

Abstract
Glomerulopathy due to dysproteinemia can have a wide spectrum of pathologic and clinical features based on specific characteristics of the abnormal protein and the response induced within the parenchymal tissue. Monoclonal immunoglobulin G (IgG) deposition can manifest as a different glomerular disease. Proliferative glomerulonephritis (GN) with monoclonal IgG deposits (PGNMID) is a unique entity mimicking immune complex GN that does not conform to any of those subtypes. IgG monoclonal granular deposition in the glomeruli with a pattern similar to immune complex disease suggested by C3 and C1q deposition should prompt consideration of PGNMID. Literature is scarce in terms of recurrence of disease in renal allografts. In this article we present the clinical–pathologic features of three cases of PGNMID in the renal allograft showing the variable course and manifestation of the disease.
Keywords: immune complex, monoclonal IgG, proliferative GN, renal allograft
Background
Various glomerular diseases can be caused by the deposition of monoclonal immunoglobulin G (IgG). Renal disease related to the deposition of monoclonal IgG containing both heavy and light chains can occur in type 1 cryoglobulinemia, Randall-type light- and heavy-chain deposition disease (LHCDD), light- and heavy-chain amyloidosis and immunotactoid glomerulonephritis [1–5]. Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) is a relatively newly described entity mimicking immune complex glomerulonephritis (GN) that does not conform to any of those subtypes. It represents a form of proliferative GN with electron-dense deposits (EDDs) localized to glomeruli and immunofluorescence (IF) findings of monoclonal IgG deposits. Recurrent PGNMID in the renal allograft has been described in a few case series [6–8]. In this article we present the clinical–pathologic features of three cases of PGNMID in the renal allograft showing the variable course and manifestation of the disease. Our three cases, like most other reported cases, have no bone marrow involvement and no evidence of monoclonal protein on electrophoresis and immunofixation studies. Thus, this makes disease activity and remission status monitoring very difficult to assess.
Case 1
A 61-year-old Caucasian female developed end-stage kidney disease (ESKD) secondary to membranoproliferative glomerulonephritis (MPGN) based on a native kidney biopsy in 2001, with no further information about IF or electron microscopy (EM). In December 2006, her serum creatinine (SCr) was 530 μmol/L and she underwent a preemptive unrelated living kidney transplantation. In 2014, she was found to have a random urine protein/gram creatinine (UPCR) of ∼452 mg/mmol and an SCr of 132 μmol/L, up from 106 μmol/L. Her workup for a lymphoproliferative disorder was negative, as was her serologic workup (see Table 1). On biopsy, IF showed diffuse granular positivity for IgG, C3 and C1q. Deposits were composed of IgG3-kappa with no peritubular C4d. EM showed EDDs, mostly in the mesangium and subendothelial regions (please see Figures 1–7). She received three cycles of bortezomib. UPCR was persistently elevated at 565 mg/mmol. She was later switched to Carfilzomib, as she developed acute kidney injury with SCr up to 247 μmol/L; after completion of therapy her SCr was 150 μmol/L.
Fig. 2.
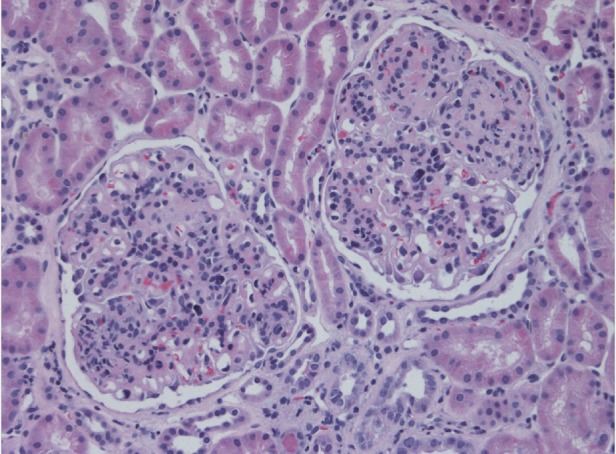
The preserved glomeruli are enlarged and hypercellular. The increase in cells includes increased mononuclear elements within the expanded mesangial areas and many mononuclear inflammatory cells within the glomerular capillary lumens. As a result, the capillary tufts have a distinctly lobulated appearance. Magnification ×200, hematoxylin and eosin stain.
