

Abstract
Chiral biphenols were found to catalyze the enantioselective asymmetric addition of aryl- or alkenyl-boronates to ortho-quinone methides. Substituted 2-styryl phenols were obtained in good yields (up to 95%) and high enantiomeric ratios (up to 98:2) in presence of 10 mol % of 3,3′-Br2-BINOL. A two step synthesis of (S)-4-Methoxy-dalbergione is achieved in good yield and selectivity.
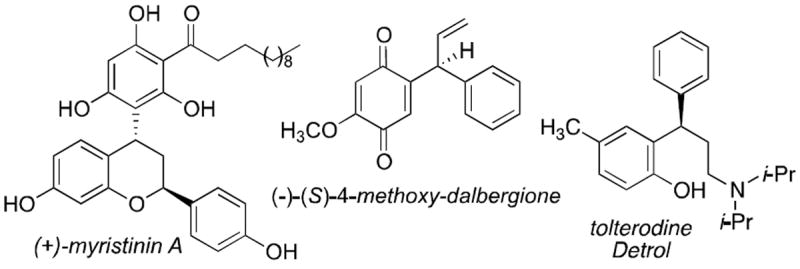
Ortho-quinone methides (oQMs) are employed as synthetic intermediates most notably in hetero-Diels-Alder reactions.1 Within this context they are proposed as reactive intermediates in a number of biomimetic natural product syntheses.2 Ortho-quinone methides exhibit other modes of reactivity including, but not limited to, 1,4-conjugate addition reactions at the exocyclic carbon with nucleophiles.3 Exploiting this reactivity, Sigman developed a Pd-catalyzed vinylphenol difunctionalization process including a nucleophilic addition to the quinone methide intermediate.4 Pettus described a mild and efficient anionic approach to generate ortho-quinone methides and used this approach in the synthesis of a collection of natural products.5 Enantioselective nucleophilic boronate chemistry has proven to be widely useful in asymmetric synthesis.6 We sought to develop a collection of enantioselective catalytic boronate addition reactions to ortho-quinone methides and related intermediates.7 Inspiration for this target class of reactions is derived from the numerous bioactive natural products and drugs, only a few depicted in Scheme 1, all of which can be made via this type of enantioselective addition (Eq. 1).8
Scheme 1.
 |
(1) |
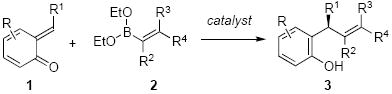
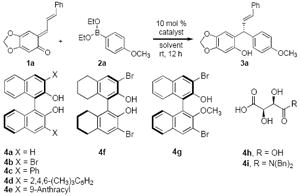
The recent work of Letka9 and Sigman10 illustrate the potential utility of these intermediates and the challenges in developing enantioselective addition reactions. Our lab11 along with others12 have developed a series of asymmetric boronate addition reactions catalyzed by chiral diols. We envisaged an enantioselective addition of vinyl- and aryl boronates to ortho-quinone methides would result in compounds with a benzylic chiral carbon center.13 By leveraging the driving force of quinone methide rearomatisation, the boronate addition could be conducted under extremely mild conditions to generate chiral phenols of general structure 3 [Eq. 1]. Herein, we describe the development of an enantioselective boronate addition to ortho-quinone methides catalyzed by chiral diols.14
We initiated our investigation by evaluating the reaction of aryl boronate 2a with ortho-quinone methide 1a in toluene at room temperature. Aryl boronate 2a is known to be a weak nucleophile, but it reacted smoothly with 1a in the presence and absence of catalyst, 54% and 36% yield respectively after 12 h. Chiral BINOLs and tartaric acid derived catalysts were evaluated as catalyst in the presence of 1a and boronate 2a (Table 1).15 3,3′-Br2-BINOL 4b afforded the product in the highest er (3:97, Table 1, entry 2) Toluene proved to be the best solvents among our solvent screens (Table 1, entries 3-5). Compared to 4b, aryl substituted BINOLs 4c and 4d gave slightly low yields and enantioselectivities (Table 1, entries 6 and 7). 9-Anthracyl substituted BINOL 4e performed similarly to 4b (entry 8). The use of H8-BINOL 4f catalyzed the reaction in much lower enantioselectivity (entry 9). Mono-protected BINOL 4g resulted in racemic product and lower yield (entry 10). Lastly, tartaric acid and its derivitives 4g and 4h, were evaluated as well, but only gave low to moderate enantioselectivies (entries 11 and 12). The commercially available 4b was chosen for use in the assessment of the substrate scope for the reaction.
