

Highlights
-
•
Retarded cleavage of abasic site when in nucleosomal DNA by AP endonuclease.
-
•
Efficient cleavage of abasic site when in nucleosomal DNA by CHO-K1 nuclear extracts.
-
•
Efficient cleavage of bistranded abasic sites in a cluster in nucleosomal DNA by cell extract.
-
•
Cleavage of abasic site in bistranded cluster with oxidized guanine in nucleosomal DNA.
Keywords: Clustered DNA damage, BER, Nucleosome, AP site, 8-oxoG
Abstract
Clustered DNA damage is a unique characteristic of radiation-induced DNA damage and the formation of these sites poses a serious challenge to the cell’s repair machinery. Within a cell DNA is compacted, with nucleosomes being the first order of higher level structure. However, few data are reported on the efficiency of clustered-lesion processing within nucleosomal DNA templates. Here, we show retardation of cleavage of a single AP site by purified APE1 when contained in nucleosomal DNA, compared to cleavage of an AP site in non-nucleosomal DNA. This retardation seen in nucleosomal DNA was alleviated by incubation with CHO-K1 nuclear extract. When clustered DNA damage sites containing bistranded AP sites were present in nucleosomal DNA, efficient cleavage of the AP sites was observed after treatment with nuclear extract. The resultant DSB formation led to DNA dissociating from the histone core and nucleosomal dispersion. Clustered damaged sites containing bistranded AP site/8-oxoG residues showed no retardation of cleavage of the AP site but retardation of 8-oxoG excision, compared to isolated lesions, thus DSB formation was not seen. An increased understanding of processing of clustered DNA damage in a nucleosomal environment may lead to new strategies to enhance the cytotoxic effects of radiotherapeutics.
1. Introduction
Many thousands of DNA modifications are induced daily in each cell through reactive oxygen species (ROS), formed as a by-product of aerobic metabolism, in contrast to the much lower levels of DNA damage induced at low doses of ionizing radiation. In contrast, the formation of clustered DNA damage sites, defined as two or more lesions formed within one or two helical turns of the DNA double helix by a single radiation track, is a characteristic of exposure to ionizing radiation when compared with their rarer occurrence when induced endogenously [1], [2]. The type of lesions produced via exposure to ionizing radiation is thought to be chemically identical to those formed by ROS [3], [4]. The formation of clustered DNA damage has been experimentally verified in both isolated DNA and mammalian cells [5], [6], [7], [8], [9], [10], [11]. The yield of bistranded non-DSB clusters is at least 4–8 times that of prompt DSB damage [7], [9], [10], with ∼10% of the total yield of non-DSB clustered damage converted into DSB at early times following γ-irradiation in xrs5 cells [12]. As such, it is the formation of these clustered damage sites, including DSB with lesions downstream of the DSB ends, that ultimately determines the severity of the biological consequences of exposure to ionizing radiation.
Within the cell, the base excision repair (BER) pathway is predominantly used for repair of base lesions, AP sites and SSB, irrespective of whether they are formed endogenously or through exposure to ionizing radiation [13], [14]. Numerous studies using oligonucleotides containing synthetic clustered damage sites have verified the hypothesis [15] that clustered DNA damage sites are more difficult to repair than single lesions, as a result of the reduced efficiency of BER (reviewed in [16], [17], [18]). Complementary investigations using plasmid-based bacterial or mammalian reporter systems confirmed the retardation of BER during the repair of clustered damage sites (reviewed in [16], [17], [18], [19], [20]) and as a consequence an increase in mutation frequency was seen resulting from the lifetime extension of lesions within clusters.
Cellular DNA must be compacted in order to be contained within the nucleus and DNA compaction is highly conserved throughout eukaryotes [21], with the first level of DNA higher order structure being the nucleosome. The nucleosome structure consists of a dimer of H3-H4 histones heterodimers with two individual heterodimers of H2A–H2B that bind to form a histone octamer. Approximately 147 base pairs of DNA associate with the histone octamer; the central 60 base pairs bound by the H3–H4 octamer whilst the H2A–H2B dimers contact 30 base pairs either side of this central region [22]. The superhelical turns of the DNA do not remain constant throughout the nucleosome, with a more relaxed structure observed towards the outer regions than at the central dyad [23]. Although DNA compaction is essential, the presence of the histone octamer has been shown to interfere with cellular processes, particularly DNA repair, as binding to the histone proteins causes DNA distortion and may also provide a physical block to interactions of other proteins. This is evident in a number of studies where reduced digestion with restriction enzymes was observed in nucleosomal templates in comparison to non-nucleosomal (“free”) DNA [24], [25], [26], [27]. Despite this level of protection offered by histone-association, the DNA is still susceptible to damaging agents.
Only a few studies have reported on the levels of DNA damage processing by BER within nucleosomes and have focused on the level of retardation of repair of DNA damage seen within nucleosomal DNA compared with free DNA, using purified proteins. Despite some variability in the extent of retardation, attributed to different DNA sequences used with varying positioning affinity, the data unequivocally show that repair occurs less efficiently within nucleosomal templates as opposed to free DNA [28], [29], [30], [31], [32], [33], [34]. Importantly, treatment of reconstituted nucleosomes with repair proteins does not lead to disruption of the structure, with the exception of the ligaseIII/XRCC1 complex [34]. Efficiency of enzymatic processing may differ between damage located close to the dyad rather than towards the outer regions of the nucleosome [26], [27], [30] in that a higher level of retardation was observed when the lesion is placed closer to the dyad. Further, the efficiency of processing is dependent upon whether a lesion was oriented “in” i.e., facing the histone octamer, or “out” i.e., facing solution, with higher levels of retardation observed with lesions facing inwards [26], [27], [28], [29], [30], [34], [35], [36], [37] and recently reviewed in ref [38]. Addition of the chromatin remodelling factor SWI/SNF leads to an increase in the rate of excision of 8-oxoG by OGG1 from native nucleosomes [31]. The activity of polymerase (pol) β within nucleosomal templates was demonstrated to be minimal [28], [29], [31] leading to a stall in the BER pathway. Despite a reduction in the level of ligation achieved within nucleosomes [33], [39], [40], this appeared to be the most efficient step during repair by BER of lesions contained within nucleosomal DNA. However, a more recent study has shown contrasting results to these earlier studies in that pol β could process its substrate in nucleosomal bound DNA but ligation could only occur if the nucleosome had been disrupted [34]. The retardation of DNA repair is not limited to the BER proteins; reports of similar levels of protein retardation have been shown in the nucleotide excision repair, mismatch repair and non-homologous end joining pathways [25], [41], [42], [43]. Few studies have been undertaken on bistranded clusters in a nucleosome environment. However, two recent studies on bistranded clustered damage showed that the efficiency of glycosylases or APE1 to cause SSB from excision of a lesion within the clusters is even more reduced when in a nucleosome environment and importantly cause suppression of BER-generated DSB [44], [45] than previously reported in ‘free’ DNA [16], [17], [18].
Building on ours and others studies of the efficiency of clustered DNA damage processing by both mammalian nuclear extract and purified proteins, within short sequence oligonucleotides (reviewed in [16]), we have now extended this present study to nucleosomal DNA containing clustered damage sites, to assess the rates of cleavage of the lesions by CHO-K1 nuclear extract, in comparison with that of purified AP endonuclease (APE1). To date, studies investigating the processing of DNA lesions within nucleosomes have generally focused on repair of single lesions. We have designed DNA sequences containing a single AP site or clustered sites containing two lesions, either bistranded AP sites or an AP site opposing an 8-oxoG residue (see Table 1). The mono-nucleosome constructs contain these DNA damage sites close to the nucleosome dyad. Through treatment of these DNA substrates with either purified APE1 or OGG1 and CHO-K1 nuclear extract, we aim to gain more insight into the processing of clustered DNA damage within the added complexity of a higher order DNA environment. Using clustered DNA damage, we identified the formation of DSB through cleavage of opposing AP sites which leads to nucleosome disruption. In contrast, an AP site opposing 8-oxoG does not lead to DSB but retardation of cleavage of the 8-oxoG lesion was observed. Significant retardation of cleavage of the single AP site contained within nucleosomes by APE1 was noted, in comparison to free DNA, which was alleviated when DNA substrates were incubated with CHO-K1 nuclear extract.