Fig. 3.
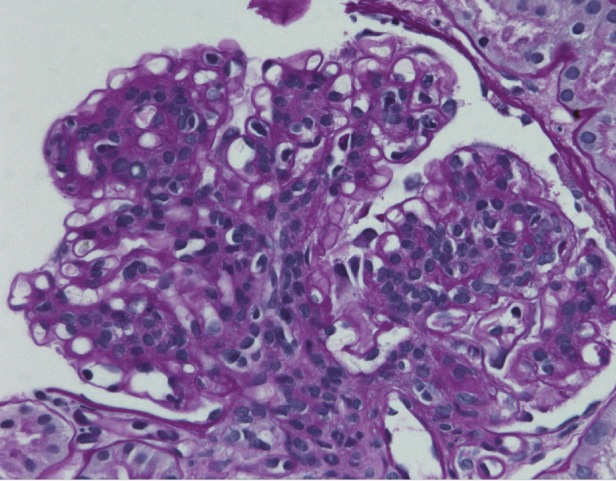
The peripheral capillary walls are distorted and thickened, often with the formation of double contours or basement membranes with a chain-like appearance. Magnification ×400, periodic acid–Schiff stain.
Fig. 4.
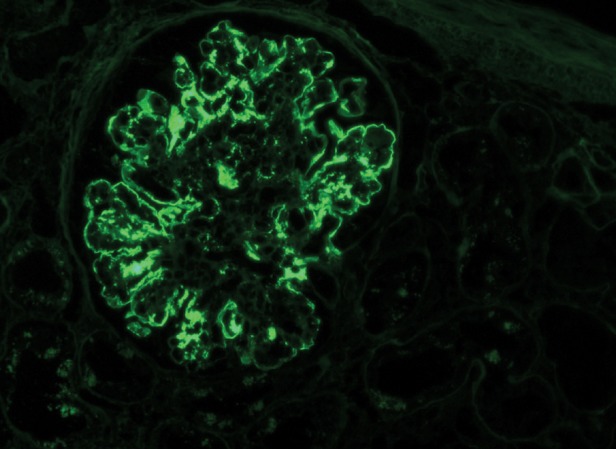
Diffuse irregular granular and pseudolinear deposition of IgG (3+/4+). No staining is found in Bowman's capsule or the tubular basement membranes. Magnification ×200, IF micrograph.
Fig. 5.
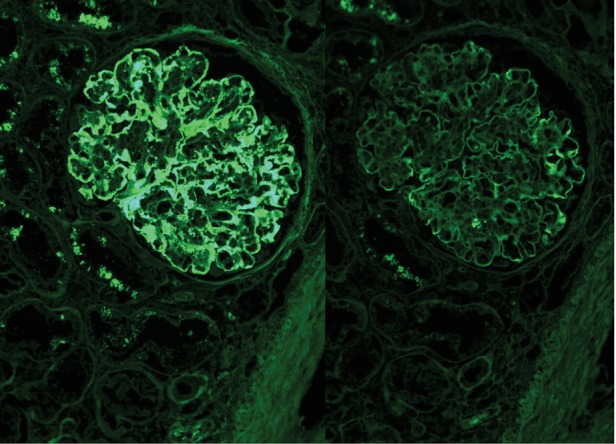
Kappa light chains stain strongly positive (3+/4+) along the peripheral capillary walls and mesangial areas. Lambda light chains are negative in the deposits. Magnification ×400, IF micrograph.
Fig. 6.

IF staining for the IgG subtypes shows intense glomerular positivity for (C) IgG3 with negative staining for (A), IgG1 (B) IgG2 and (D) IgG3.
Table 1.