Table 1.
Asymmetric Arylboration Catalyzed by Chiral Brønsted Acidsa
| ||||
|---|---|---|---|---|
| entry | catalyst | solvent | yieldb | e. r.c |
| 1 | 4a | PhCH3 | 54% | 86:14 |
| 2 | 4b | PhCH3 | 76% | 97:3 |
| 3 | 4b | PhCF3 | 70% | 96:4 |
| 4 | 4b | CH2Cl2 | 75% | 95:5 |
| 5 | 4b | THF | 72% | 90:10 |
| 6 | 4c | PhCH3 | 65% | 92.5:7.5 |
| 7 | 4d | PhCH3 | 65% | 90:10 |
| 8 | 4e | PhCH3 | 73% | 97:3 |
| 9 | 4f | PhCH3 | 66% | 74:26 |
| 10 | 4g | PhCH3 | 57% | 50:50 |
| 11 | 4h | PhCH3 | 47% | 52:48 |
| 12 | 4i | PhCH3 | 72% | 77:23 |
Reactions were run with 0.25 mmol of oQM, 0.5 mmol of boronate in solvent (2.5mL, 0.1M)
Isolated yields.
Enantiomeric ratios determined by HPLC analysis using a chiral stationary phase.
The aforementioned conditions were successfully applied to a range of boronate additions to ortho-quinone methides. Similar yields and enantioselectivities were observed with oQMs bearing both electron-deficient and electron-rich vinyl groups (Table 2, entries 1-4).
Table 2.
Enantioselective Addition of Boronates (2) to Vinyl Ortho-Quinone Methides (1)a
| ||||
|---|---|---|---|---|
| entry | R1b | Ar | yield | e. r.c |
| 1 | Ph (1a) | 4-CH3OC6H4 | 76% (3a) | 97:3 |
| 2 | 4-CH3OC6H4 (1b) | 4-CH3OC6H4 | 74% (3b) | 96.5:3.5 |
| 3 | 4-FC6H4 (1c) | 4-CH3OC6H4 | 85% (3c) | 96.5:3.5 |
| 4 | 4-BrC6H4 (1d) | 4-CH3OC6H4 | 84% (3d) | 97:3 |
| 5 | CH3 (1e) | 4-CH3OC6H4 | 71% (3e) | 96:4 |
| 6 | Ph (1a) |
|
82% (3f) | 95:5 |
| 7 | Ph (1a) | 2,4-CH3OC6H3 | 76% (3g) | 90:10 |
| 8 | Ph (1a) |
|
62% (3h) | 96:4 |
| 9 | Ph (1a) |
|
70% (3i) | 98:2 |
| 10 | Ph (1a) |
|
71% (3j) | 97.5:2.5 |
| 11 | Ph (1a) |
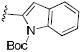
|
46% (3k) | 96:4 |
Isolated yields.
R2 = H, except for 1e.
Enantiomeric ratios determined by HPLC analysis using a chiral stationary phase.
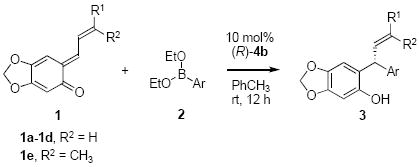
Preparation of quinone methide 1e with a terminal prenyl group was also successful.16 Quinone methide 1e provided the desired aryl addition product under the same conditions in good yield and excellent enantioselectivity (Table 2, entry 5) Good yields and high enantioselectivities were achieved with different aryl boronate nucleophiles (Table 2, entries 6-7). Reactions with hetero-aryl boronates also successful in terms of yield and selectivity (Table 2, entries 8-9). Benzo-2-thiophene boronate 2j and N-boc-indole-2-boronate 2k gave lower yield but similar enantioselectivities (Table 2, entries 10-11). The vinyl group on the ortho-quinone methide could also be replaced by an electron rich aromatic group without destabilizing the quinone methide moiety.17 We evaluated the oQM 5, in combination with alkenyl boronate 6a, under the same conditions. This alkenylation of 5 furnished the S-enantiomer of 3a in good yield and selectivity in a shorter reation time (Scheme 2).