Table 1.
Oligonucleotide sequences.
| Cluster | Sequence | Strand |
|---|---|---|
| AP control | 5′ TTGGTGCGTTTAAGCCGTGC 3′ | 1 |
| 3′ CGCAACCACYCAAATTCGGC 5′ | 2 | |
| THF control | 5′ TTGGTGCGTTTAAGCCGTGC 3′ | 1 |
| 3′ CGCAACCACZCAAATTCGGC 5′ | 2 | |
| 8-oxoG control | 5′ TTGGTGCGTTTAAGCCGTGC 3′ | 1 |
| 3′ CGCAACCACXCAAATTCGGC 5′ | 2 | |
| AP/AP + 1 | 5′ TTGGTGCYTTTAAGCCGTGC 3′ | 1 |
| 3′ CGCAACCACYAAAATTCGGC 5′ | 2 | |
| AP/AP − 3 | 5′ TTGYTGCGTTTAAGCCGTGC 3′ | 1 |
| 3′ CGCAACAACYCAAATTCGGC 5′ | 2 | |
| AP/8-oxoG + 1 | 5′ TTGGTGCXTTTAAGCCGTGC 3′ | 1 |
| 3′ CGCAACCACYCAAATTCGGC 5′ | 2 |
Oligonucleotide sequences: Y represents a uracil residue converted to an AP site before use and X represents 8-oxoG. The lesions on strand 1 have been given a number relating their positions to the AP site on the complementary strand 2. The number denotes the base separation. A positive number is given if the lesion on strand 1 is in the 5′ direction to that on strand 2 and a negative number is given if the lesion on strand 1 is in the 3′ direction to that on strand 2. The AP, THF and 8-oxoG control oligonucleotides consists of a single lesion on strand 2 with no lesion on strand 1.
2. Materials and methods
2.1. Substrate oligonucleotides
The oligonucleotide sequences, as depicted in Table 1, were purchased PAGE purified with 5′ phosphorylated termini from Eurogentec. Strand 1 contains either uracil (Y) or 8-oxoG (X) at variable positions. Strand 2 contains a single uracil (Y) at a fixed position. The control oligonucleotides contain a single uracil (Y), tetrahydrofuran (Z) or 8-oxoG (X) present in strand 2 and no lesion in strand 1. Based on the previous nomenclature [46] lesions situated on strand 1, 3′ to the single uracil found on strand 2 within the cluster are given a negative number and 5′ lesions a positive number, the number relating to the base separation of the lesions.
2.2. Preparation of lesion-containing DNA for nucleosome reconstitution
The plasmid pGEM3Z-601 contains the strong rotational positioning 601 DNA sequence [47], [48]. In order to incorporate restriction site sequences at each termini of the 601 sequence, plasmid DNA was used as a PCR template using the following primers: forward 5′ – CTCGGAATTCTATCCGACTGGCACCGGCAAG – 3′ (EcoRI) and reverse 5′ – GCATGATTCTTAAGACCGAGTTCATCCCTTATGTG – 3′ (Bst98I). Following purification, a central 20 bp fragment was released from the 601 sequence by incubation with Van91I (Fermentas) and BglI (Fermentas) at 37 °C for 16 h. Fragments of size 126 bp and 106 bp were gel purified from a 3% agarose (Lonza) gel using a Qiagen MinElute gel extraction kit, according to manufacturer’s instructions. Lesion-containing 20 bp fragments (see Table 1) were hybridized in Tris–EDTA buffer pH 8.0 by heating at 90 °C for 5 min before being left to cool slowly over 2–3 h. In a two-step ligation, double-stranded lesion-containing oligonucleotides were ligated to the 126 bp fragment by incubation with 30 U T4 DNA ligase (Roche) in 150 μL buffer (66 mM Tris–HCl, 5 mM MgCl2, 10 mM DTT, 10 mM ATP pH 7.5 at 20 °C) for 16 h at 4 °C. Excess 20 bp oligonucleotides were removed by passage through a Qiagen QIAquick nucleotide removal kit, according to manufacturer’s instructions. The ligation product was incubated with a 1.5-fold excess of 106 bp DNA in 120 μl buffer (66 mM Tris–HCl, 5 mM MgCl2, 10 mM DTT, 10 mM ATP pH 7.5 at 20 °C) with 30 U T4 DNA ligase (Roche) for 16 h at 4 °C. The resultant 252 bp full length product was purified from a 3% agarose (Lonza) gel using a Qiagen MinElute gel extraction kit, according to manufacturer’s instructions.
2.3. Preparation of 5′-end labelled oligonucleotides
To prepare strand 1 (see Table 1) for radiolabelling, 100 ng DNA was digested with 20 U Bst98I (Fermentas) in 20 μL buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 100 mM NaCl, 0.1 mg/mL BSA) for 16 h at 37 °C. To prepare strand 2 for radiolabeling, 100 ng DNA was digested with 40 U EcoRI (NEB) in 20 μL buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 100 mM NaCl, 0.02% Triton X-100, 0.1 mg/mL BSA) for 16 h at 37 °C. For 5′ 32P-end labelling, 100 ng DNA was incubated with 15 U T4 polynucleotide kinase (invitrogen) and 15 μCi of [γ-32P] ATP (6000Ci/mmol, 10 mCi/mL, PerkinElmer LAS) in 20 μL forward reaction buffer (70 mM Tris–HCl pH 7.6, 10 mM MgCl2, 100 mM KCl, 1 mM 2-mercaptoethanol) for 1 h at 37 °C. Unincorporated radionuclide was subsequently removed by ethanol precipitation and the resultant DNA pellet was resuspended in 30 μL Tris–EDTA pH 8.0. The DNA was then incubated with 1 U uracil DNA glycosylase (invitrogen) in 50 μL buffer (10 mM Tris–HCl pH 7.5, 50 mM NaCl, 1 mM EDTA) at 37 °C for 2 h, to convert the uracil residue within the radiolabeled 601 sequence to an AP site. The percentage conversion of uracil to AP site was confirmed as previously described [49].
2.4. Nucleosome reconstitution
Nucleosomes were reconstituted using the standard salt-step dialysis method. In brief, nucleosomes were reconstituted with 1.5 μg carrier DNA (approximately 150 bp length), 1 μg un-labelled 255 bp 601 oligonucleotide, 20 ng 32P-5′ radiolabelled 601 oligonucleotide and ∼2.5 μg native chicken erythrocyte histone mix in 100 μL buffer (2 M NaCl, 10 mM Tris–HCl pH 7.4, 1 mM EDTA, 5 mM 2-mercaptoethanol, 0.1 mg/mL BSA, 0.01% Nonidet P-40). The exact ratio of DNA: histones was determined experimentally to ensure <5% free DNA remained after reconstitution. The mixture was dialyzed at 4 °C using dialysis tubing (Spectra-por, MW cut-off 6000–8000) against decreasing salt buffers containing 10 mM Tris–HCl pH 7.4, 5 mM 2-mercaptoethanol, 1 mM EDTA: 1.6 M NaCl for 30 min; 1.2 M, 1.0 M, 0.8 M and 0.6 M NaCl for 90 min and 0.3 M NaCl for 30 min. Finally the reconstitution was dialyzed against 10 mM NaCl (10 mM Tris–HCl pH 7.4, 0.25 mM EDTA) for 14–18 h. Verification of reconstitution was performed by subjecting a 5 μL sample, with a non-reconstituted (free) DNA sample for comparison, to electrophoresis on a 5% native polyacrylamide gel in 0.3× TBE at a constant 12 V/cm for 80 min at room temperature.
