

Abstract
Background. Secondary bacterial infections after influenza can be a serious problem, especially in young children and the elderly, yet the efficacy of current vaccines is limited. Earlier work demonstrated that a replication-incompetent PB2-knockout (PB2-KO) influenza virus possessing a foreign gene in the coding region of its PB2 segment can serve as a platform for a bivalent vaccine.
Methods. In the current study, we generated the PB2-KO virus expressing pneumococcal surface protein A (PspA), PB2-KO-PspA virus, the replication of which is restricted to PB2-expressing cells. We then examined the protective efficacy of intranasal immunization with this virus as a bivalent vaccine in a mouse model.
Results. High levels of influenza virus–specific and PspA-specific antibodies were induced in the serum and airways of immunized mice. The intranasally immunized mice were protected from lethal doses of influenza virus or Streptococcus pneumoniae. These mice were also completely protected from secondary pneumococcal pneumonia after influenza virus infection.
Conclusions. These findings indicate that our recombinant influenza virus serves as a novel and powerful bivalent vaccine against primary and secondary pneumococcal pneumonia as well as influenza.
Keywords: secondary pneumococcal pneumonia, replication-incompetent influenza virus, bivalent vaccine
Secondary bacterial infections after influenza account for a sizeable proportion of the deaths associated with influenza pandemics [1, 2]. Most secondary bacterial infections are caused by Streptococcus pneumoniae.
S. pneumoniae is one of the major causes of disease and death resulting from pneumonia, bacteremia, and meningitis worldwide [3]. Pneumococcal carriage is considered to be an important source of the horizontal spread of this pathogen, because preceding nasopharyngeal colonization with this bacterium is essential for pneumococcal diseases [4]. S. pneumoniae is classified into >90 serotypes defined based on the antigenicity of their capsular polysaccharide, which is a virulence factor of this pathogen [5].
The current first line of defense against pneumococcal pneumonia is vaccination: pneumococcal polysaccharide conjugate vaccines derived from multiple serotypes of S. pneumoniae are used globally to provide protective immunity in infants [6, 7]. Although the introduction of the 7-valent pneumococcal conjugate vaccine resulted in significant declines in the incidence of invasive pneumococcal disease (IPD) caused by the serotypes covered by the vaccine, serotype replacement in carriage and IPD occurred [8–13]. Moreover, since the introduction of a 13-valent pneumococcal conjugate vaccine for use in children, the frequency of serotypes not included in a 13-valent pneumococcal conjugate vaccine has increased in pediatric and adult patients with IPD [14, 15]. Therefore, an alternative vaccine format is desired for the control of S. pneumoniae infection.
Recent studies on pneumococcal vaccine development have focused on pneumococcal surface protein A (PspA), a choline-binding protein exposed on the cell surface of all pneumococcal strains [16–20]. Anti-PspA antibodies are known to overcome the anticomplementary effect of PspA, allowing for increased complement activation and C3 deposition on PspA-bearing bacteria [21–23]. In addition, anti-PspA antibodies enhance bacterial clearance and induce cross-serotype immunity [24–27]. Collectively these data suggest that PspA is a promising vaccine candidate against pneumococcal infection.
To prevent influenza, both inactivated and live attenuated vaccines are available [28, 29]. Inactivated vaccines present few safety concerns and are used globally; however, they do not induce the mucosal immune responses that play important roles in preventing influenza virus replication [30, 31]. Live attenuated vaccines elicit mucosal immune responses more efficiently than inactivated vaccine; however, their usage is restricted because of safety concerns [32–34]. To overcome the limitations of the current influenza vaccines, Ozawa et al [35] previously generated a replication-incompetent influenza virus that does not express the PB2 protein, an influenza virus polymerase subunit that is essential for virus replication. Mice intranasally immunized with PB2-knockout (PB2-KO) virus efficiently elicited mucosal immunity and were protected from challenge with a lethal dose of influenza virus [36, 37]. Uraki et al [37] also demonstrated the protective efficacy as bivalent vaccines of PB2-KO viruses by introducing foreign genes into their PB2-coding region. Together, these findings suggest that PB2-KO influenza virus is a novel platform for a bivalent influenza vaccine that is safe and efficacious.
In the current study, we generated PB2-KO virus expressing PspA as a bivalent vaccine for influenza and pneumococcal pneumonia and examined whether intranasal immunization with this bivalent vaccine could induce influenza virus–specific and PspA-specific antibodies and afford protection from lethal infection with influenza virus or S. pneumoniae in a mouse model.
METHODS
Cells
Human embryonic kidney cell (293T cell) were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum (Gibco). Madin-Darby canine kidney (MDCK) cells were maintained in minimum essential medium (MEM) supplemented with 5% newborn calf serum (NCS) (Equitech-Bio). AX4 cells, an MDCK-derived cell line with enhanced expression of human-type receptors for influenza virus [38], were maintained in 5% NCS-MEM supplemented with puromycin (2 µg/mL). AX4/PB2 cells, which are AX4 cells stably expressing the PB2 protein derived from A/Puerto Rico/8/34 (H1N1; PR8) [35], were maintained in 5% NCS-MEM supplemented with puromycin (2 µg/mL) and blasticidin (10 µg/mL). All cells were maintained in a humidified incubator at 37°C with 5% carbon dioxide.
Viral and Bacterial Strains
H1N1 subtype influenza virus PR8 strain was propagated in MDCK at 37°C for 48 hours and harvested as culture supernatants. A/New Caledonia/20/99 (H1N1; NC) virus was obtained from the Research Foundation for Microbial Diseases, Osaka University. S. pneumoniae WU2 strain (serotype 3) [39], which expresses PspA (family 1, clade 2) and is virulent in mice, and EF3030 strain (serotype 19F) [40], which expresses PspA (family 1, clade 1) and is relatively avirulent in mice, were grown in Todd-Hewitt broth (BD) supplemented with 0.5% yeast extract (THY) at 37°C with 5% carbon dioxide. The stocks of the bacterial strains for the challenge experiments were collected at an optical density (OD) at a wavelength of 600 nm (OD600) of 0.3–0.4, washed with fresh THY, resuspended in fresh THY with 10% glycerol, and stored at −80°C until use.