Scheme 2.
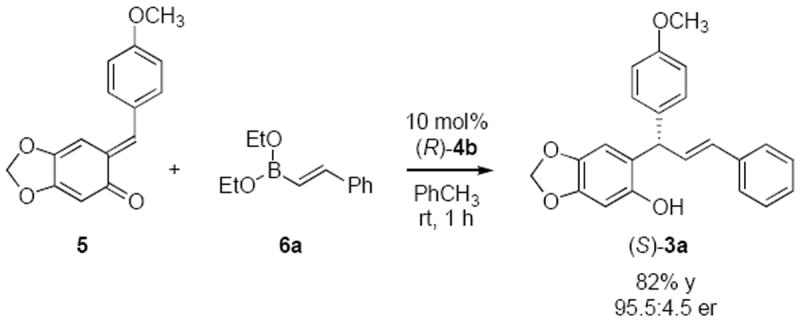
Alkenylation of Ortho-quinone Methides (5) Catalyzed by (R)-4b
At this stage in our investigations, the substrate scope was limited by the number of isolable ortho-quinone methides. Our attention then turned to the development of a method for the generation of in-situ ortho-quinone methide under mild acidic conditions. Reactive oQMs would ideally be trapped by nucleophilic boronates before dimerization or polymerization can occur. Of the methods for oQM generation,18 use of hydroxybenzyl alcohol as the oQM precursor, substrate alcoholysis may be possible by the boronate. Hydroxy-substituted benzyl alcohols have been employed in the photolytic generation of oQMs.19 Scott Snyder completed the total synthesis of resveratrol-based natural products employing in-situ generated quinone methides.20 Thermal generation of ortho-quinone methides from hydroxybenzyl alcohol precursors has also been studied21 used in the synthesis of natural products.22-25 Substitution reactions using vinylboron dihalides have been achieved with n-BuLi activation.26 Alkenyl-,27 allylsilanes28 and enol acetates29 perform substitution of secondary benzylic alcohols.
At the outset of our investigation using hydroxybenzyl alcohol 7a, boronate 6a was acidic enough to promote oQM formation in the presence of BINOL catalysts. (R)-4b was once again the best catalyst in terms of yields and enantiopurities in our initial screens (Table 3, entries 1-5). The addition of 4Å molecular sieves improved the yield (Table 3, entry 6). Switching from hydroxybenzyl alcohol 7a to its ethyl ether 7b gave an er of 95:5, together with 95% isolated yield (Table 3, entry 7). Use of BINOL 4g once again resulted in no enantioselectivity and lower yield. The optimal conditions were determined to be 4 °C with 1.5 equivalent of boronate and 10 mol % of (R)-4b as catalyst (Table 3, entry 9). The 2-hydroxy group was determined crucial for reactivity; absence results in no product (Table 3, entry 10). Also, no product was observed when 2-OCH3 7d substrate was used (Table 3, entry 11) consistent with a quinone methide intermediate. Constituent electron-rich arenes of the hydroxybenzyl ethers proved important for rate and selectivity.
Table 3.
