

Abstract
Metformin, an established first-line treatment for patients with type 2 diabetes, has been associated with gastrointestinal (GI) adverse effects that limit its use. Histamine and serotonin have potent effects on the GI tract. The effects of metformin on histamine and serotonin uptake were evaluated in cell lines overexpressing several amine transporters (OCT1, OCT3 and SERT). Metformin inhibited histamine and serotonin uptake by OCT1, OCT3 and SERT in a dose-dependent manner, with OCT1-mediated amine uptake being most potently inhibited (IC50 = 1.5 mM). A chemoinformatics-based method known as Similarity Ensemble Approach predicted diamine oxidase (DAO) as an additional intestinal target of metformin, with an E-value of 7.4 × 10−5. Inhibition of DAO was experimentally validated using a spectrophotometric assay with putrescine as the substrate. The Ki of metformin for DAO was measured to be 8.6 ± 3.1 mM. In this study, we found that metformin inhibited intestinal amine transporters and DAO at concentrations that may be achieved in the intestine after therapeutic doses. Further studies are warranted to determine the relevance of these interactions to the adverse effects of metformin on the gastrointestinal tract.
Keywords: Organic cation transporter, Histamine, Serotonin, Putrescine, Diamine oxidase, Metformin
Introduction
Metformin, a biguanide, is used worldwide as first-line therapy for the treatment of type 2 diabetes, and acts primarily in the liver to reduce gluconeogenesis [1, 2]. However, 30–50 % of patients on metformin report gastrointestinal (GI) side effects, such as diarrhea and bloating [3–6]. These GI side effects are generally dose-dependent, and for the majority of patients, subside after several doses. However, in approximately 5 % of patients, these side effects are severe enough to warrant discontinuation of the drug, and these patients switch to other anti-diabetic drugs. Several hypotheses as to the cause of these metformin-induced GI side effects have been proposed, including mal-absorption of bile salts in the ileum [7], increased serotonin release from human gut mucosa [8], increased gastrointestinal hormone levels and increased gastric acid secretion [9].
Signaling molecules such as histamine and serotonin can bind to receptors in the GI tract to regulate normal physiologic function. Histamine receptors are highly expressed in the gastrointestinal tract [10, 11]. Interactions of histamine with H1 receptors regulate intestinal smooth muscle contraction, and H2 receptors stimulate gastric acid secretion. In contrast, histamine interactions with H3 receptors may inhibit gastric acid secretion, whereas histamine interactions with H4 receptors have been implicated in immune-mediated responses in gut inflammation [10–12]. In the intestine, histaminergic signalling is terminated by the inactivation of histamine by the enzymes diamine oxidase (DAO) and N-methyltransferase (HNMT) [13]. Given the important roles that histamine plays in the gastrointestinal tract, it is not surprising that when histamine levels increase due to impaired histamine degradation or ingestion, GI effects such as nausea, vomiting and diarrhea can occur [14].
Most of the serotonin in the body is produced in the gastrointestinal tract, where the monoamine plays an important role in the regulation of gastrointestinal physiology [15]. Primarily stored in enterochromaffin (EC) cells of the mucosal epithelium, serotonin release activates intrinsic and extrinsic sensory GI neurons, leading to gut motility, secretion and sensation [15, 16]. Defective serotonergic signaling has been implicated in a number of GI disorders, and small molecules that interact with serotonin receptors have been developed to treat GI symptoms [17]. The primary serotonin transporter, SERT, is responsible for the reuptake of extracellular serotonin in order to terminate serotonergic signaling. SERT knockout mice were observed to have increased stool water and colon motility when compared to wildtype mice [18]. Further, many drugs, including many antidepressants that inhibit SERT, cause GI adverse effects, suggesting that inhibition of the transporter may produce undesired GI effects [19]. Though less permissive in its substrate selectivity, SERT shares some overlapping substrates with organic cation transporters (OCTs). Moreover, SERT knockout mice show upregulation of OCT transporters [18].