Plasmid-Driven Reverse Genetics
The wild-type PR8 and PB2-KO viruses were engineered by using reverse genetics, as described elsewhere [41]. For the expression of viral RNA, plasmids containing the cloned complementary DNAs of PR8 genes between the human RNA polymerase I promoter and the mouse RNA polymerase I terminator (referred to as PolI plasmids) were used. To generate the PR8-based PB2-KO virus possessing the antigenic portion of PspA in the recombinant PB2 gene (PR8/PB2-PspA virus) in place of the authentic PB2 gene, pPolIPB2(120)PspA(336) (Figure 1A) and the remaining 7 PolI plasmids were cotransfected into 293T cells along with eukaryotic protein expression plasmids for PB2, PB1, PA, and NP proteins derived from PR8 virus by use of the TransIT 293 transfection reagent (Mirus), according to the manufacturer's instructions as described elsewhere [37]. The inserted antigenic portion of PspA was 302 amino acids (positions 32–333) of mature PspA belonging to family 1, clade 2 [42]. At 48 hours after transfection, the supernatants containing the PB2-KO virus were harvested and propagated in AX4/PB2 cells. Viruses were titrated by means of plaque assays with AX4/PB2 cells.
Figure 1.
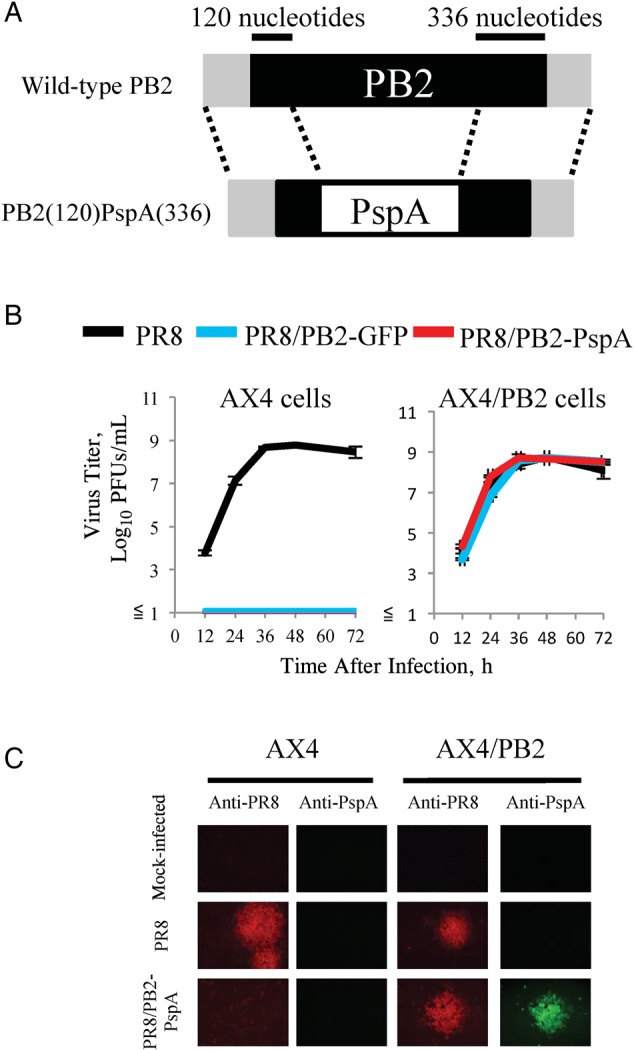
Characterization of PR8/PB2–pneumococcal surface protein A (PspA) virus. A, Schematic diagrams of wild-type PB2 and PB2(120)PspA(336) viral RNAs (vRNAs). PB2(120)PspA(336) vRNA possesses the 3′ noncoding region, 120 nucleotides of the coding sequence of PB2 vRNA, the PspA gene, and 336 nucleotides of the 3′ and 5′ noncoding regions of PB2 vRNA. The noncoding region and coding regions of PB2 vRNA are represented by gray and black bars, respectively. B, Growth kinetics of PR8/PB2–PspA virus. AX4 and AX4/PB2 cells were infected with PR8, PR8/PB2–green fluorescent protein (GFP), or PR8/PB2–PspA virus at a multiplicity of infection of 0.001. Supernatants were collected at 12, 24, 36, 48, and 72 hours after infection for virus titration by plaque assays in AX4/PB2 cells. C, PspA expression in AX4/PB2 cells infected with PR8/PB2–PspA virus. AX4 and AX4/PB2 cells were mock-infected or infected with PR8 or PR8/PB2–PspA virus. At 48 hours after infection, the cells were fixed and stained with anti-influenza polyclonal (R309) or anti-PspA polyclonal antibodies. Abbreviation: PFUs, plaque-forming units.
Growth Kinetics and Virus Titration
To determine virus growth kinetics, triplicate wells of confluent AX4 or AX4/PB2 cells were infected with viruses at a multiplicity of infection of 0.001 at 33°C. After 1 hour of virus adsorption, cells were washed in MEM containing 0.3 % bovine serum albumin and overlaid with MEM containing L-(tosylamido-2-phenyl) ethyl chloromethyl ketone-treated trypsin (1.0 µg/mL). Supernatants were collected at 12, 24, 36, 48, and 72 hours after infection and assayed for virus titers by means of plaque assays in AX4/PB2 cells.
Immunofluorescence Assay
Twenty-four hours after virus infection, cells were washed twice with phosphate-buffered saline and fixed with 4% paraformaldehyde for 15 minutes at room temperature. These cells were then stained with anti-influenza virus rabbit polyclonal (R309) and anti-PspA mouse polyclonal antibodies prepared in our laboratory.