2.5. Preparation of nuclear extract
The nuclear extracts were prepared as previously described [46] from CHO-K1 cells. In summary, cells were harvested in exponential phase and the pelleted cells were resuspended in an equal volume of buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT). To lyse the cytoplasmic membranes the cells were drawn through a 0.5 mm diameter needle 10 times. Following a brief centrifugation at 18,000 × g at 4 °C the nuclei were collected and resuspended in two-third volume of high salt buffer (20 mM HEPES pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF) for 30 min with agitation, on ice. Following centrifugation at 18,000 × g for 10 min at 4 °C, the supernatant was dialyzed for 16 h against 1 L of buffer (20 mM HEPES pH 7.9, 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF). The protein concentration was determined using the Bradford colorimetric technique and was found to be between 6.6 and 11.1 mg/mL. Aliquots of nuclear extracts were stored at −80 °C.
2.6. Processing of lesion-containing 601 DNA
Radiolabelled nucleosomal DNA (0.25 ng, equivalent to 30 ng of reconstituted nucleosome) or radiolabelled free DNA (0.25 ng, in presence of 30 ng of native chicken erythrocyte nucleosomes) was incubated with varying amounts of purified APE1 (human origin); CHO-K1 nuclear extract; Fpg (E. coli origin), Nth (E. coli origin) or OGG1 together with 10 ng purified APE1 or 1 μg CHO-K1 nuclear extract, in 5 μL of buffer (1 mM MgCl2, 0.2 mM EDTA, 30 mM KCl) for 30 min at 37 °C. To stop the reaction 90 μL buffer (0.1% SDS, 25 mM EDTA) was added.
2.7. Repair of lesion-containing 601 DNA by CHO-K1 nuclear extract
Nucleosomal DNA (0.25 ng, equivalent to 30 ng of reconstituted nucleosome) or free DNA (0.25 ng), was incubated with 3.0 μg CHO-K1 nuclear extract in 5 μL buffer (70 mM Tris–HCl pH 7.5, MgCl2 10 mM, DTT 10 mM, ATP 4 mM, phosphocreatine 40 mM, phosphocreatine kinase 3.2 μg/mL, 2 mM NAD, 0.1 mM each of dATP, dCTP, dTTP and dGTP) for 0, 1, 5, 20, 60 min to investigate lesion repair. The reaction was stopped by the addition of 95 μL buffer (1 μg proteinase K, 0.1% SDS, 25 mM EDTA).
2.8. Purification and electrophoresis of nucleosome reactions
A 100 μL phenol chloroform (25:24:1 phenol:chloroform:isoamyl alcohol, Fluka) was added to each reaction, at room temperature. All samples were vortexed and centrifuged at 17,950 × g for 10 min to collect the DNA and the DNA was isolated by ethanol precipitation. Where the cleavage of the lesion was to be assessed, the DNA pellet was resuspended in 4 μL denaturing loading buffer (98% formamide, 2 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol). DNA was then denatured by heating at 90 °C for 60 s. The samples were immediately put on ice then subjected to electrophoresis on an 8% denaturing polyacrylamide gel at a constant 30 V/cm for 30 min at room temperature. For non-denaturing samples where DSB formation was investigated, 4 μL native loading buffer (40% sucrose, 5 mM EDTA, 0.025% bromophenol, 0.025% xylene cyanol) was used to resuspend the DNA pellets after ethanol precipitation and samples were subjected to electrophoresis on a 5% native polyacrylamide gel at a constant 12 V/cm for 45 min at room temperature. Dried gels were exposed to a Bio-Rad Phosphorimager screen for visualization of repair products using phosphorimaging technology (Bio-Rad, Molecular Imager FX) and the image was quantified with Quantity One software (Bio-Rad, Hercules, CA). The intensity of the band representing cleaved DNA (either as a single lesion or DSB formation) was expressed as a percentage of the total intensities of all bands within each reaction. The error bars represent standard error of the mean from at least three independent experiments.
3. Results
3.1. Inhibition of purified APE1 to incise a single AP site within nucleosomal substrates
We first investigated the level of cleavage of a single AP site by incubation with increasing amounts of APE1 for 30 min, in both nucleosomal and free DNA templates. A single AP site within the DNA template was situated 10 bp from the nucleosome dyad and mid-way between facing solution and the histone octamer. Following incubation of both DNA templates with varying amounts of APE1, reaction products (seen as SSB) were separated by PAGE (Fig. 1A).
Fig. 1.

APE1 cleavage of a single AP site within free and nucleosomal DNA substrates. (A) Representative phosphorimaging scan of a denaturing polyacrylamide gel showing the cleavage of a single AP site within free (left) or nucleosomal-bound (right) DNA by APE1. (B) Cleavage of the AP site within free DNA (square) or nucleosomal-bound DNA (triangle) by purified APE1. (*) The cleavage of nucleosomal-bound DNA was compared to free DNA for each amount of APE (ng) used. Student t-tests were performed to identify statistically significant differences in cleavage. P-values <0.05 were considered to be significant. (C) Representative phosphorimaging scan of a denaturing polyacrylamide gel showing the cleavage of a single AP site within free (left) or nucleosomal-bound DNA (right) by CHO-K1 nuclear extract. (D) Cleavage of the AP site within free DNA (square) or nucleosomal-bound DNA (triangle) by CHO-K1 nuclear extract. Error bars represent standard deviation determined from at least three independent experiments. In (B) and (D) the background has been subtracted from the data prior to normalization to 100% for each experiment.
The incision of the AP site by various amounts of APE1 was assessed by comparing the levels of DNA found in the intact or cleaved bands following quantification. Cleavage of the free DNA, (evident at the lowest concentration of enzyme used), becomes saturated ≥0.8 ng APE1 (Fig. 1B). In contrast, incision of the AP site in nucleosomal DNA does not become saturated until APE1 concentrations are >1 ng and even at saturation, a 3.3 fold lower level of AP site incision was seen than that in free DNA (Fig. 1B). At low concentrations of APE1 (<0.4 ng), the level of AP site incision is reduced by 3.6 fold in nucleosomal DNA compared to free DNA, consistent with the reduction reported in [45]. When AP site cleavage becomes saturated in the free DNA (at 0.8 ng APE1) an approximately 4.7 fold decrease was seen in the level of APE1 cleavage of the AP site in nucleosomal-bound DNA.
3.2. Efficient cleavage of single AP sites within a nucleosomal template by nuclear extract
In contrast, an AP site is cleaved to comparable levels by 1 μg CHO-K1 nuclear extract when present in free DNA or nucleosomal-bound DNA (Fig. 1D). Fig. 1C shows a representative phosphorimaging scan of a polyacrylamide gel with quantification of the level of cleavage of the AP site expressed graphically in Fig. 1D. At the lower concentrations of nuclear extract used (<300 ng), the efficiency of cleavage of the AP site in nucleosomal bound DNA is approximately 2 fold lower when compared with free DNA. However at higher concentrations, the reduction in efficiency of AP site cleavage is only 1.4 fold. It was determined that 1 μg CHO-K1 nuclear extract contains approximately 3 ng APE1 (Supplementary Fig. 1). Thus, in the nucleosome, the AP site incision at the highest concentration (1 μg) of CHO-K1 nuclear extract used (77% relative% DNA cleaved) is 2.5 fold greater than with 3 ng APE1 (30% relative % DNA cleaved). Therefore, the difference in the extent of cleavage of the nucleosomal-bound AP site by either CHO-K1 nuclear extract or purified APE1 is not due to a greater abundance of APE1 in CHO-K1 nuclear extract. In addition, it was verified that the CHO-K1 nuclear extract incubation does not disrupt the nucleosome structure (Supplementary Fig. 2) and therefore, the difference in cleavage levels is not due to either DNA degradation or nucleosome disruption by the extract.