Clinical data of post–kidney transplant recipients with PGNMID
| Patient | # 1 | # 2 | # 3 | # 4 [6] | # 5 [6] | # 6 [6] |
|---|---|---|---|---|---|---|
| Age (years) | 61 | 74 | 40 | 61 | 68 | 68 |
| Race/sex | Caucasian/female | Caucasian/male | Hispanic/female | Caucasian/male | Caucasian/female | Caucasian/female |
| Cause of ESKD | MPGN | PGMID | MPGN | PGNMID | Unclear—nephrotic range proteinuria | Polycystic kidney disease (PKD) |
| Transplant type | Unrelated living donor | Deceased donor | Deceased donor | Deceased donor | Related living donor | Deceased donor |
| IS | FK506, prednisone, MMF | FK506, prednisone, MMF | FK506 and azathioprine | MMF, rapamycin | MMF, rapamycin | Prednisone, cyclosporine |
| Baseline SCr (μmol/L) | 106 | 264 | 96.8 | 114 | 132 | 132 |
| SCr at biopsy (μmol/L) | 132 | 396 | 114 | 237 | 281 | 228 |
| Time from transplant to diagnosis of recurrent or de novo PGNMID (months) | 98 | 6 | 132 | 13 | 21 | 158 |
| Proteinuria (mg/mmol) | 400 | 100 | 20 | 870 | 50 | 150 |
| Hematuria | 1+ | 3+ | 2+ | 2+ | 2+ | 1+ |
| Serum complement | Normal | Normal | Normal | Normal | Normal | Normal |
| Cryoglobulin | Negative | Negative | Negative | n/a | n/a | n/a |
| Hepatitis B | Negative | Negative | Negative | Negative | Negative | Negative |
| Hepatitis C | Negative | Negative | Negative | Negative | Negative | Negative |
| HIV | Non-reactive | Non-reactive | Non-reactive | Non-reactive | Non-reactive | Non-reactive |
| Monoclonal protein spike | Negative | Negative | Negative | Negative | Negative | Negative |
| Pattern of injury | EC on both biopsies with a crescent on the second biopsy | Native biopsy: 1st – ATN 2nd – EC Transplant biopsy: MP |
MP | MPGN-like | MP | MPGN-like |
| Number of glomeruli sclerosed | None on first, 3 of 20 on second | 13/36 | 4 of 10 | 1 of 9 | 1 of 27 | 5 of 8 |
| Interstitial fibrosis | None on first and 15% on second | 50% | 15% | 1+ | 3+ | 3+ |
| IgG subtype kappa or lambda | IgG3-kappa | Native: IgG lambda (subtype not available) 2nd transplant: IgG3-lambda |
IgG3-kappa | IgG3-kappa, mild lambda | IgG3-kappa | IgG1-kappa |
| Electron microscopy deposits | EDM, EDS on both biopsies | Native biopsy: EDS, EDM Transplant biopsy: EDM |
EDM, EDS | MPGN-like | MP | MPGN-like |
| Bone marrow biopsy | Positive stain for plasma cells for CD138 ∼5% cellularity; no staining for kappa and lambda light chain by in situ hybridization | Positive stain for plasma cells for CD138 <5% cellularity; no staining for kappa and lambda light chain by in situ hybridization (pre-transplant) | Not done due to stability of kidney function | None | None | None |
| Treatment | Bortezomib ×3 doses, dexamethasone ×3 doses | Patient opted for none | No change | No change | Change to MMF and prednisone | No change |
| Outcome after allograft biopsy: SCr (μmol/L) | 149, proteinuria 600 mg/mmol Remains on IS as above |
End-stage kidney disease | 114 proteinuria 10 mg/mmol Remains on IS as above |
Fatal MI after 20 months of kidney biopsy | Died with cryptococcal meningitis 15 months after biopsy | End-stage kidney disease |
| Follow-up (months) after biopsy | 17 | 3 | 36 | 20 | 15 | 20 |
| Patient | # 7 [6] | # 8 [7] | # 9 [7] | # 10 [7] | # 11 [7] | # 12 [8] |
| Age | 24 | 55 | 60 | 74 | 38 | 66 |
| Race/sex | Caucasian/female | Caucasian/male | Caucasian/female | Caucasian/female | Caucasian/female | Caucasian/female |