Effect of Primary PR8/PB2-PspA Virus Inoculation of Mice on Secondary Infection
Five-week-old female C57BL/6 mice (Japan SLC) were anesthetized with isoflurane and inoculated with 50 µL of medium (MEM containing 0.3% bovine serum albumin fraction V), PR8/PB2–green fluorescent protein (GFP) virus (1.0 × 106 plaque-forming units [PFUs]) [35–37], PR8/PB2-PspA virus (1.0 × 106 PFUs), or NC virus (1.0 × 103 PFUs). Five days after virus inoculation, mice were intranasally challenged with 6.0 × 103 colony-forming units (CFUs) of S. pneumoniae WU2 strain in 30 µL of phosphate-buffered saline. Five days after infection with the WU2 strain, 6 mice were euthanized for quantitative bacterial cultures of lung tissue on sheep blood agar (BD). The survival of the remaining challenged mice (10 mice per group) was monitored daily for 10 days.
Immunization of Mice
Mice were intranasally inoculated with medium, PR8/PB2-GFP virus (1.0 × 106 PFUs), or PR8/PB2-PspA virus (1.0 × 106 PFUs) 3 times at 2-week intervals.
Detection of Virus-Specific Antibodies
Virus-specific antibodies in nasal wash, bronchoalveolar lavage fluid (BALF), and serum samples were detected using an enzyme-linked immunosorbent assay, as described elsewhere [36, 43]. The end-point titers were expressed as the reciprocal log2 of the last dilution, giving an OD405 of 0.1 OD unit above the OD405 of the negative control samples. Hemagglutination inhibition titers were determined as described elsewhere [44, 45].
Detection of PspA-Specific Antibodies
PspA-specific antibodies in serum, BALF, and nasal wash samples were detected using an enzyme-linked immunosorbent assay, as described elsewhere [46]. The end-point titers were expressed as the reciprocal log2 of the last dilution, giving an OD405 of 0.1 OD unit above the OD405 of the negative control samples.
Protection Tests
Lethal Influenza Virus Challenge
Two weeks after the final immunization, the immunized mice were intranasally challenged with 1.0 × 104 PFUs (10-fold 50% mouse lethal dose [10 MLD50]) of PR8 virus (50 µL). Three and 6 days after the viral challenge, we euthanized 3 mice per group, collected the nasal turbinates and lungs, and determined the viral titers using plaque assays in AX4/PB2 cells. The survival of the remaining challenged mice (8 mice per group) was monitored daily for 14 days.
Lethal Pneumococcal Challenge
Two weeks after the final immunization, the immunized mice were intranasally challenged with 2.0 × 107 CFUs (3 MLD50) of S. pneumoniae WU2 strain (30 µL). The survival of the challenged mice (10 mice per group) was monitored daily for 10 days.
Secondary Pneumococcal Pneumonia
Two weeks after the final immunization, the immunized mice were intranasally infected with 1.0 × 103 PFUs of NC virus (30 µL). Three days after the viral challenge, we euthanized 6 mice per group, collected the nasal turbinates and lungs, and determined the viral titers using plaque assays in AX4/PB2 cells. Five days after the viral infection, the remaining mice were intranasally infected with 2.0 × 107 CFUs (3 MLD50) or 6.0 × 103 CFUs (sublethal dose) of S. pneumoniae WU2 strain (30 µL). Five days after this bacterial challenge, we euthanized 6 mice per group, collected their lungs, and determined the bacterial densities on sheep blood agar, as described elsewhere [32, 47]. The survival of the remaining challenged mice (10 mice per group) was monitored daily for 10 days after the bacterial challenge.
Nasal Colonization
Two weeks after the final immunization, the immunized mice were intranasally challenged with 1.0 × 102 CFUs (sublethal dose) of S. pneumoniae EF3030 strain (30 µL). Five days after the bacterial challenge, we euthanized 10 mice per group, collected nasal wash samples, and determined the bacterial densities on sheep blood agar, as described elsewhere [32, 47].
Ethics Statement
Our research protocol for the use of mice followed the University of Tokyo's Regulations for Animal Care and Use, which was approved by the Animal Experiment Committee of the Institute of Medical Science, the University of Tokyo (approval No. PA10–15) and the Animal Care and Use Committee of the Research Institute for Microbial Diseases, Osaka University (approval No. Biken-AP-H23-05-0).
Statistical Analysis
Statistically significant differences in the bacterial densities or viral titers were assessed using 1-way analysis of variance followed by a Dunnett test. Log-rank statistical analysis was performed to determine significant differences in the survival rates of the immunized mice.
RESULTS
Characterization of the PR8/PB2-PspA Virus in Vitro
PR8-based PB2-KO virus expressing PspA (PR8/PB2-PspA virus) was generated by using plasmid-driven reverse genetics (Figure 1A). We first determined whether the PR8/PB2-PspA virus replicates only in PB2 protein-expressing and not normal cells. Although no replication of the PR8/PB2-PspA virus was detected in wild-type AX4 cells, as was also the case with the control PR8/PB2-GFP virus (Figure 1B, left), the replication of the PR8/PB2-PspA virus in AX4/PB2 cells was comparable to that of wild-type PR8 virus (Figure 1B, right). These results indicate that PR8/PB2-PspA virus replication was restricted to PB2 protein-expressing cells, as demonstrated elsewhere [35–37].
We then attempted to confirm that the PR8/PB2-PspA virus expressed PspA encoded in its PB2 gene in virus-infected cells. Using an immunofluorescence assay, we detected both PspA and influenza viral proteins in AX4/PB2 cells, but not wild-type AX4 cells, infected with PR8/PB2-PspA virus (Figure 1C). Wild-type PR8 virus expressed viral proteins but not PspA in both wild-type AX4 and AX4/PB2 cells. These results demonstrate that the PspA in the PB2 gene of the PR8/PB2-PspA virus was expressed in the virus-infected PB2-expressing cells.