Fig. 2 shows glycosylase mediated incision of an AP site to determine if higher efficiency of incision of an AP site from nucleosomal-bound DNA using CHO-K1 nuclear extracts than purified APE1 is due to glycosylase cleavage.
Fig. 2.

Cleavage of a THF lesion and an AP site in free and nucleosomal bound DNA. (A) Cleavage of an AP site site by purified Fpg. (B) Cleavage of an AP site site by purified Nth. (C) Cleavage of an AP site site by purified APE1. (D) Cleavage of a THF lesion by purified APE1. (E) Cleavage of a THF lesion by CHO-K1 nuclear extract. (Square) free DNA, (triangle) nucleosomal-bound DNA. For each experiment, the background has been subtracted from the data prior to normalization to 100%. Error bars represent standard deviation determined from at least three independent experiments.
The incision of an AP site by Fpg is similar to that by CHO-K1 nuclear extract. As AP site incision in free DNA approaches saturation (at 1 ng) there is ∼1.5 fold less incision of an AP site with nucleosomal-bound DNA, by 10 ng the difference is 1.3 fold (Fig. 2A). In contrast, a larger differential was seen between the incision of an AP site from free DNA and nucleosomal-bound DNA by Nth. Nth incision of an AP site from free DNA approaches saturation at 0.1 ng Nth, whereas the incision of an AP site from nucleosomal-bound DNA is 2.7 fold less. However, by 10 ng Nth there is only a 1.3 fold decrease in the level of AP site incised from nucleosomal-bound DNA compared to free DNA (Fig. 2B). The histone proteins used to reconstitute mononucleosomes for the glycosylase experiments were from a different preparation than that used for all the other experiments in this current study, therefore the level of AP site incision with APE1 was reassessed. Similar levels of AP site were incised by APE1 and Nth in both free and nucleosomal-bound DNA (Fig. 2B and C). Thus it appears as if certain, but not all, glycosylases (possibly the Fpg functional homologue OGG1) could contribute to AP site incision in CHO-K1 nuclear extract.
To explore this possibility further, a tetrahydrofuran (THF) lesion was substituted for an AP site and cleavage of the THF lesion was measured. A THF lesion is an analogue of a reduced AP site and is thought to be refractory to incision by DNA glycosylases, i.e., it is predominantly be cleaved by APE1. Therefore, if there is a large reduction in the level of THF cleaved by CHO-K1 nuclear extract when it is in nucleosomal bound DNA, compared to free DNA, as seen for an AP site using purified APE1 (Fig. 1B), it can be inferred that the AP site is incised, in part, by glycosylases present in the nuclear extract. Fig. 2D and E show that, whilst at lower levels of purified APE1 and CHO-K1 nuclear extract the THF is incised less efficiently when nucleosomal-bound than when in free DNA, at higher levels THF lesion is incised from nucleosomal-bound DNA to a comparable level by both purified APE1 and CHO-K1 nuclear extract. THF incision by APE1 approaches saturation at 0.4 ng when in free DNA but with a 2.1 fold lower incision of the THF when nucleosomal-bound (Fig. 2D). THF cleavage steadily increases as the amount of APE1 increases, until no difference was seen in the incision levels of a THF from free or nucleosomal-bound DNA at 10 ng APE1. A similar trend was seen with CHO-K1 nuclear extract, but at the highest amount of nuclear extract used there remains a 1.2 fold decrease in the level of THF cleaved from nucleosomal-bound DNA than from free DNA (Fig. 2E). From the similarity in the extent of cleavage by APE1 and with APE1 or nuclear extracts at the higher concentrations, it is inferred that the contribution of cleavage of THF by glycosylases or polynucleotide kinase phosphatase [50] is minimally.
3.3. Cleavage of AP sites within clustered damage sites in nucleosomal templates by CHO-K1 nuclear extract
Having determined that cleavage of a single AP site is comparable within free and nucleosomal-bound DNA, following treatment with CHO-K1 nuclear extract, we wished to investigate the efficiency of cleavage of AP sites when present within a clustered damaged site and whether any DSB result. We designed two bistranded AP site clusters, AP/AP + 1 and AP/AP − 3, in addition to a cluster with an 8-oxoG substituting one AP site, AP/8-oxoG + 1 (see Table 1).
3.3.1. Cleavage of bistranded AP sites
With AP/AP − 3, cleavage of both AP sites by nuclear extract occurs within 1 min in both free and nucleosomal bound DNA, leading to extensive DSB formation, as shown in a representative phophorimaging scan of a native polyacrylamide gel (Fig. 3A) and graphically (Fig. 3B). In contrast, such prompt DSB formation was not seen with the AP/AP + 1 cluster (Fig. 3C). The efficiency of cleavage of the AP site within strand 2 (*AP/AP + 1; see Table 1) in the first 10 min is about 1.5 fold lower than that of the control AP site for both free and nucleosomal-bound DNA (Supplementary Fig. 3), whereas the efficiency of incision of the AP site in strand 1 (AP/*AP + 1; see Table 1) is comparable to the control (Supplementary Fig. 3). Eighty percent of the AP sites on strand 2 within free DNA are cleaved, compared to only 63% of the AP sites when contained in a nucleosomal template. These values should be compared with the conversion of 55% and 73% of the AP/AP + 1 cluster into DSB within 30 min (Fig. 3C) in nucleosomal and free DNA, respectively. These percentage yields of DSB compare with the level of cleavage of the AP site located in strand 2 of the cluster. Additionally, this level of DSB formation is 1.5 fold (nucleosomal-bound DNA) and 1.2 fold (free DNA) lower than that seen with the AP/AP − 3 cluster, reflecting the reduction in the efficiency of AP site incision on strand 2 in the AP/AP + 1 cluster. Thus, the incision of the AP site on strand 1 may confer retardation on the incision of the AP site on strand 2 in the AP/AP + 1 cluster.
Fig. 3.

Cleavage of two bistranded AP sites by 1 μg CHO-K1 nuclear extract. (A) Representative phosphorimaging scans of native polyacrylamide gels showing the formation of DSB in bistranded cluster (AP/AP − 3) in both free and nucleosomal-bound DNA substrates following treatment with 1 μg CHO-K1 nuclear extract for given times. Strand 2 within the DNA duplex was 5′-end labelled with 32P. The DSB band represents DNA that has been cleaved at both AP sites. A single AP site in strand 2 (AP con Table 1) does not give DSB when incubated with extract. (B) Percentage formation of DSB from AP/AP − 3 substrate: (open square) AP/AP − 3 free DNA, (open triangle) AP/AP − 3 nucleosomal-bound DNA and no extract control for both AP/AP − 3 free DNA (filled triangle) and nucleosomal-bound DNA, (filled square). (C) Percentage formation of DSB from AP/AP + 1 substrate: (open square) AP/AP + 1 free DNA, (open triangle) AP/AP + 1 nucleosomal-bound DNA and no extract control for both AP/AP + 1 free DNA (filled triangle) and nucleosomal-bound DNA, (filled square). (D) Representative phosphorimaging scan of a native polyacrylamide gel showing the nucleosome stability after DSB formation resulting from treatment with 1 μg CHO-K1 nuclear extract for 0 and 30 min. The positions of the nucleosome, free DNA remaining intact and free DNA, that has been cleaved by the nuclear extract, are indicated. A single AP site in strand 2 (AP con Table 1) is stable when incubated with extract. The nucleosomes were loaded onto the gel without deproteinization. A sample of free DNA was included for comparison. Error bars represent standard deviation determined from at least three independent experiments.