| Cause of ESKD | Diabetic nephropathy | PGNMID | PGNMID | PGNMID | PGNMID | PGNMID |
| Transplant type | Simultaneous kidney–pancreas | Unrelated living donor | Deceased donor | Unrelated living donor | Unrelated living donor | Deceased donor |
| IS | FK506, prednisone, MMF | FK506, prednisone, MMF | FK506, prednisone, MMF | FK506, prednisone, MMF | FK506, prednisone, MMF | FK506, prednisone, MMF stopped at 1 month |
| Baseline SCr (μmol/L) | 158 | 167 | 106 | 123 | 79.2 | 60.7 |
| SCr at biopsy (μmol/L) | 290 | 246 | 326 | 422 | 106 | 176 |
| Time from transplant to diagnosis of recurrent or de novo PGNMID (months) | 43 | 12 | 22 | 156 | 43 | 18 |
| Proteinuria (mg/mmol) | 170 | 80 | 740 | 580 | 6 | 330 |
| Hematuria | 3+ | Yes | Yes | Yes | No | 3+ |
| Serum complement | Normal | Normal | Low C3 and C4 | Low C3 and C4 | Normal | n/a |
| Cryoglobulin | n/a | Negative | Negative | Negative | Not done | Negative |
| Hepatitis B | Negative | Negative | Negative | Negative | Negative | Negative |
| Hepatitis C | Negative | Negative | Negative | Negative | Negative | Negative |
| HIV | Non-reactive | Non-reactive | Non-reactive | Non-reactive | Non-reactive | n/a |
| Monoclonal protein spike | Negative | Negative | Negative | Negative | Negative | |
| Pattern of injury | MPGN-like | First two biopsies showed MP, third showed diffuse EC and fourth showed focal proliferative | Diffuse EC on both with diffuse on first and focal on second biopsy | First biopsy showed diffuse EC and exudative GN, next two biopsies with MP | Minimal to mild MP | EC with crescents |
| Number of glomeruli sclerosed | 8 of 13 | 3/37 sclerosed, 16/27 crescents | ||||
| Interstitial fibrosis | 2+ | Mild | Mild | Mild then moderate | Mild then moderate | |
| IgG subtype kappa or lambda | IgG3-kappa | IgG3-kappa | IgG3-kappa | IgG3-lambda | IgG3-kappa | Kappa (subtype not available) |
| Electron microscopy deposits | MPGN-like | First two biopsies showed EDM, and EDM and EDS on last two | EDM, EDS | First and third showed EDS and second showed EDM and EDS | EDM in first two biopsies and EDM and EDS in others; third and fifth biopsies showed Banff 1A | EDM and parietal deposits |
| Bone marrow biopsy | None | None | None | None | None | None |
| Treatment | No change | Prednisone, oral cyclophosphamide for 6 months | Methylprednisolone with prednisone, rituximab ×2 doses, lisinopril | Methylprednisolone with prednisone, rituximab ×1 dose, with plasmapheresis ×4 and HD ×3 | Methylprednisolone for ACR, rituximab ×2 doses | Plasmapheresis, IVIG 0.5 g/kg/day with methylprednisolone total 850 mg, MMF 1.5 g/day |
| Outcome after allograft biopsy: SCr (μmol/L) | 290 | 202 | 96.8 | 114 | 202 | 96.8, transplant nephrectomy in the setting of sepsis 6 months post-treatment with biopsy showing resolution of disease despite nephrectomy, disappearance of EC and cellular crescents, IF staining remained kappa |
| Follow-up (months) after biopsy | ‘Short’ | 15 | 11 | 83 | 63 | 6 |
MPGN, membranoproliferative glomerulonephritis; PGNMID, proliferative glomerulonephritis and monoclonal immunoglobulin deposits; MP, mesangial proliferative; EP, endothelial proliferative; EC, endocapillary proliferative; SCr, serum creatinine; MMF, mycophenolate mofetil; EDM, electron-dense mesangial deposits; EDS, electron-dense subendothelial deposits; FK506, prograf/tacrolimus.
Fig. 1.