Effect of Intranasal Inoculation of PB2-KO Virus on Susceptibility to S. pneumoniae
Previously, Ezoe et al [46] demonstrated that preinfection of mice with A/New Caledonia/20/99 (H1N1) (NC virus) increased their susceptibility to S. pneumoniae by about 3000-fold. We therefore examined whether preinoculation with either PR8/PB2-GFP virus or PR8/PB2-PspA virus increased the susceptibility of mice to the S. pneumonia WU2 strain. The survival rates of mice infected with PR8/PB2-GFP virus or PR8/PB2-PspA virus and then infected with a sublethal dose of S. pneumoniae WU2 strain (Figure 2A; 100% each) were significantly higher than that of mice preinfected with NC virus mice (10%). On day 5 after pneumococcal infection, the bacterial densities in the lungs of mice preinoculated with PR8/PB2-PspA virus were remarkably lower than those of the NC virus–preinfected mice and comparable to those in mock-inoculated and PR8/PB2-GFP virus–infected mice (Figure 2B). These results indicate that the PB2-KO virus inoculation did not alter the susceptibility of mice to S. pneumoniae.
Figure 2.

Survival of mice inoculated with PR8/PB2–pneumococcal surface protein A (PspA) virus and then infected with WU2 strain. Mice (n = 16) were mock-inoculated with medium, PR8/PB2–green fluorescent protein (GFP) (1.0 × 106 plaque-forming units [PFUs]), PR8/PB2-PspA (1.0 × 106 PFUs) or A/New Caledonia/20/99 (NC) (1.0 × 103 PFUs) virus. Five days after inoculation, the mice were intranasally challenged with WU2 strain (6.0 × 103 colony-forming units [CFUs]). A, Survival was monitored in 10 mice for 10 days after bacterial challenge. B, Lung tissues were collected 5 days after the bacterial challenge and subjected to bacterial titration on sheep blood agar. Values represent the mean (standard deviation) log10 CFUs per lung for 6 mice per group. Because 5 of the 6 mice inoculated with NC virus died after the challenge with WU2 strain, the lung bacterial density of the sole surviving mouse is shown.
Antibody Responses Induced by Intranasal Immunization With the PR8/PB2-PspA Virus
We examined the antibody responses in PR8/PB2-PspA virus–inoculated mice to PR8 virus, a backbone strain of PR8/PB2-PspA virus. As expected, both immunoglobulin (Ig) G and IgA against PR8 virus were efficiently elicited in the serum, BALF, and nasal wash samples of mice immunized with either PR8/PB2-GFP or PR8/PB2-PspA virus (Figure 3A and 3B).
Figure 3.

Antibody responses and survival of mice after influenza virus challenge. Mice were mock-immunized with medium or immunized with the PR8/PB2–green fluorescent protein (GFP) or PR8/PB2–pneumococcal surface protein A (PspA) virus 3 times at 2-week intervals. A, B, PR8-specific antibodies raised by the immunized mice were detected by means of an enzyme-linked immunosorbent assay with purified PR8 virus as an antigen. Immunoglobulin (Ig) G titers in the serum (A), IgG and IgA titers in bronchoalveolar lavage fluid (BALF) (B, left panel), and nasal wash samples (B, right panel) from mice intranasally mock-immunized with medium or immunized with the PR8/PB2-GFP or PR8/PB2-PspA virus were measured. Values are expressed as the mean (standard deviation [SD]) reciprocal log2 titer for 6 samples. *P < .05. C, Two weeks after the final vaccination, mice were intranasally challenged with 10-fold 50% mouse lethal dose of PR8 virus. Survival was monitored in 8 mice for 14 days after challenge. †P < .01.
We also determined whether PspA-specific antibodies were induced in mice immunized with the PR8/PB2-PspA virus. Although a negligible amount of PspA-specific IgG or IgA was detected in the serum, BALF, and nasal wash samples from the control mice, significantly higher levels of PspA-specific IgG or IgA were found in the serum, BALF, or nasal wash samples from mice immunized with the PR8/PB2-PspA virus (Figure 4A and 4B). These results indicate that PR8/PB2-PspA virus efficiently induced antibodies specific for influenza virus and PspA in the immunized mice.
Figure 4.

Antibody responses and survival of mice after Streptococcus pneumoniae challenge. Mice were mock-immunized with medium or immunized with the PR8/PB2–green fluorescent protein (GFP) or PR8/PB2–pneumococcal surface protein A (PspA) virus 3 times at 2-week intervals. A. B, PspA-specific antibodies raised by the immunized mice were detected by means of an enzyme-linked immunosorbent assay with PspA as an antigen. Immunoglobulin (Ig) G titers in serum (A), IgG and IgA titers in bronchoalveolar lavage fluid (BALF) (B, left panel), and nasal wash samples (B, right panel) from mice intranasally mock-immunized with medium or immunized with the PR8/PB2-GFP or PR8/PB2-PspA virus were measured. Values are expressed as the mean (standard deviation [SD]) reciprocal log2 titer of 6 samples. *P < .05. C, Two weeks after the final vaccination, mice were intranasally challenged with 3-fold 50% mouse lethal dose of WU2 strain. Survival was monitored in 10 mice for 10 days after challenge. †P < .01.
Effect of Intranasal Immunization With the PR8/PB2-PspA Virus on Bacterial Clearance
We examined the bacterial colonization in the nasopharynx of the immunized mice by infecting them with an avirulent S. pneumoniae EF3030 strain. On day 5 after the intranasal challenge, the bacterial densities in the nasal wash samples from mice immunized with the PR8/PB2-PspA virus (mean [standard deviation], 2.4 [0.9] log10 CFUs/mL) were significantly lower than those of mock-immunized or PR8/PB2-GFP virus–immunized mice (3.6 [0.7] and 3.6 [0.8] log10 CFUs/mL, respectively). These results indicate that the PR8/PB2-PspA virus immunization enhanced the clearance of S. pneumoniae from the nasopharynx.