To determine if the formation of DSB would affect the stability of the nucleosome, nucleosomes reconstituted with DNA containing either an AP control or clusters containing AP/AP + 1 or AP/AP − 3 were incubated with 1 μg CHO-K1 nuclear extract for 0 or 30 min before being subjected to native PAGE without deproteinization to visualize reaction products. Fig. 3D shows a representative phosphorimaging scan of a native polyacrylamide gel. The nucleosome reconstituted with AP control DNA remains intact after incubation with the nuclear extract whereas with DNA containing clustered AP sites the nucleosome structure is disrupted. In the case of the AP/AP − 3 cluster, which forms DSB within five minutes (Fig. 3B), the nucleosome structure is completely disrupted at 30 min, in contrast to ∼40% of the DNA nucleosomal structure remaining intact with the AP/AP + 1 cluster at 30 min. The disrupted DNA containing the clusters is cleaved, as indicated in Fig. 3D, which is in agreement with the formation of DSB presented in Fig. 3A–C. The rapid formation of DSB seen with the AP/AP − 3 cluster is reflected in the dissolution of the nucleosome structure. Where DSB are slower to form as with the AP/AP + 1 cluster, the structure remains intact longer and even at 30 min some nucleosome-bound DNA is still visible.
3.3.2. Cleavage of an AP site or an 8-oxoG lesion within a bistranded AP/8-oxoG cluster
We also wanted to explore the effect of 8-oxoG on the efficiency of cleavage of an AP site. The extent of cleavage of the AP site within a bistranded AP/8-oxoG + 1 cluster at 30 min in the presence of nuclear extract is comparable to that of a single AP site in both free and nucleosomal templates (Fig. 4A). In contrast, the efficiency of excision of the 8-oxoG lesion when an AP site is position +1 is severely retarded compared to an 8-oxoG lesion present in isolation (Fig. 4B and C).
Fig. 4.

Cleavage the lesions within an AP/8-oxoG + 1 clustered damaged site. (A) Incision of AP site by 1 μg CHO-K1 nuclear extract. (Filled square) AP control free DNA, (filled triangle) AP control nucleosomal-bound DNA, (open square) AP/8-oxoG + 1 free DNA, (open triangle) AP/8-oxoG + 1 nucleosomal-bound DNA. (B and C) Excision of 8-oxoG by increasing amounts of OGG1 and 10 ng APE1 (B) or 1 μg CHO-K1 nuclear extract (C) (to incise AP site resulting from OGG1 activity). (Filled square) 8-oxoG control free DNA, (filled triangle) 8-oxoG control nucleosomal-bound DNA, (open square) AP/8-oxoG + 1 free DNA, (open triangle) AP/8-oxoG + 1 nucleosomal-bound DNA. For each experiment, the background has been subtracted from the data prior to normalization to 100%. Error bars represent standard deviation determined from at least three independent experiments.
Thus, DSB are not formed from the cleavage of the AP/8-oxoG + 1 cluster when in nucleosome-bound DNA (data not shown). In order to investigate the excision of 8-oxoG purified OGG1 was used. However, OGG1 has a poor AP lyase activity thus OGG1 was used in combination with APE1 or CHO-K1 nuclear extract to cleave the AP site resulting from the excision of the damaged base by OGG1. The efficiency of 8-oxoG excision, both in isolation and in a cluster with an AP site, is reduced when the lesion is within nucleosomal-bound DNA compared to when present in free DNA and this affect is most dramatic when APE1 is used in combination with OGG1 (Fig. 4B and C). It has been well documented in previous studies using free DNA and purified proteins or mammalian cell extracts that lesion hierarchy limits the formation of DSB [46], [51], [52]. That DSB are not induced is consistent with inefficient excision of 8-oxoG from the AP/8-oxoG + 1 cluster, most likely due to the AP site being cleaved initially and the resulting SSB conferring an inhibition on the excision of 8-oxoG.
3.4. Retardation of repair of a single AP site within nucleosomal templates by CHO-K1 nuclear extract
As incubation of nucleosome-bound DNA templates containing an AP site with nuclear extract leads to comparable cleavage to that in free DNA, we next wished to ascertain the level of repair possible within such templates. As demonstrated in Fig. 5, no further repair is seen of the SSB resulting from AP site cleavage within nucleosomal templates, in comparison to efficient repair of the resulting SSB within free DNA. This contrasts with comparable cleavage levels between the two substrates using the same extract, as shown in Fig. 1.
Fig. 5.
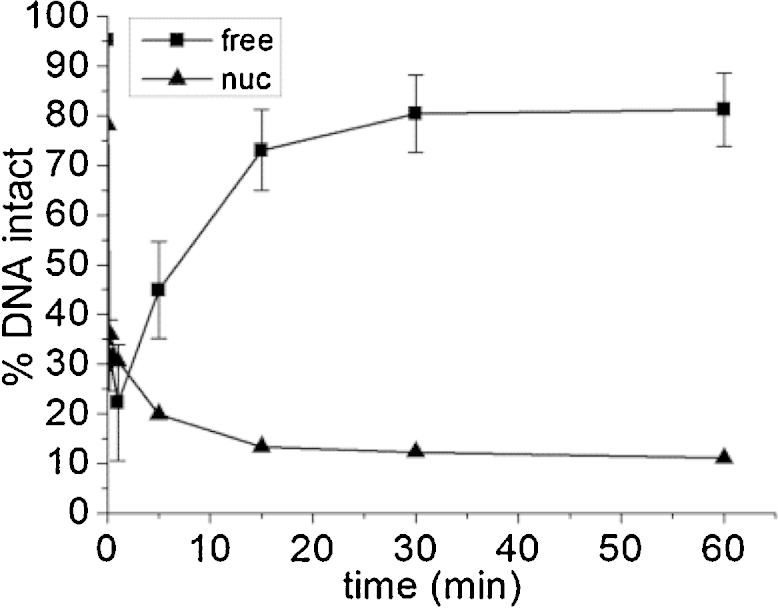
Repair of an AP site within free and nucleosomal-bound DNA by CHO-K1 nuclear extract. (Square) free DNA, (triangle) nucleosomal-bound DNA. Error bars represent standard deviation determined from at least three independent experiments.