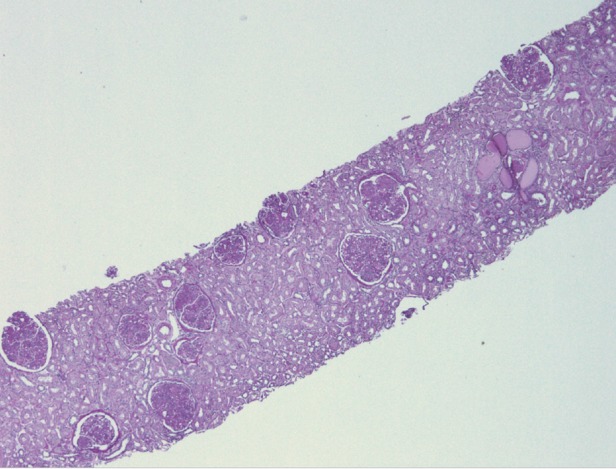
Mild chronic changes of tubular atrophy and interstitial fibrosis. Low power light microscopy, periodic acid–Schiff stain.
Fig. 7.

Glomerular capillaries are greatly distorted and thickened by the presence of numerous, sometimes large and/or confluent subendothelial electron-dense deposits. The electron-dense deposits have a variegated (‘two-toned’) appearance and are finely granular, but they do not show organized substructures. Magnification ×12 000, electron micrograph.
Case 2
A 74-year-old Caucasian male developed ESKD secondary to PGNMID seen on a native biopsy on September 2009. He received bortezomib and dexamethasone; however, he developed ESKD requiring hemodialysis (HD) after three doses. He received a diseased donor transplant (DDT) in May 2013, with his course complicated by biopsy-proven acute tubular injury. His SCr improved to 265 μmol/L until 6 months post-transplant, at which time his SCr was ∼ 530 μmol/L. He had a negative M spike and negative serologies. IF from his biopsy showed positivity for IgG3, C3, C1q and lambda light chains. Kappa was negative, as were peritubular capillaries for C4d (see Table 1). His kidney disease progressed and he declined therapy and restarted HD 9-month post-transplantation.
Case 3
A 40-year-old Hispanic female received a DDT in June 2001 after developing ESKD. In 1995, she had biopsy-proven type 1 MPGN. There were no IF data for reevaluation. In June 2012, she had hematuria and her SCr was 114 μmol/L, up from her baseline SCr of 97 μmol/L. UPCR was less than 20 mg/mmol. Her allograft biopsy showed a mesangioproliferative pattern of injury by light microscopy (see Table 1). IF revealed mesangial deposits restricted to IgG3-kappa. Her plasma cell dyscrasia workup was negative as was her serologic workup. SCr is stable at 115 μmol/L with UPCR at 10 mg/mmol.
Discussion
PGNMID, a novel type that does not conform to any subtypes, was described initially in 2004 [9] followed by the largest series of 37 cases in 2009 [10]. The disease manifests with proteinuria, hematuria and variable degrees of renal impairment. Disease incidence is 0.17% [10]. Most of the patients have a normal workup for paraproteinemia. Histologic patterns by light microscopy were mostly membranoproliferative with a lobular nodular pattern of injury followed by an endocapillary proliferative pattern. EM revealed granular, nonorganized deposits mostly in the mesangium and subendothelium. IF demonstrated granular glomerular deposits that stained for a single light-chain isotype and a single heavy-chain subtype, most commonly IgG3-kappa [7, 10]. More than 40 additional cases were identified by other groups [6, 11–19].
Recently, recurrent PGNMID after kidney transplantation has been reported as case reports or small series of one to four patients [6–8]. The recurrent disease manifested clinically as allograft dysfunction, hematuria and proteinuria. No patient had a detectable M spike in serum or urine. Complements were normal. Hepatitis B, hepatitis C and cryoglobulins were negative. Histologic features were mostly endocapillary proliferative GN or mesangial proliferative GN [7].
Of the patients reported (see Table 1), the average age was 57 years. Patients tended to be Caucasian (11/12) and female (10/12). Disease diagnosis averaged 60 months from transplantation. The baseline SCr following transplantation was 128 μmol/L and at disease diagnosis was 246 μmol/L. IgG3-kappa was the predominant IgG identified (IgG3 11/12 and kappa 10/12). Three patients ended up with ESKD and two died related to infectious complications. There was an average of 24 months of follow-up after diagnosis.