Enantioselective Alkenylation of Hydroxybenzyl Alcohols and Ethersa
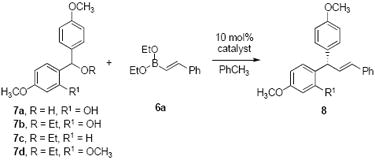
| ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | 7 | R | R1 | yieldb | e. rc |
| 1 | 4a | 7a | H | OH | 46% | 59:41 |
| 2 | 4b | 7a | H | OH | 53% | 75:25 |
| 3 | 4c | 7a | H | OH | 26% | 70:30 |
| 4 | 4d | 7a | H | OH | 40% | 58:42 |
| 5 | 4e | 7a | H | OH | 11% | 66:34 |
| 6d | 4b | 7a | H | OH | 71% | 80:20 |
| 7 | 4b | 7b | Et | OH | 95% | 95:5 |
| 8 | 4g | 7b | Et | OH | 85% | 50:50 |
| 9e | 4b | 7b | Et | OH | 92% | 97.5:2.5 |
| 10 | 4b | 7c | Et | H | 0% | — |
| 11 | 4b | 7d | Et | OCH3 | 0% | — |
Reactions were run with 0.25 mmol of 7, 0.5 mmol of boronate in solvent (2.5mL, 0.1M)
Isolated yields.
Enantiomeric ratios determined by HPLC analysis using a chiral stationary phase.
With 4Å molecular sieves
4 °C.
With these optimal conditions in hand, 2-hydroxybenzyl ethers and alkenyl boronate nucleophiles were evaluated in the reaction. Electron rich and poor substrates (Table 4, products 8a-8c) were effective in the reaction. The absolute stereochemistry was determined by X-ray analysis of compound 8c.30 Simple alkenyl boronate nucleophiles also reacted well (Table 4, products 8f and 8g), elevated temperatures were necessary for reaction (Table 4, products 8h and 8i).
Table 4.
Enantioselective Addition of Vinyl Boronates to Hydroxybenzyl Ethyl Ethersa
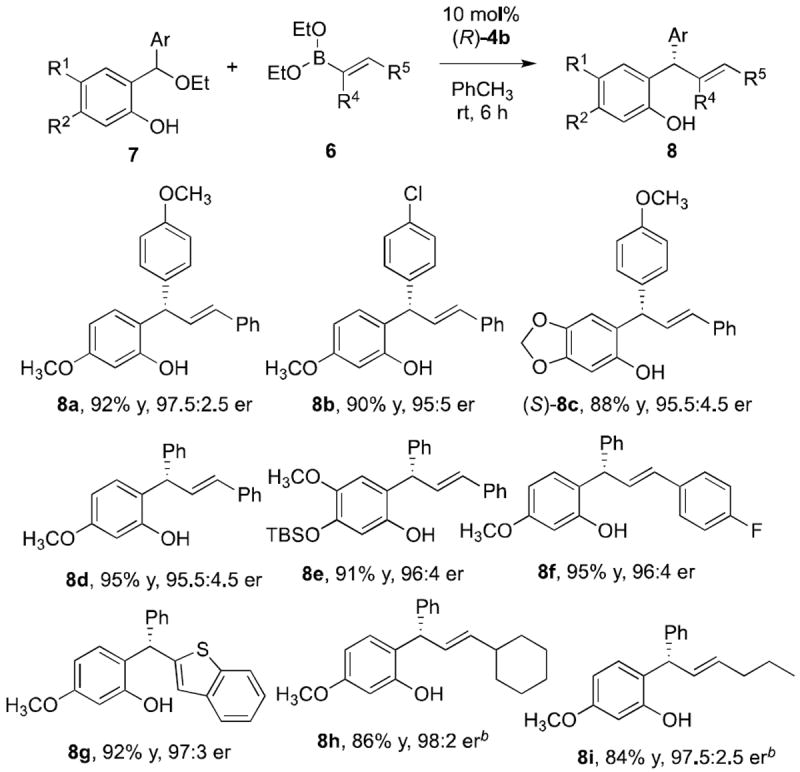
|
Reactions were run with 0.25 mmol of 7, 0.5 mmol of boronate in solvent (2.5mL, 0.1M) at 4 °C for 12h.
60 °C for 12 h.
A short synthesis of the natural product (S)-4-methoxy-dalbergione was accomplished using vinyl boronate as the nucleophile (Scheme 3). Good yield and enantioselectivity of vinyl adduct 9 was achieved at 80 °C and prolonged reaction times using 10 mol% of the catalyst. (S)-4-Methoxy-dalbergione was obtained in 97:3 er by oxidation of phenol 9 with salcomine in DMF under oxygen.32 The stereochemical outcome of the reaction is consistent with the selectivity observed in 1,4-conjugate addition reactions12 and additions to acyl imines.11e The biphenol character of the catalyst is paramount for selectivity. The mechanistic details of the reaction are currently under investigation and will be disclosed.