Effect of Intranasal Immunization With the PR8/PB2-PspA Virus on a Lethal Infection With Influenza Virus and S. pneumoniae
We examined the protective efficacy of intranasal immunization with the PR8/PB2-PspA virus in a commonly used mouse influenza virus challenge model [48]. All mice immunized with either PR8/PB2-GFP or PR8/PB2-PspA virus survived (Figure 3C) after the challenge with 10 MLD50 of PR8 virus, whereas all mice mock-immunized with medium succumbed to the lethal infection with wild-type PR8 virus. In addition, mice immunized with either PR8/PB2-GFP or PR8/PB2-PspA virus showed an appreciable reduction in viral shedding in the lungs and nasal turbinates on days 3 and 6 after challenge, compared with those inoculated with medium (Table 1). These results show that immunization with the PR8/PB2-PspA virus conferred protective immunity against the lethal challenge with influenza virus.
Table 1.
Viral Titers in the Respiratory Tract of PR8/PB2-PspA Virus–Immunized Mice Challenged With PR8 Virusa
| Immunization | Time After Challenge, d | Viral Titer, Mean (SD) log10 PFUs/g |
|
|---|---|---|---|
| Nasal Turbinates | Lungs | ||
| Medium | 3 | 7.4 (0.2) | 8.5 (0.4) |
| 6 | 7.0, NA, NAb | 7.1, NA, NAb | |
| PR8/PB2-GFP virus | 3 | 6.9 (0.2) | 7.8 (0.9) |
| 6 | 5.8 (0.3) | 6.4, 4.5 | |
| PR8/PB2-PspA virus | 3 | 7.8, 7.9 | 8.5 |
| 6 | NDc | ND | |
Abbreviations: GFP, green fluorescent protein; NA, not applicable; ND, not detected; PFUs, plaque-forming units; PspA, pneumococcal surface protein A; SD, standard deviation.
a Mice were intranasally immunized with the indicated agents (50 µL) 3 times at 2-week intervals. Two weeks after the final vaccination, the immunized mice were challenged with 10-fold 50% mouse lethal dose of PR8 virus (50 µL). Nasal turbinates and lungs were harvested from the challenged mice (n = 3) on days 3 and 6 after challenge and subjected to virus titration by plaque assays in AX4/PB2 cells. When virus was not recovered from all 3 mice, individual titers are provided.
b NA because the mice died.
c The detection limit was 1.5 log10 PFUs/g.
Next, we assessed the protective efficacy of intranasal immunization with the PR8/PB2-PspA virus in a commonly used S. pneumoniae challenge model [20, 39, 46]. All mice mock-immunized with medium or immunized with the PR8/PB2-GFP virus died after the S. pneumoniae challenge (Figure 4C). By contrast, all mice immunized with the PR8/PB2-PspA virus were protected from the lethal challenge with S. pneumoniae (Figure 4C). These results demonstrate that the PR8/PB2-PspA virus conferred protective immunity against the lethal challenge with S. pneumoniae.
Effect of Intranasal Immunization With the PR8/PB2-PspA Virus on Secondary Pneumococcal Pneumonia
We next examined the protective efficacy of the PR8/PB2-PspA virus against secondary infection with a sublethal dose of S. pneumoniae after influenza virus infection [46]. The survival rate of mice immunized with the PR8/PB2-PspA virus (100%) was significantly higher than that of the PR8/PB2-GFP virus–immunized (50%) and the mock-immunized (20%) mice after secondary pneumococcal pneumonia (Figure 5A).
Figure 5.

Survival of immunized mice after secondary pneumococcal pneumonia challenge and bacterial densities in mice. Mice were mock-immunized with medium or immunized with the PR8/PB2–green fluorescent protein (GFP) or PR8/PB2–pneumococcal surface protein A (PspA) virus 3 times at 2-week intervals. Two weeks after the final vaccination, mice were intranasally challenged with 1.0 × 103 plaque-forming units (PFUs) of A/New Caledonia/20/99 (NC) virus. Ten days after the viral challenge, mice were intranasally challenged with 6.0 × 103 colony-forming units (CFUs) (sublethal dose; A, B) or 2.0 × 107 CFUs (3-fold 50% mouse lethal dose; C, D). A, C, Survival was monitored in 10 mice for 10 days after the bacterial challenge. *P < .05; †P < .01. B, D, Lung tissues were harvested 5 days after bacterial challenge and subjected to bacterial titration on sheep blood agar. The detection limit of Streptococcus pneumoniae is 10 CFUs/mL. Values represent mean (standard deviation) log10 CFUs per lung for 6 mice per group. *P < .05; †P < .01. Abbreviation: NS, not statistically significant.
We also determined viral titers and bacterial densities in the respiratory organs of the mice subjected to secondary pneumococcal pneumonia after influenza virus infection. Viral titers in the lungs, but not those in the nasal turbinates, of mice immunized with either the PR8/PB2-GFP virus or the PR8/PB2-PspA virus were significantly lower than those in mice mock-immunized with medium (Table 2). The bacterial densities in mice immunized with either the PR8/PB2-GFP (P < .05) or the PR8/PB2-PspA virus (P < .01) were significantly lower than those in mock-immunized mice (Figure 5B). In addition, the bacterial densities in the lungs of the PR8/PB2-PspA virus–immunized mice were significantly lower than those in the PR8/PB2-GFP virus–immunized mice.
Table 2.
