapidly declining course was reversed with the use of eculizumab. We administered eculizumab for a total of 8 months before discontinuing it. More than 3 years after her admission, she remains well off eculizumab. This is the first reported instance where eculizumab has successfully treated SRC complicating SSc/PM overlap syndrome.
Conflicts of interest statement
My co-authors and I do not hold stock in Alexion Pharmaceuticals, the maker of eculizumab, nor are we conducting research for or are on the speakers bureau for Alexion. The results presented in this paper have not been published previously in whole or part.
References
- 1.Penn H, Howie A, Kingdon E, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. Q J Med 2007; 100: 485–494 [DOI] [PubMed] [Google Scholar]
- 2.Guillevin L, Berezne A, Seror R, et al. Scleroderma renal crisis: a retrospective multicenter study on 91 patients and 427 controls. Rheumatology 2012; 51: 460–467 [DOI] [PubMed] [Google Scholar]
- 3.Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: What is it, how is it diagnosed and how is it treated? Am Soc Hematol 2012; 2012: 617–625 [DOI] [PubMed] [Google Scholar]
- 4.Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum 2005; 35: 35–42 [DOI] [PubMed] [Google Scholar]
- 5.Balbir-Gurman A, Braun-Moscovici Y. Scleroderma overlap syndrome. Isr Med Assoc J 2011; 13: 14–20 [PubMed] [Google Scholar]
- 6.Hanke K, Bruckner C, Dahnrich C, et al. Antibodies against PM/Scl-75 and PM/Scl-100 are independent markers for different subsets of systemic sclerosis patients. Arthritis Res Ther 2009; 11: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maes L, Blockmans D, Verschueren P, et al. Anti-PM/Scl-100 and anti-RNA-polymerase III antibodies in scleroderma. Clin Chim Acta 2010; 411: 965–971 [DOI] [PubMed] [Google Scholar]
- 8.Mahler M, Raijmakers R. Novel aspects of autoantibodies to the PM/Scl complex: clinical, genetic and diagnostic insights. Autoimmun Rev 2007; 6: 432–437 [DOI] [PubMed] [Google Scholar]
- 9.Sanders PW, Herrera GA, Ball GV. Acute renal failure without fibrotic skin changes in progressive systemic sclerosis. Nephron 1988; 48: 121–125 [DOI] [PubMed] [Google Scholar]
- 10.Zwettler U, Andrassy K, Waldherr R. Scleroderma renal crisis as a presenting feature in the absence of skin involvement. Am J Kidney Dis 1993; 22: 53–56 [DOI] [PubMed] [Google Scholar]
- 11.Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 26–2001. Hypertensive encephalopathy with impaired renal function in a 67-year-old woman with polymyositis. N Engl J Med 2001; 345: 596–605 [DOI] [PubMed] [Google Scholar]
- 12.Shanmugam VK, Steen VD. Renal disease in scleroderma: an update on evaluation, risk stratification, pathogenesis and management. Curr Opin Rheumatol 2012; 24: 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwagami M, Kubo K, Tanaka R. Thrombotic thrombocytopenia purpura with severe hypertension in a patient with systemic sclerosis sine scleroderma and polymyositis. Intern Med 2011; 50: 2413–2416 [DOI] [PubMed] [Google Scholar]
- 14.Hudson M, Walker JG, Fritzler M, et al. Hypocomplementemia in systemic sclerosis—clinical and serological correlations. J Rheumatol 2007; 34: 2218–2223 [PubMed] [Google Scholar]
- 15.Foocharoen C, Distler O, Becker M, et al. Clinical correlations of hypocomplementaemia in systemic sclerosis: an analysis of the EULAR Scleroderma Trial and Research group (EUSTAR) database. Scand J Rheumatol 2012; 41: 243–246 [DOI] [PubMed] [Google Scholar]
- 16.Wild G, Watkins J, Ward AM, et al. Complement activation in systemic sclerosis. J Clin Lab Immunol 1990; 31: 39–41 [PubMed] [Google Scholar]
- 17.Hadaya K, Ferrari-Lacraz S, Fumeaux D, et al. Eculizumab in acute recurrence of thrombotic microangiopathy after renal transplantation. Am J Transplant 2011; 11: 2523–2527 [DOI] [PubMed] [Google Scholar]
- 18.Stewart Z, Collins T, Schlueter A, et al. Case report: eculizumab rescue of severe accelerated antibody-mediated rejection after ABO-incompatible kidney transplant. Transplant Proc 2012; 44: 3033–3036 [DOI] [PubMed] [Google Scholar]