Disease recurrence can range from an average of 3.8 months in some series [7] up to 2 years after kidney transplantation [5, 7]. The risk of recurrence is difficult to estimate based on the small number of patients and the absence of protocol biopsies in most cases. Moreover, the immunosuppressive treatment may have an impact on disease course that may be difficult to isolate. Disease recurrence can have a variable outcome, with some patients having persistent renal impairment and some progressing to end-stage renal disease. Our three cases reflect this variable disease course.
PGNMID is often missed or underdiagnosed, with diagnoses of MPGN or even light- and heavy-chain deposition disease (LHCDD). Some cases like case 1 and case 3 were originally diagnosed as MPGN type 1 before this entity was well established. In many instances, IF and EM were not available, nor was staining for light chains. PGNMID and LHCDD are similar in many aspects, however, the pathogenesis is different. LHCDD results from the deposition of heavy and light chains in the glomerular and tubular basement membrane, as opposed to PGNMID, where the deposits are intact monoclonal immunoglobulin affecting only the glomeruli. PGNMID is characterized by granular EDDs, in contrast to LHCDD, where those are mostly powdery deposits. IgG3 was the IgG isoform identified most often in prior studies to be deposited in the glomeruli despite comprising only 8% of the total immunoglobulin in the serum [6, 7]. Limitation to IgG3 in our three cases confirms the specific predilection for IgG3 to precipitate and self-aggregate in the glomeruli given its properties of high molecular weight, positive charge and complement activation capacity [7].
Monoclonal gammopathy of renal significance is an increasingly recognized entity in patients who are found to have monoclonal protein with renal involvement. Patients who do not fulfill the criteria for multiple myeloma have traditionally been labeled as monoclonal gammopathy of undetermined significance given the limited diagnostic schema, and consequently no therapy has been offered. However, it is more established now that addressing this clonal proliferation may preserve the kidneys and potentially prevent disease recurrence. There was no monoclonal protein identified in our patients, nor the other cases. Nevertheless, a thorough workup is strongly recommended as it may impact the therapeutic plan [20].
Cases of de novo PGNMID on kidney allografts have been reported. One case series described the occurrence of PGNMID in two patients after transplantation. One patient with polycystic kidney disease was diagnosed with PGNMID 13 years after kidney transplantation. Another patient had diabetes nephropathy leading to ESKD and developed de novo PGNMID 3 years after transplantation [6]. While having different primary pathology supports that allograft disease is de novo, late occurrence does not exclude recurrence, as kidney transplant recipients may have a different disease course and disease activity due to immunosuppressive drugs.
Based on a pathogenesis hypothesis, it would seem rational to treat with immunomodulating therapy. Unfortunately, there is no proven effective treatment. Transplant recipients are on continuous immunosuppressive therapy. Aggressive immunosuppression with rituximab/or cyclophosphamide combined with steroids has been used [7]. Bortezomib was tried in one patient with native disease in one series [10], however, not in any transplant patients. Given the role of monoclonal Ig in the disease pathogenesis, bortezomib was tried in two of our patients, one with disease in the native kidney prior to transplant (case 2) and one with disease recurrence following transplant (case 1), with no favorable response.
Conclusion
PGNMID is becoming a more recognized entity, with more patients being evaluated for renal transplant. This disease has a wide spectrum of clinical presentations. Distinguishing this entity from other diseases may have an impact on outcome and treatment. Evaluating these patients for transplant eligibility may be challenging in the absence of distinct data of remission given that most patients have normal bone marrow biopsy and no monoclonal band on serum and urine testing. Data are scarce in terms of follow-up and treatment. More long-term studies will be needed to further investigate disease course and response to therapy.
Conflict of interest statement
None declared.