Viral Titers in the Respiratory Tract of PR8/PB2-PspA Virus–Immunized Mice Challenged With NC Virusa
| Immunization | Viral Titer, Mean (SD), log10 PFUs/g |
|
|---|---|---|
| Nasal Turbinates | Lungs | |
| Medium | 6.3 (0.4) | 6.1 (0.3) |
| PR8/PB2-GFP virus | 6.0 (0.4) | 4.0 (1.4)b |
| PR8/PB2-PspA virus | 5.9 (0.4) | 3.7 (1.3)c |
Abbreviations: GFP, green fluorescent protein; NC, A/New Caledonia/20/99; PFUs, plaque-forming units; PspA, pneumococcal surface protein A; SD, standard deviation.
a Mice were intranasally immunized 3 times with the indicated agents (50 µL) and challenged with NC virus (50 µL) 2 weeks after the final vaccination. Nasal turbinates and lungs were harvested from mice (n = 6) 3 days after challenge and subjected to virus titration by use of plaque assays in AX4/PB2 cells. Statistically significant differences in the viral titers compared with the medium (control) group were assessed using 1-way analysis of variance followed by a Dunnett test.
b P < .05.
c P < .01.
Finally, we examined the protective efficacy of the PR8/PB2-PspA virus against secondary infection with a lethal dose of S. pneumoniae. The survival rate of mice immunized with the PR8/PB2-PspA virus (100%) was significantly higher than that of the PR8/PB2-GFP virus–immunized (10%) or mock-immunized (0%) mice after secondary pneumococcal pneumonia (P < .01; Figure 5C). The bacterial density in the lungs of mice immunized with the PR8/PB2-PspA virus was significantly lower than that of the mock-immunized or the PR8/PB2-GFP virus–immunized mice (P < .01; Figure 5D). These results demonstrate that immunization with the PR8/PB2-PspA virus protects mice from secondary pneumococcal pneumonia.
DISCUSSION
In the current study, we generated a PB2-KO virus expressing the PspA of S. pneumoniae as a foreign protein in the coding region of its PB2 segment. We found no evidence of increased susceptibility of mice to pneumococcal pneumonia after intranasal inoculation of the PR8/PB2-PspA virus (Figure 2), indicating that our PB2-KO virus can be safely used in mice as a vaccine against S. pneumoniae.
Intranasal immunization with the PR8/PB2-PspA virus could induce influenza virus–specific IgG in the airways and serum, and influenza virus–specific IgA in the airways of mice without the need for adjuvant (Figure 3A and 3B). We found that hemagglutination inhibition titers in mice immunized with the PR8/PB2-PspA virus were higher than those in PR8/PB2-GFP virus–immunized or mock-immunized mice (data not shown), suggesting that PspA might affect virus-specific immune responses. Similarly, intranasal immunization with the PR8/PB2-PspA virus could induce PspA-specific IgG in the airways and serum, and PspA-specific IgA in the lower airways of mice without the need for adjuvant (Figure 4A and 4B). We also demonstrated that intranasal immunization with 2.5 µg of PspA and 10 µg of a Toll-like receptor agonist were required to induce PspA-specific IgA in the airways [20]. Collectively, these results imply that the PR8/PB2-PspA virus may stimulate innate immunity, leading to the effective production of PspA-specific IgA in the airways.
The PR8/PB2-PspA virus elicited PR8-specific antibodies efficiently and protected mice from a lethal challenge with PR8 virus (Figure 3), as demonstrated elsewhere [35–37]. In addition, we found an inhibitory effect of intranasal immunization with PR8/PB2-KO virus on the replication of NC virus in the lungs of mice (Table 2). We also demonstrated the effect of intranasal immunization with PR8/PB2-PspA virus on bacterial clearance in the nasopharynx of mice.
The protective effect of intranasal immunization with PR8/PB2-PspA virus against pneumococcal pneumonia can be explained by the critical role of PspA-specific IgA for clearance in pneumococcal infection [49] and by the PspA-specific IgG-mediated opsonophagocytic killing activity [27, 50]. Importantly, we found no statistically significant difference in protection, as measured by the survival rate after infection with a low-dose of S. pneumoniae (Figure 5A), for mice intranasally immunized with the PR8/PB2-GFP virus in the secondary infection model, although this was not the case with the high-dose challenge with S. pneumoniae (Figure 5C). Our data suggest that intranasal immunization with the PR8/PB2-GFP virus partly diminished the effect of preinfection with influenza virus on the enhanced susceptibility to pneumococcal infection. Of note, although intranasal immunization with the PR8/PB2-GFP virus did not protect the mice after infection with a high-dose of S. pneumoniae, all of the mice immunized with the PR8/PB2-PspA virus survived after infection with not only a low dose, but also a high dose of S. pneumoniae after influenza virus infection (Figure 5A and 5C). Taken together, these data suggest that the PR8/PB2-PspA virus is a powerful bivalent vaccine for both influenza virus and S. pneumoniae infections.
It is possible that PB2-KO vaccines may not induce immunity in those who have immunity to influenza viruses. Therefore, further studies are needed to examine whether the PR8/PB2-PspA virus can serve as a bivalent vaccine for mice previously infected with a homologous or heterologous influenza A virus strain. However, as the antigenicity of circulating influenza viruses changes, the backbone of the PB2-KO viruses could be replaced with that of the circulating strains.
In conclusion, in the current study we demonstrated the potential of a PB2-KO influenza virus expressing PspA from the recombinant PB2 gene as a novel bivalent vaccine against influenza virus and S. pneumoniae, respectively, but also against secondary pneumococcal pneumonia after influenza virus infection. Our PB2-KO virus therefore provides a promising option to control respiratory infections caused by common respiratory pathogens.
Notes
Acknowledgments. We thank Dr Shin Murakami, Dr Seiya Yamayoshi, Dr Hiroaki Katsuta, and Mr Hirofumi Kobayashi for fruitful discussions. We also thank Dr Susan Watson for editing the manuscript.