Scheme 3.
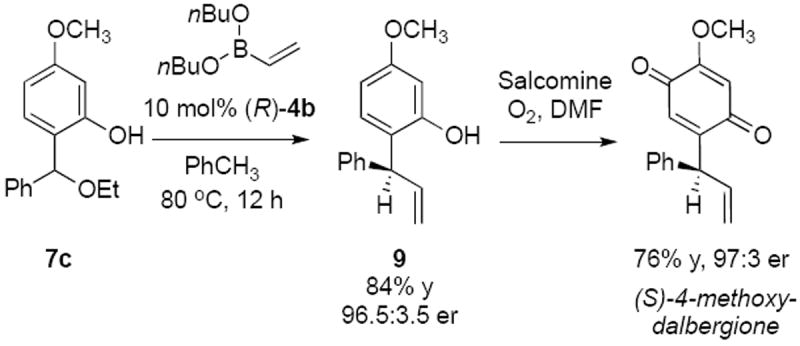
Synthesis of (S)-4-Methoxy-dalbergione
In summary, we have developed a novel enantioselective addition of boronates to ortho-quinone methides catalyzed by chiral biphenols. Good yields and selectivities were achieved using stable oQM with aryl or alkenyl boronates. An in-situ generation of oQMs has also been developed under extremely mild conditions. The method was applied to an improved synthesis of natural product (S)-4-methoxy-dalbergione. Continued investigations include detailed mechanistic studies and further reaction development.
Supplementary Material
Acknowledgments
This research was supported by the NIH (R01 GM078240, S.E.S. and Y.L.) The author thanks Dr. Lauren Brown and Dr. Philip Moquist for helpful discussions concerning this work.
Footnotes
SUPPORTING INFORMATION
Experimental procedures, structural proofs, and spectral data for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.(a) Rokita SE, editor. Quinone Methides. John Wiley & Sons, Inc.; New York: 2009. [Google Scholar]; (b) Boger DL, Weinreb, Hetero SM. Diels-Alder Methodology in Organic Synthesis. Academic Press; San Diego: 1987. [Google Scholar]
- 2.(a) Bulger PG, Bagal SK, Marquez R. Nat Prod Rep. 2008;25:254–297. doi: 10.1039/b705909b. [DOI] [PubMed] [Google Scholar]; (b) Chapman OL, Engel MR, Springer JP, Clardy JC. J Am Chem Soc. 1971;93:6696–6698. [Google Scholar]
- 3.(a) Selenski C, Pettus TRR. J Org Chem. 2004;69:9196–9203. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]; (b) Córdova A, editor. Catalytic Asymmetric Conjugate Reactions. John Wiley & Sons, Inc.; New York: 2010. [Google Scholar]
- 4.(a) Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870–7871. doi: 10.1021/ja103472a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jensen KH, Pathak TP, Zhang Y, Sigman MS. J Am Chem Soc. 2009;131:17074–17075. doi: 10.1021/ja909030c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang Y, Sigman MS. J Am Chem Soc. 2007;129:3076–3077. doi: 10.1021/ja070263u. [DOI] [PubMed] [Google Scholar]; (d) Gligorich KM, Schultz MJ, Sigman MS. J Am Chem Soc. 2006;128:2794–2795. doi: 10.1021/ja0585533. [DOI] [PubMed] [Google Scholar]
- 5.(a) Marsini MA, Huang YD, Lindsey CC, Wu KL, Pettus TRR. Org Lett. 2008;10:1477–1480. doi: 10.1021/ol8003244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Green JC, Pettus TRR. J Am Chem Soc. 2011;133:1603–1608. doi: 10.1021/ja109925g. [DOI] [PubMed] [Google Scholar]; (c) Tuttle K, Rodriguez AA, Pettus TRR. Synlett. 2003:2234–2236. [Google Scholar]
- 6.(a) Hall DG. Boronic Acids Preparations and Applications in Organic Synthesis and Medicine. Wiley-VCH; Weinheim: 2005. [Google Scholar]; (b) Wu TR, Chong JM. Org Lett. 2006;8:15–18. doi: 10.1021/ol0523087. [DOI] [PubMed] [Google Scholar]; (c) Jain P, Wang H, Houk KN, Antilla JC. Angew Chem Int Ed. 2012;51:1391–1394. doi: 10.1002/anie.201107407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044–8049. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira SB, Silva FC, Pinto AC, Gonzaga DTG, Ferreira VF. J Heterocycl Chem. 2009;46:1080. [Google Scholar]