4. Discussion
Previous in vitro studies investigating the processing of clustered DNA damaged sites have constructed these sites only in “naked” or “free” DNA, which is DNA that is not associated with histone proteins. This current study was designed to investigate how such clustered damaged sites are processed in DNA that is associated with histones. DNA containing bistranded clustered AP sites within reconstituted mononucleosomes resulted in rapid formation of DSB through cleavage of the AP sites, when treated with nuclear extracts. DSB formation from the AP/AP − 3 cluster (Table 1) was more rapid and complete than that from the AP/AP + 1 cluster (Table 1). This DSB formation is comparable to that seen in free DNA and to previous published studies [51], [53], [54], [55]. With the cluster containing AP/AP − 3, the AP site on strand 1 (Table 1) is three base pairs closer to the histone core and is almost at the DNA/histone interface, so that of the two AP sites this one would be expected to be removed more slowly. However, no retardation of cleavage of either AP site was apparent in the rapid DSB formation. This lack of any significant differential removal of the AP sites agrees with previous studies using free DNA [49], [51], [54], [55], [56], [57]. In contrast, cleavage of one of the AP sites within AP/AP + 1 is retarded to a greater extent when within nucleosomal DNA than when in free DNA. The AP site positioned on strand 1 (Table 1) within the clustered damage site is more accessible in the nucleosome as it faces the solution. This orientation causes cleavage to the same extent as that contained within free DNA. However, cleavage of the AP site located in strand 2 (Table 1) is retarded and to a greater extent when present in a nucleosomal context. Since the structure of the control nucleosome remains intact in the presence of the nuclear extract, it is suggested that the DNA remains loosely associated to the histone core during incision of the second lesion. This relaxation of the nucleosome structure could explain the increased cleavage of the AP site that is further from the histone core within nucleosomal templates. Structural studies have shown that when two bistranded AP sites are in position −1 to each other, the AP sites bulge out into the major groove in a conformation close to that of APE1/DNA, so that only small structural changes will be required for APE1 incision of the AP sites [58]. In contrast, two opposing AP sites in the +1 position remain aligned with the sugar-phosphate backbone and are thus less exposed than two AP sites in the −1 position and will require major structural changes for APE1 incision [58]. When bistranded AP site clusters are converted into DSB, the DNA dissociates from the nucleosome. As this study uses only mononucleosomes it is not yet known whether the structure would remain within larger nucleosome arrays. In contrast, a previous study [44] with bistranded thymine glycol and purified BER proteins, the ensuing DSB did not lead to dissociation of the nucleosome.
Again in agreement with previous studies using free DNA [59], [60], the efficiency of incision of the AP site present as an isolated lesion or in a cluster with 8-oxoG (AP/8-oxoG + 1 cluster) is comparable in both free and nucleosomal templates, after treatment with CHO-K1 nuclear extract without the formation of DSB. In contrast, excision of 8-oxoG within this cluster is impaired in both free and nucleosomal DNA as also shown recently [44] but using hOGG1 with a bistranded cluster containing two 8-oxoG spaced 5 bp apart in a nucleosome but further away from the dyad, namely a distance of 50/51 bp. These observations now extend our understanding of the hierarchy of repair of clusters, previously developed for clusters in free DNA, which tend to limit DSB formation [46], [49], [52], [54]. OGG1 is unable to bind to an 8-oxoG lesion when it is positioned at +1 to an AP site in free DNA [59], consistent with initial excision of the AP site. Additionally, in agreement with this current study, Menoni et al. [31], [32] and Cannan et al. [44] have shown that the excision of an isolated 8-oxoG lesion by OGG1 was also significantly reduced in nucleosomal DNA compared to free DNA.
Cleavage of a single AP site within the 601 DNA sequence but in the absence of histone-association, is efficient by APE1 at a concentration similar to that noted previously with shorter DNA sequences [49]. However, when the 601 DNA sequence containing an AP site in the same position is used to reconstitute nucleosomes, a drastic reduction in the efficiency of cleavage was seen. It was previously shown that the location of the lesion on the DNA sequence used for reconstitution is important for lesion processing [27], [40], in that the closer the lesion is located to the nucleosome dyad the higher the level of retardation for removal of the lesion. The position of the lesion with respect to the histone core is also vitally important [26], [30], [35] as lesions facing “out”, away from the histone, are processed with almost comparable efficiency to lesions contained in free DNA but lesions inward-facing and mid-way (meaning half-way between the histone core and solution) lesions are generally excised with much lower efficiency. The inserted lesion within our chosen sequence is located 10 bp from the nucleosome dyad and is precisely halfway between the most- and least-exposed DNA to the water. It would have been predicted [26], [30], [35] that retardation of cleavage would occur once the DNA is associated with the histone core, particularly as the position of the AP site is close to the dyad.
In contrast to the reduced efficiency of the nucleosome on incision of the AP site by APE1, the efficiency of cleavage of the AP site in nucleosomal DNA and free DNA by CHO-K1 nuclear extract is comparable. The increased efficiency of cleavage of an AP site in the nucleosome by nuclear extract relative to that with purified APE1 is not due to disruption of the nucleosome structure by nuclear extract to give free DNA containing an AP site. Cleavage of the AP site in free and nucleosomal-bound DNA by Fpg suggests that glycoslases present in the nuclear extract may contribute to this increased cleavage. In addition the increase in cleavage could be due to the action of a chromatin remodeler, many of which are capable of disruption of DNA-histone contacts and nucleosome translocation (reviewed in [61], [62]). SWI/SNF, RSC, ACF, are chromatin remodelers that have been shown to interact with the BER pathway [31], [32], [63], [64] and can be further facilitated by HMGB1 protein [65], [66]. Alternatively, PARP1 which acts rapidly and has roles in the regulation of chromatin structure, DNA repair and transcription (reviewed in [67]) may facilitate enzymatic access. However, we found that the PARP inhibitor, KU-0058684, had no effect on the incision or repair of an AP site in either free or nucleosomal DNA in our in vitro BER assays (data not shown).
Despite the comparable levels of AP site cleavage between free and nucleosomal DNA templates after treatment with nuclear extract for given times, repair of the resultant SSB was not seen in nucleosomal templates. Potentially, the pol β or ligase III step of the BER pathway could be the position at which repair stalls. Due to experimental constraints, we are unable to detect single base addition into the DNA template, an indication of the efficiency of pol β activity. Previously reported levels of ligase retardation [33], [39], [40] are minimal in comparison to the inhibition of pol β observed [28], [29], [31]. Conversely; however, the repair gaps have recently been shown to be processed by pol β whereas ligation by ligase III/XRCC1 only takes place following nucleosome disruption [34].
Sczepanski et al. reported that DNA protein cross links between AP sites and histone H4 tails, leading to strand scission, were much more rapidly formed in nucleosomal-bound DNA than free DNA and that two closely opposed AP sites could result in a DSB, without the requirement of incision by APE1 [68]. However, the time scale for this reaction is hours, rather than minutes it takes to form a DSB from two opposing AP sites by APE1 incision. After formation of the mononucleosome the histone tails are excluded from the core structure and interact with linker DNA, the DNA that connects two nucleosomes within an array. If this linker DNA is absent the histone tails can associate with the DNA surrounding the dyad and retard lesion processing [40], [69]. In our present study the central 147 bp DNA that associates with the histone proteins to form the nucleosome was flanked by linker DNA. The presence of the linker DNA, together with a time frame of up to 30 min for the completion of the experiments, may explain why no DNA protein crosslinks and subsequent AP site scission was observed.
5. Conclusions
In summary, novel insights are presented into the repair of both single and clustered DNA lesions placed within nucleosome-bound regions of DNA. This gives a greater reflection on how damage is processed in vivo as DNA is found condensed into chromatin, with nucleosomes as the first level of higher order DNA structure. It is not only the types of lesions found within a cluster that affects repair but the orientation of lesions to each other and also their orientation to the solution or towards the histone octamer. These additional factors need now to be taken into consideration when predicting the outcomes of BER. The inefficient repair of such clustered damage sites has great biological significance due to the ultimate risk of tumourigenesis.
Conflict of interest
None declared.
Funding
This work was supported by the Medical Research Council, UK (MC_PC_12001 and studentship to L.J.E.); the Association pour la Recherche sur le Cancer, France (SFI20101201424 to D.A.) and the Agence Nationale de la Recherche, France (ANR-09-BLAN-NT09-485720 ‘CHROREMBER’ to D.A.).
Acknowledgements
The authors wish to thank Dr. Grigory Dianov (CR-UK/MRC Oxford Institute of Radiation Biology, Department of Oncology, University of Oxford, UK) for providing the purified APE1 used in this study. They would also like to thank Herve Menoni and other members of the Angelov laboratory for technical assistance.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.dnarep.2015.08.003.