Financial support. This work was supported by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation; the Biomedical Cluster Kansai project, promoted by the Regional Innovation Cluster Program; a grant-in-aid for Specially Promoted Research; the Japan Initiative for the Global Research Network on Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; the Ministry of Health, Labour, and Welfare, Japan (grants-in-aid); ERATO and Strategic Basic Research Programs, Japan Science and Technology Agency; Advanced Research & Development Programs for Medical Innovation, the Japan Agency for Medical Research and Development; National Institute of Allergy and Infectious Diseases (NIAID) (Public Health Service research grants); the NIAID-funded Center for Research on Influenza Pathogenesis (grant HHSN266200700010C); and Japan Society for the Promotion of Science (research fellowships for young scientists to R. U.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 2008; 198:962–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chien YW, Klugman KP, Morens DM. Bacterial pathogens and death during the 1918 influenza pandemic. N Engl J Med 2009; 361:2582–3. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien KL, Wolfson LJ, Watt JP et al. . Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 2009; 374:893–902. [DOI] [PubMed] [Google Scholar]
- 4.Bogaert D, De Groot R, Hermans PW. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 2004; 4:144–54. [DOI] [PubMed] [Google Scholar]
- 5.Isaacman DJ, McIntosh ED, Reinert RR. Burden of invasive pneumococcal disease and serotype distribution among Streptococcus pneumoniae isolates in young children in Europe: impact of the 7-valent pneumococcal conjugate vaccine and considerations for future conjugate vaccines. Int J Infect Dis 2010; 14:e197–209. [DOI] [PubMed] [Google Scholar]
- 6.Cutts FT, Zaman SM, Enwere G et al. . Efficacy of nine-valent pneumococcal conjugate vaccine against pneumonia and invasive pneumococcal disease in The Gambia: randomised, double-blind, placebo-controlled trial. Lancet 2005; 365:1139–46. [DOI] [PubMed] [Google Scholar]
- 7.Klugman KP, Madhi SA, Huebner RE, Kohberger R, Mbelle N, Pierce N. A trial of a 9-valent pneumococcal conjugate vaccine in children with and those without HIV infection. N Engl J Med 2003; 349:1341–8. [DOI] [PubMed] [Google Scholar]
- 8.Ardanuy C, Tubau F, Pallares R et al. . Epidemiology of invasive pneumococcal disease among adult patients in Barcelona before and after pediatric 7-valent pneumococcal conjugate vaccine introduction, 1997–2007. Clin Infect Dis 2009; 48:57–64. [DOI] [PubMed] [Google Scholar]
- 9.Burgos J, Falco V, Borrego A et al. . Impact of the emergence of non-vaccine pneumococcal serotypes on the clinical presentation and outcome of adults with invasive pneumococcal pneumonia. Clin Microbiol Infect 2013; 19:385–91. [DOI] [PubMed] [Google Scholar]
- 10.Hicks LA, Harrison LH, Flannery B et al. . Incidence of pneumococcal disease due to non-pneumococcal conjugate vaccine (PCV7) serotypes in the United States during the era of widespread PCV7 vaccination, 1998–2004. J Infect Dis 2007; 196:1346–54. [DOI] [PubMed] [Google Scholar]
- 11.Pilishvili T, Lexau C, Farley MM et al. . Sustained reductions in invasive pneumococcal disease in the era of conjugate vaccine. J Infect Dis 2010; 201:32–41. [DOI] [PubMed] [Google Scholar]
- 12.Singleton RJ, Hennessy TW, Bulkow LR et al. . Invasive pneumococcal disease caused by nonvaccine serotypes among Alaska native children with high levels of 7-valent pneumococcal conjugate vaccine coverage. JAMA 2007; 297:1784–92. [DOI] [PubMed] [Google Scholar]
- 13.Weinberger DM, Malley R, Lipsitch M. Serotype replacement in disease after pneumococcal vaccination. Lancet 2011; 378:1962–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croney CM, Coats MT, Nahm MH, Briles DE, Crain MJ. PspA family distribution, unlike capsular serotype, remains unaltered following introduction of the heptavalent pneumococcal conjugate vaccine. Clin Vaccine Immunol 2012; 19:891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regev-Yochay G, Rahav G, Riesenberg K et al. . Initial effects of the National PCV7 Childhood Immunization Program on adult invasive pneumococcal disease in Israel. PloS One 2014; 9:e88406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Briles DE, Tart RC, Swiatlo E et al. . Pneumococcal diversity: considerations for new vaccine strategies with emphasis on pneumococcal surface protein A (PspA). Clin Microbiol Rev 1998; 11:645–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jedrzejas MJ, Lamani E, Becker RS. Characterization of selected strains of pneumococcal surface protein A. J Biol Chem 2001; 276:33121–8. [DOI] [PubMed] [Google Scholar]
- 18.Nabors GS, Braun PA, Herrmann DJ et al. . Immunization of healthy adults with a single recombinant pneumococcal surface protein A (PspA) variant stimulates broadly cross-reactive antibodies to heterologous PspA molecules. Vaccine 2000; 18:1743–54. [DOI] [PubMed] [Google Scholar]
- 19.Jedrzejas MJ. Unveiling molecular mechanisms of pneumococcal surface protein A interactions with antibodies and lactoferrin. Clin Chim Acta 2006; 367:1–10. [DOI] [PubMed] [Google Scholar]
- 20.Oma K, Zhao J, Ezoe H et al. . Intranasal immunization with a mixture of PspA and a Toll-like receptor agonist induces specific antibodies and enhances bacterial clearance in the airways of mice. Vaccine 2009; 27:3181–8. [DOI] [PubMed] [Google Scholar]
- 21.Ochs MM, Bartlett W, Briles DE et al. . Vaccine-induced human antibodies to PspA augment complement C3 deposition on Streptococcus pneumoniae. Microb Pathog 2008; 44:204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren B, Szalai AJ, Hollingshead SK, Briles DE. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect Immun 2004; 72:114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu AH, Fulgham RL, McCrory MA, Briles DE, Szalai AJ. Pneumococcal surface protein A inhibits complement activation by Streptococcus pneumoniae. Infect Immun 1999; 67:4720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crain MJ, Waltman WD 2nd, Turner JS et al. . Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect Immun 1990; 58:3293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briles DE, Tart RC, Wu HY, Ralph BA, Russell MW, McDaniel LS. Systemic and mucosal protective immunity to pneumococcal surface protein A. Ann N Y Acad Sci 1996; 797:118–26. [DOI] [PubMed] [Google Scholar]