- 8.Myristinin A: Maloney DJ, Deng JZ, Starck SR, Gao Z, Hecht SM. J Am Chem Soc. 2005;127:4140–4141. doi: 10.1021/ja042727j.. Deng JZ, Starck SR, Li SS, Hecht SM. J Nat Prod. 2005;68:1625–1628. doi: 10.1021/np058064g.. Dalbergione: Beldjoudi N, Mambu L, Labaied M, Grellier P, Ramanitrahasimbola D, Rasoanaivo P, Martin MT, Frappier F. J Nat Prod. 2003;66:1447–1450. doi: 10.1021/np030008x.. Tolterodine: Srinivas K, Srinivasan N, Reddy KS, Ramakrishna M, Reddy CR, Arunagiri M, Kumari RL, Venkataraman S, Mathad VT. Org Process Res Dev. 2005;9:314–318.
- 9.(a) Alden-Danforth E, Scerba MT, Lectka T. Org Lett. 2008;10:4951–4953. doi: 10.1021/ol802029e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bekele T, Shah MH, Wolfer J, Abraham CJ, Weatherwax A, Lectka T. J Am Chem Soc. 2006;128:1810–1811. doi: 10.1021/ja058077g. [DOI] [PubMed] [Google Scholar]
- 10.(a) Pathak TP, Sigman MS. Org Lett. 2011;13:2774–2777. doi: 10.1021/ol200913r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pathak TP, Sigman MS. J Org Chem. 2011;76:9210–9215. doi: 10.1021/jo201789k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660–12661. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]; (b) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398–15404. doi: 10.1021/ja075204v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barnett DS, Moquist PN, Schaus SE. Angew Chem Int Ed. 2009;48:8679–8682. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Barnett DS, Schaus SE. Org Lett. 2011;13:4020–4023. doi: 10.1021/ol201535b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bishop JA, Lou S, Schaus SE. Angew Chem Int Ed. 2009;48:4337–4340. doi: 10.1002/anie.200901023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lou S, Schaus ES. J Am Chem Soc. 2008;130:6922–6923. doi: 10.1021/ja8018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Chong JM, Shen LX, Taylor NJ. J Am Chem Soc. 2000;122:1822–1823. [Google Scholar]; (b) Wu TR, Chong JM. J Am Chem Soc. 2005;127:3244–3245. doi: 10.1021/ja043001q. [DOI] [PubMed] [Google Scholar]; (c) Wu TR, Chong JM. J Am Chem Soc. 2007;129:4908–4909. doi: 10.1021/ja0713734. [DOI] [PubMed] [Google Scholar]; (d) Turner HM, Patel J, Niljianskul N, Chong JM. Org Lett. 2011;13:5796–5799. doi: 10.1021/ol202391r. [DOI] [PubMed] [Google Scholar]
- 13.(a) Lv H, You L, Ye S. Adv Synth Catal. 2009;351:2822–2826. [Google Scholar]; (b) Petasis NA, Butkevich AN. J Organomet Chem. 2009;694:1747–1753. doi: 10.1016/j.jorganchem.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Han WY, Wu ZJ, Zhang XM, Yuan WC. Org Lett. 2012;14:976–979. doi: 10.1021/ol203109a. [DOI] [PubMed] [Google Scholar]
- 14.Brunel JM. Chem Rev. 2005;105:857–898. doi: 10.1021/cr040079g. [DOI] [PubMed] [Google Scholar]
- 15.(a) Moquist PN, Kodama T, Schaus SE. Angew Chem Int Ed. 2010;49:7096–7100. doi: 10.1002/anie.201003469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kodama T, Moquist PN, Schaus SE. Org Lett. 2011;13:6316–6319. doi: 10.1021/ol2028702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop LM, Winkler M, Houk KN, Bergman RG, Trauner D. Chem Eur J. 2008;14:5405–5408. doi: 10.1002/chem.200800662. [DOI] [PubMed] [Google Scholar]
- 17.Jurd L. Tetrahedron. 1977;33:163–168. [Google Scholar]
- 18.Van de Water RW, Pettus TRR. Tetrahedron. 2002;58:5367–5405. [Google Scholar]
- 19.For a brief review, see: Wan P, Barker B, Diao L, Fischer M, Shi Y, Yang C. Can J Chem. 1996;74:465–475.. Also see: Chiang Y, Kresge AJ, Zhu Y. Pure Appl Chem. 2000;72:2299–2308.. Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2000;122:9854–9855.. Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2001;123:8089–8094. doi: 10.1021/ja010826g.