Appendix A. Supplementary data
The following are Supplementary data to this article:
References
- 1.Bennett P.V., Cintron N.S., Gros L., Laval J., Sutherland B.M. Are endogenous clustered DNA damages induced in human cells? Free Radical Biol. Med. 2004;37:488–499. doi: 10.1016/j.freeradbiomed.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Sutherland B.M., Bennett P.V., Cintron N.S., Guida P., Laval J. Low levels of endogenous oxidative damage cluster levels in unirradiated viral and human DNAs. Free Radical Biol. Med. 2003;35:495–503. doi: 10.1016/s0891-5849(03)00327-7. [DOI] [PubMed] [Google Scholar]
- 3.Cadet J., Carell T., Cellia L., Chatgilialoglu C., Gimisis T., Miranda M., O’Neill P., Ravanat J.-L., Robert M. DNA damage and radical reactions: mechanistic aspects, formation in cells and repair studies. Chimia. 2008;62:742–749. [Google Scholar]
- 4.O’Neill P., Wardman P. Radiation chemistry comes before radiation biology. Int. J. Radiat. Biol. 2009;85:9–25. doi: 10.1080/09553000802640401. [DOI] [PubMed] [Google Scholar]
- 5.Blaisdell J.O., Wallace S.S. Abortive base-excision repair of radiation-induced clustered DNA lesions in E. coli. Proc. Natl. Acad. Sci. U. S. A. 2001;98:7426–7430. doi: 10.1073/pnas.131077798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Georgakilas A.G. Processing of DNA damage clusters in human cells: current status of knowledge. Mol. Biosyst. 2008;4:30–35. doi: 10.1039/b713178j. [DOI] [PubMed] [Google Scholar]
- 7.Gulston M., Fulford J., Jenner T., de Lara C., O’Neill P. Clustered DNA damage induced by gamma radiation in human fibroblasts (HF19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic Acids Res. 2002;30:3464–3472. doi: 10.1093/nar/gkf467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prise K.M., Pullar C.H., Michael B.D. A study of endonuclease III-sensitive sites in irradiated DNA: detection of alpha-particle-induced oxidative damage. Carcinogenesis. 1999;20:905–909. doi: 10.1093/carcin/20.5.905. [DOI] [PubMed] [Google Scholar]
- 9.Sutherland B.M., Bennett P.V., Sidorkina O., Laval J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. U. S. A. 2000;97:103–108. doi: 10.1073/pnas.97.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutherland B.M., Bennett P.V., Sutherland J.C., Laval J. Clustered DNA damages induced by X rays in human cells. Radiat. Res. 2002;157:611–616. doi: 10.1667/0033-7587(2002)157[0611:cddibx]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Tsao D., Kalogerinis P., Tabrizi I., Dingfelder M., Stewart R.D., Georgakilas A.G. Induction and processing of oxidative clustered DNA lesions in 56Fe-ion-irradiated human monocytes. Radiat. Res. 2007;168:87–97. doi: 10.1667/RR0865.1. [DOI] [PubMed] [Google Scholar]
- 12.Gulston M., de Lara C., Jenner T., Davis E., O’Neill P. Processing of clustered DNA damage generates additional double-strand breaks in mammalian cells post-irradiation. Nucleic Acids Res. 2004;32:1602–1609. doi: 10.1093/nar/gkh306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegde M.L., Hazra T.K., Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zharkov D.O. Base excision DNA repair. Cell Mol. Life Sci. 2008;65:1544–1565. doi: 10.1007/s00018-008-7543-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodhead D.T. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int. J. Radiat. Biol. 1994;65:7–17. doi: 10.1080/09553009414550021. [DOI] [PubMed] [Google Scholar]
- 16.Eccles L.J., O’Neill P., Lomax M.E. Delayed repair of radiation induced clustered DNA damage: friend or foe? Mutat. Res. 2011;711:134–141. doi: 10.1016/j.mrfmmm.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shikazono N., O’Neill P. Biological consequences of potential repair intermediates of clustered base damage site in E. coli. Mutat. Res. 2009;669:162–168. doi: 10.1016/j.mrfmmm.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Georgakilas A.G., O’Neill P., Stewart R.D. Induction and repair of clustered DNA lesions: what do we know so far? Radiat. Res. 2013;180:100–109. doi: 10.1667/RR3041.1. [DOI] [PubMed] [Google Scholar]
- 19.Magnander K., Hultborn R., Claesson K., Elmroth K. Clustered DNA damage in irradiated human diploid fibroblasts: influence of chromatin organization. Radiat. Res. 2010;173:272–282. doi: 10.1667/RR1891.1. [DOI] [PubMed] [Google Scholar]
- 20.Sage E., Harrison L. Clustered DNA lesion repair in eukaryotes: relevance to mutagenesis and cell survival. Mutat. Res. 2011;711:123–133. doi: 10.1016/j.mrfmmm.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGhee J.D., Felsenfeld G., Eisenberg H. Nucleosome structure and conformational changes. Biophys. J. 1980;32:261–270. doi: 10.1016/S0006-3495(80)84955-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 23.Hayes J.J., Tullius T.D., Wolffe A.P. The structure of DNA in a nucleosome. Proc. Natl. Acad. Sci. U. S. A. 1990;87:7405–7409. doi: 10.1073/pnas.87.19.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoyagi S., Hayes J.J. hSWI/SNF-catalyzed nucleosome sliding does not occur solely via a twist-diffusion mechanism. Mol. Cell Biol. 2002;22:7484–7490. doi: 10.1128/MCB.22.21.7484-7490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li F., Tian L., Gu L., Li G.M. Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J. Biol. Chem. 2009;284:33056–33061. doi: 10.1074/jbc.M109.049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Odell I.D., Newick K., Heintz N.H., Wallace S.S., Pederson D.S. Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair (Amsterdam) 2010;9:134–143. doi: 10.1016/j.dnarep.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prasad A., Wallace S.S., Pederson D.S. Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol. Cell Biol. 2007;27:8442–8453. doi: 10.1128/MCB.00791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beard B.C., Stevenson J.J., Wilson S.H., Smerdon M.J. Base excision repair in nucleosomes lacking histone tails. DNA Repair (Amsterdam) 2005;4:203–209. doi: 10.1016/j.dnarep.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 29.Beard B.C., Wilson S.H., Smerdon M.J. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7465–7470. doi: 10.1073/pnas.1330328100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cole H.A., Tabor-Godwin J.M., Hayes J.J. Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J. Biol. Chem. 2010;285:2876–2885. doi: 10.1074/jbc.M109.073544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menoni H., Gasparutto D., Hamiche A., Cadet J., Dimitrov S., Bouvet P., Angelov D. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A. Bbd nucleosomes. Mol. Cell Biol. 2007;27:5949–5956. doi: 10.1128/MCB.00376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menoni H., Shukla M.S., Gerson V., Dimitrov S., Angelov D. Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res. 2012;40:692–700. doi: 10.1093/nar/gkr761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nilsen H., Lindahl T., Verreault A. DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 2002;21:5943–5952. doi: 10.1093/emboj/cdf581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Odell I.D., Barbour J.E., Murphy D.L., Della-Maria J.A., Sweasy J.B., Tomkinson A.E., Wallace S.S., Pederson D.S. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell Biol. 2011;31:4623–4632. doi: 10.1128/MCB.05715-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinz J.M., Rodriguez Y., Smerdon M.J. Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4646–4651. doi: 10.1073/pnas.0914443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriquez Y., Smerdon M.J. The structural location of DNA lesions in nuvleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem. 2013;288:13863–13875. doi: 10.1074/jbc.M112.441444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinz J.M. Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutat. Res. 2014;766–767:19–24. doi: 10.1016/j.mrfmmm.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Rodriquez Y., Hinz J.M., Smerdon M.J. Accessing DNA damage in chromatin: preparing the chromatin landscape for base excision repair. DNA Repair. 2015;32:113–119. doi: 10.1016/j.dnarep.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chafin D.R., Hayes J.J. Site-directed cleavage of DNA by linker histone-Fe(II) EDTA conjugates. Methods Mol. Biol. 2001;148:275–290. doi: 10.1385/1-59259-208-2:275. [DOI] [PubMed] [Google Scholar]