References
- 1.Lin J, Markowitz GS, Valeri AM, et al. Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol 2001; 12: 1482–1492 [DOI] [PubMed] [Google Scholar]
- 2.Nasr SH, Colvin R, Markowitz GS. IgG1λ light and heavy chain renal amyloidosis. Kidney Int 2006; 70: 7. [DOI] [PubMed] [Google Scholar]
- 3.Nasr SH, Markowitz GS, Reddy BS, et al. Dysproteinemia, proteinuria, and glomerulonephritis. Kidney Int 2006; 69: 772–775 [DOI] [PubMed] [Google Scholar]
- 4.Rosenstock JL, Markowitz GS, Valeri AM, et al. Fibrillary and immunotactoid glomerulonephritis: distinct entities with different clinical and pathologic features. Kidney Int 2003; 63: 1450–1461 [DOI] [PubMed] [Google Scholar]
- 5.Nasr SH, Said SM, Valeri AM, et al. The diagnosis and characteristics of renal heavy-chain and heavy/light-chain amyloidosis and their comparison with renal light-chain amyloidosis. Kidney Int 2013; 83: 463–470 [DOI] [PubMed] [Google Scholar]
- 6.Albawardi A, Satoskar A, Von Visger J, et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs or may develop de novo in kidney allografts. Am J Kidney Dis 2011; 58: 276–281 [DOI] [PubMed] [Google Scholar]
- 7.Nasr SH, Sethi S, Cornell LD, et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol 2011; 6: 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranghino A, Tamagnone M, Messina M, et al. A case of recurrent proliferative glomerulonephritis with monoclonal IgG deposits after kidney transplant treated with plasmapheresis. Case Rep Nephrol Urol 2012; 2: 46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nasr SH, Markowitz GS, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int 2004; 65: 85–96 [DOI] [PubMed] [Google Scholar]
- 10.Nasr SH, Satoskar A, Markowitz GS, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol 2009; 20: 2055–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans DJ, Macanovic M, Dunn MJ, et al. Membranous glomerulonephritis associated with follicular B-cell lymphoma and subepithelial deposition of IgG1-kappa paraprotein. Nephron Clin Pract 2003; 93: c112–c118 [DOI] [PubMed] [Google Scholar]
- 12.Komatsuda A, Masai R, Ohtani H, et al. Monoclonal immunoglobulin deposition disease associated with membranous features. Nephrol Dial Transplant 2008; 23: 3888–3894 [DOI] [PubMed] [Google Scholar]
- 13.Lee JG, Moon K, Lee JE, et al. A case of proliferative glomerulonephritis with monoclonal IgG deposits. Korean J Nephrol 2004; 23: 987–991 [Google Scholar]
- 14.Bridoux F, Zanetta G, Mougenot B, et al. Glomerulopathy with non-organized and non-Randall type monoclonal immunoglobulin deposits. J Am Soc Nephrol 2001; 12: 94A [Google Scholar]
- 15.Geldenhuys L, Jones B. Glomerulonephritis with monoclonal immunoglobulin deposits. J Am Soc Nephrol 2008; 19: 671A [Google Scholar]
- 16.Masai R, Wakui H, Komatsuda A, et al. Characteristics of proliferative glomerulonephritis with monoclonal IgG deposits associated with membranoproliferative features. Clin Nephrol 2009; 72: 46–45 [DOI] [PubMed] [Google Scholar]
- 17.Alpers CE, Tu WH, Hopper J, Jr, et al. Single light chain subclass (kappa chain) immunoglobulin deposition in glomerulonephritis. Hum Pathol 1985; 16: 294–304 [DOI] [PubMed] [Google Scholar]
- 18.Komatsuda A, Wakui H, Ohtani H, et al. Steroid-responsive nephrotic syndrome in a patient with proliferative glomerulonephritis with monoclonal IgG deposits with pure mesangial proliferative features. NDT Plus 2010; 3: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Seigneux S, Bindi P, Debiec H, et al. Immunoglobulin deposition disease with a membranous pattern and a circulating monoclonal immunoglobulin G with charge-dependent aggregation properties. Am J Kidney Dis 2010; 56: 117–121 [DOI] [PubMed] [Google Scholar]
- 20.Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 2012; 120: 4292–4295 [DOI] [PubMed] [Google Scholar]