- 26.McDaniel LS, McDaniel DO, Hollingshead SK, Briles DE. Comparison of the PspA sequence from Streptococcus pneumoniae EF5668 to the previously identified PspA sequence from strain Rx1 and ability of PspA from EF5668 to elicit protection against pneumococci of different capsular types. Infect Immun 1998; 66:4748–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDaniel LS, Sheffield JS, Delucchi P, Briles DE. PspA, a surface protein of Streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect Immun 1991; 59:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nabel GJ, Fauci AS. Induction of unnatural immunity: prospects for a broadly protective universal influenza vaccine. Nat Med 2010; 16:1389–91. [DOI] [PubMed] [Google Scholar]
- 29.Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med 2010; 363:2036–44. [DOI] [PubMed] [Google Scholar]
- 30.Cox RJ, Brokstad KA, Ogra P. Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand J Immunol 2004; 59:1–15. [DOI] [PubMed] [Google Scholar]
- 31.Rimmelzwaan GF, Fouchier RA, Osterhaus AD. Influenza virus-specific cytotoxic T lymphocytes: a correlate of protection and a basis for vaccine development. Curr Opin Biotechnol 2007; 18:529–36. [DOI] [PubMed] [Google Scholar]
- 32.Fiore AE, Uyeki TM, Broder K et al. . Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep 2010; 59:1–62. [PubMed] [Google Scholar]
- 33.Khan AS, Polezhaev F, Vasiljeva R et al. . Comparison of US inactivated split-virus and Russian live attenuated, cold-adapted trivalent influenza vaccines in Russian schoolchildren. J Infect Dis 1996; 173:453–6. [DOI] [PubMed] [Google Scholar]
- 34.Palese P, Garcia-Sastre A. Influenza vaccines: present and future. J Clin Invest 2002; 110:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozawa M, Victor ST, Taft AS et al. . Replication-incompetent influenza A viruses that stably express a foreign gene. J Gen Virol 2011; 92:2879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Victor ST, Watanabe S, Katsura H, Ozawa M, Kawaoka Y. A replication-incompetent PB2-knockout influenza A virus vaccine vector. J Virol 2012; 86:4123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uraki R, Kiso M, Iwatsuki-Horimoto K et al. . A novel bivalent vaccine based on a PB2-knockout influenza virus protects mice from pandemic H1N1 and highly pathogenic H5N1 virus challenges. J Virol 2013; 87:7874–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatakeyama S, Sakai-Tagawa Y, Kiso M et al. . Enhanced expression of an alpha2,6-linked sialic acid on MDCK cells improves isolation of human influenza viruses and evaluation of their sensitivity to a neuraminidase inhibitor. J Clin Microbiol 2005; 43:4139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Briles DE, Hollingshead SK, Paton JC et al. . Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J Infect Dis 2003; 188:339–48. [DOI] [PubMed] [Google Scholar]
- 40.Briles DE, Novak L, Hotomi M, van Ginkel FW, King J. Nasal colonization with Streptococcus pneumoniae includes subpopulations of surface and invasive pneumococci. Infect Immun 2005; 73:6945–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neumann G, Watanabe T, Ito H et al. . Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 1999; 96:9345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briles DE, Ades E, Paton JC et al. . Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect Immun 2000; 68:796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kida H, Brown LE, Webster RG. Biological activity of monoclonal antibodies to operationally defined antigenic regions on the hemagglutinin molecule of A/Seal/Massachusetts/1/80 (H7N7) influenza virus. Virology 1982; 122:38–47. [DOI] [PubMed] [Google Scholar]
- 44.Jia N, Wang SX, Liu YX et al. . Increased sensitivity for detecting avian influenza-specific antibodies by a modified hemagglutination inhibition assay using horse erythrocytes. J Virol Methods 2008; 153:43–8. [DOI] [PubMed] [Google Scholar]
- 45.Kayali G, Setterquist SF, Capuano AW, Myers KP, Gill JS, Gray GC. Testing human sera for antibodies against avian influenza viruses: horse RBC hemagglutination inhibition vs. microneutralization assays. J Clin Virol 2008; 43:73–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ezoe H, Akeda Y, Piao Z et al. . Intranasal vaccination with pneumococcal surface protein A plus poly(I:C) protects against secondary pneumococcal pneumonia in mice. Vaccine 2011; 29:1754–61. [DOI] [PubMed] [Google Scholar]
- 47.Ferreira DM, Darrieux M, Silva DA et al. . Characterization of protective mucosal and systemic immune responses elicited by pneumococcal surface protein PspA and PspC nasal vaccines against a respiratory pneumococcal challenge in mice. Clin Vaccine Immunol 2009; 16:636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YH, Kim JK, Kim DJ et al. . Diet-induced obesity dramatically reduces the efficacy of a 2009 pandemic H1N1 vaccine in a mouse model. J Infect Dis 2012; 205:244–51. [DOI] [PubMed] [Google Scholar]
- 49.Fukuyama Y, King JD, Kataoka K et al. . Secretory-IgA antibodies play an important role in the immunity to Streptococcus pneumoniae. J Immunol 2010; 185:1755–62. [DOI] [PubMed] [Google Scholar]
- 50.Arulanandam BP, Lynch JM, Briles DE, Hollingshead S, Metzger DW. Intranasal vaccination with pneumococcal surface protein A and interleukin-12 augments antibody-mediated opsonization and protective immunity against Streptococcus pneumoniae infection. Infect Immun 2001; 69:6718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]