- 40.Chafin D.R., Vitolo J.M., Henricksen L.A., Bambara R.A., Hayes J.J. Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J. 2000;19:5492–5501. doi: 10.1093/emboj/19.20.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kosmoski J.V., Smerdon M.J. Synthesis and nucleosome structure of DNA containing a UV photoproduct at a specific site. Biochemistry. 1999;38:9485–9494. doi: 10.1021/bi990297h. [DOI] [PubMed] [Google Scholar]
- 42.Kysela B., Chovanec M., Jeggo P.A. Phosphorylation of linker histones by DNA-dependent protein kinase is required for DNA ligase IV-dependent ligation in the presence of histone H1. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1877–1882. doi: 10.1073/pnas.0401179102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X., Smerdon M.J. Nucleotide excision repair of the 5 S ribosomal RNA gene assembled into a nucleosome. J. Biol. Chem. 2000;275:23729–23735. doi: 10.1074/jbc.M002206200. [DOI] [PubMed] [Google Scholar]
- 44.Cannan W.J., Tsang B.P., Wallace S.S., Pederson D.S. Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J. Biol. Chem. 2014;289:19881–19893. doi: 10.1074/jbc.M114.571588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh V., Kumari B., Das P. Repair efficiency of clustered abasic sites by APE1 in nucleosome core particles is sequence and position dependent. RSC Adv. 2015;5:23691–23698. [Google Scholar]
- 46.David-Cordonnier M.H., Laval J., O’Neill P. Clustered DNA damage, influence on damage excision by XRS5 nuclear extracts and E. coli Nth and Fpg proteins. J. Biol. Chem. 2000;275:11865–11873. doi: 10.1074/jbc.275.16.11865. [DOI] [PubMed] [Google Scholar]
- 47.Anderson J.D., Thastrom A., Widom J. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol. Cell Biol. 2002;22:7147–7157. doi: 10.1128/MCB.22.20.7147-7157.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 49.David-Cordonnier M.H., Cunniffe S.M., Hickson I.D., O’Neill P. Efficiency of incision of an AP site within clustered DNA damage by the major human AP endonuclease. Biochemistry. 2002;41:634–642. doi: 10.1021/bi011682l. [DOI] [PubMed] [Google Scholar]
- 50.Lebedeva N.A., Rechkunova N.I., Ishchenko A.A., Saparbaev M., Lavrik O.I. The mechanism of human tyrosyl-DNA phosphdiesterase 1 in the cleavage of AP site and its synthetic analogs. DNA Repair. 2013;12:1037–1042. doi: 10.1016/j.dnarep.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Chaudhry M.A., Weinfeld M. Reactivity of human apurinic/apyrimidinic endonuclease and E. coli exonuclease III with bistranded abasic sites in DNA. J. Biol. Chem. 1997;272:15650–15655. doi: 10.1074/jbc.272.25.15650. [DOI] [PubMed] [Google Scholar]
- 52.Harrison L., Hatahet Z., Wallace S.S. In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J. Mol. Biol. 1999;290:667–684. doi: 10.1006/jmbi.1999.2892. [DOI] [PubMed] [Google Scholar]
- 53.Eccles L.J., Lomax M.E., O’Neill P. Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage. Nucleic Acids Res. 2010;38:1123–1134. doi: 10.1093/nar/gkp1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lomax M.E., Cunniffe S., O’Neill P. Efficiency of repair of an abasic site within DNA clustered damage sites by mammalian cell nuclear extracts. Biochemistry. 2004;43:11017–11026. doi: 10.1021/bi049560r. [DOI] [PubMed] [Google Scholar]
- 55.Paap B., Wilson D.M., 3rd, Sutherland B.M. Human abasic endonuclease action on multilesion abasic clusters: implications for radiation-induced biological damage. Nucleic Acids Res. 2008;36:2717–2727. doi: 10.1093/nar/gkn118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kozmin S.G., Sedletska Y., Reynaud-Angelin A., Gasparutto D., Sage E. The formation of double-strand breaks at multiply damaged sites is driven by the kinetics of excision/incision at base damage in eukaryotic cells. Nucleic Acids Res. 2009;37:1767–1777. doi: 10.1093/nar/gkp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Z., de los Santos C. NMR characterization of clustered bistrand abasic site lesions: effect of orientation on their solution structure. J. Mol. Biol. 2001;308:341–352. doi: 10.1006/jmbi.2001.4587. [DOI] [PubMed] [Google Scholar]
- 58.Hazel R.D., Tian K., de Los Santos C. NMR solution structures of bistranded abasic site lesions in DNA. Biochemistry. 2008;47:11909–11919. doi: 10.1021/bi800950t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.David-Cordonnier M.H., Boiteux S., O’Neill P. Efficiency of excision of 8-oxo-guanine within DNA clustered damage by XRS5 nuclear extracts and purified human OGG1 protein. Biochemistry. 2001;40:11811–11818. doi: 10.1021/bi0112356. [DOI] [PubMed] [Google Scholar]
- 60.Lomax M.E., Cunniffe S., O’Neill P. 8-OxoG retards the activity of the ligase III/XRCC1 complex during the repair of a single-strand break, when present within a clustered DNA damage site. DNA Repair. 2004;3:289–299. doi: 10.1016/j.dnarep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Choudhary P., Varga-Weisz P. ATP-dependent chromatin remodelling: action and reaction. Subcell. Biochem. 2007;41:29–43. doi: 10.1007/1-4020-5466-1_2. [DOI] [PubMed] [Google Scholar]
- 62.Saha A., Wittmeyer J., Cairns B.R. Chromatin remodelling: the industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006;7:437–447. doi: 10.1038/nrm1945. [DOI] [PubMed] [Google Scholar]
- 63.Langst G., Becker P.B. ISWI induces nucleosome sliding on nicked DNA. Mol. Cell. 2001;8:1085–1092. doi: 10.1016/s1097-2765(01)00397-5. [DOI] [PubMed] [Google Scholar]
- 64.Liu Y., Prasad R., Wilson S.H. HMGB1: roles in base excision repair and related function. Biochim. Biophys. Acta. 2010;1799:119–130. doi: 10.1016/j.bbagrm.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonaldi T., Langst G., Strohner R., Becker P.B., Bianchi M.E. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 2002;21:6865–6873. doi: 10.1093/emboj/cdf692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Racki L.R., Yang J.G., Naber N., Partensky P.D., Acevedo A., Purcell T.J., Cooke R., Cheng Y., Narlikar G.J. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature. 2009;462:1016–1021. doi: 10.1038/nature08621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beneke S. Regulation of chromatin structure by poly(ADP-ribosyl) ation. Front. Genet. 2012;3:169. doi: 10.3389/fgene.2012.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sczepanski J.T., Wong R.S., McKnight J.N., Bowman G.D., Greenberg M.M. Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc. Natl. Acad. Sci. U. S. A. 2010;107:22475–22480. doi: 10.1073/pnas.1012860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Angelov D., Vitolo J.M., Mutskov V., Dimitrov S., Hayes J.J. Preferential interaction of the core histone tail domains with linker DNA. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6599–6604. doi: 10.1073/pnas.121171498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.