- 20.(a) Snyder SA, Gollner A, Chiriac MI. Nature. 2011;474:461–466. doi: 10.1038/nature10197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Snyder SA, Zografos AL, Lin YQ. Angew Chem Int Ed. 2007;46:8186–8191. doi: 10.1002/anie.200703333. [DOI] [PubMed] [Google Scholar]
- 21.(a) Chiba K, Hirano T, Kitano Y, Tada M. Chem Commun. 1999:691–692. [Google Scholar]; Merijan A, Gardner PD. J Org Chem. 1965;30:3965–3967. [Google Scholar]; (b) Gardner PD, Sarrafizadeh RH, Brandon RL. J Am Chem Soc. 1959;81:5515–5515. [Google Scholar]
- 22.Ghisalberti EL. Phytochemistry. 1996;41:7–22. doi: 10.1016/0031-9422(95)00484-x. [DOI] [PubMed] [Google Scholar]
- 23.Tatsuta K, Tamura T, Mase T. Tetrahedron Lett. 1999;40:1925–1928. [Google Scholar]
- 24.Lawrence AL, Adlington RM, Baldwin JE, Lee V, Kershaw JA, Thompson AL. Org Lett. 2010;12:1676–1679. doi: 10.1021/ol100138k. [DOI] [PubMed] [Google Scholar]
- 25.Spence JTJ, George JH. Org Lett. 2011;13:5318–5321. doi: 10.1021/ol202181g. [DOI] [PubMed] [Google Scholar]
- 26.(a) Kabalka GW, Yao ML, Borella S, Wu ZZ. Org Lett. 2005;7:2865–2867. doi: 10.1021/ol050778v. [DOI] [PubMed] [Google Scholar]; (b) Rueping M, Uria U, Lin MY, Atodiresei I. J Am Chem Soc. 2011;133:3732–3735. doi: 10.1021/ja110213t. [DOI] [PubMed] [Google Scholar]
- 27.Nishimoto Y, Kajioka M, Saito T, Yasuda M, Baba A. Chem Commun. 2008:6396–6398. doi: 10.1039/b816072d. [DOI] [PubMed] [Google Scholar]
- 28.Rubin M, Gevorgyan V. Org Lett. 2001;3:2705–2707. doi: 10.1021/ol016300i. [DOI] [PubMed] [Google Scholar]
- 29.Nishimoto Y, Onishi Y, Yasuda M, Baba A. Angew Chem Int Ed. 2009;48:9131–9134. doi: 10.1002/anie.200904069. [DOI] [PubMed] [Google Scholar]
- 30.CCDC 879792 contains the crystallographic data for this paper (also available as Supporting Information). These data can be obtained free of charge from The Cambridge Crystallographic Centre via www.ccdc.cam.ac.uk/data_request/cif
- 32.(a) De Jonge CR, Hageman HJ, Hoentjen G, Mijs WJ. Org Synth. 1983;57:78. [Google Scholar]; (b) Bissel P, Nazih A, Sablong R, Lepoittevin JP. Org Lett. 1999;1:1283–1285. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.