Organic cation transporters, such as OCT1 and OCT3, are known to transport a variety of drugs and endogenous molecules such as serotonin and histamine [20–24]. OCT1 is primarily expressed in the liver and expressed at lower levels in other tissues, whereas OCT3 is expressed ubiquitously in various tissues [25]. In the intestine, OCT3 has been localized to the apical membrane of enterocytes [26, 27], whereas there are contrasting reports about the localization of OCT1 in enterocytes [28–31], with the transporter localized to the apical membrane in some reports and to the basolateral membrane in others [29–34]. Typically prescribed at high doses (500–1000 mg), metformin is a known substrate of organic cation transporters [32, 35–39]. A recent study showed that patients on metformin with concomitant use of other medications known to inhibit OCT1 were more likely to be metformin-intolerant, and that people carrying reduced-function mutations in OCT1 were also more likely to be intolerant to metformin [40]. Though metformin is clearly a substrate of organic cation transporters, its interactions on transporters is poorly understood.
The goal of this study was to determine the effect of metformin on modulating the uptake of histamine and serotonin through amine transporters expressed in intestinal cells, particularly OCT1, OCT3 and SERT. Our data suggest that at expected concentrations of metformin in the intestine after therapeutic doses, metformin may inhibit OCT1, OCT3 and SERT, thus modulating intestinal levels of histamine and serotonin (Fig. 1). In addition, using the Similarity Ensemble Approach (SEA) [41–45], a chemoinformatic method, we predicted that metformin may interact with another protein involved in intestinal histamine disposition, DAO. This prediction was confirmed through experimental methods. Collectively, our study suggests that metformin interacts with proteins involved in the intestinal disposition of serotonin and histamine at clinically relevant intestinal concentrations, and we hypothesize that such interactions may ultimately contribute to the GI side effects of the drug.
Fig. 1.
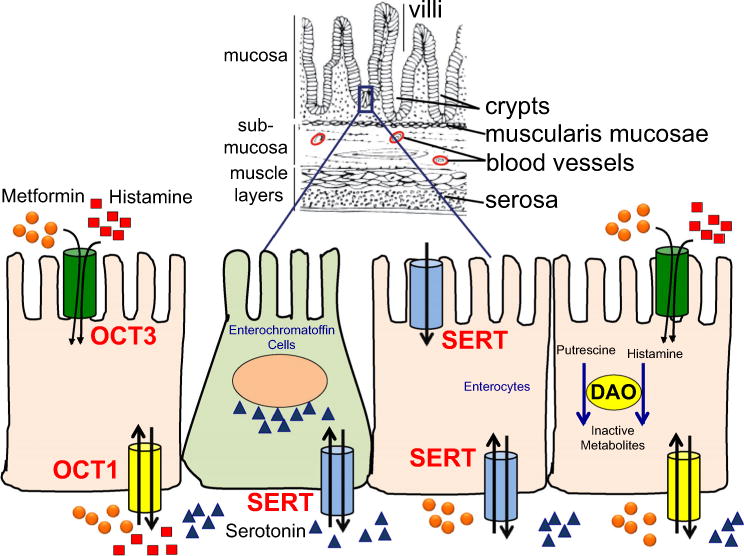
Overall figure showing the localization of various transporters (OCT1, OCT3 and SERT) and DAO, which are the subject of the current study, in enterocytes and enterochromaffin cells. This figure shows that histamine (red squares) from diet and metformin (orange circles) are transported into cells via OCT3. Transport of histamine, serotonin (blue triangles) or metformin across the basolateral membrane occurs by OCT1. Histamine, from the diet or synthesized from L-histidine, is inactivated by DAO and HNMT (not shown) to imidazole acetaldehyde and N-methylhistamine, respectively. Serotonin, which is synthesized from tryptophan (from diet), is stored in enterochromaffin cells and released into the blood by the serotonin transporter, SERT (SLC6A4). A recent study suggests that SERT is localized on the apical and basolateral membrane of enterocytes and plays a role in metformin and serotonin uptake [27]. Diamine oxidase (DAO) plays an important role in enterocytes to remove excess histamine and putrescine from diet. Other transporters not shown may also contribute to serotonin or histamine disposition. The bidirectional arrows show that the molecules could be transported into or out of the cells depending on the concentrations of the molecules inside and outside the cells. Note the upper part of the figure was modified from an image obtained from http://theibdimmunologist.com/Review/wp-content/uploads/colon-and-small-intestine-wall-01.png (Color figure online)
Methods
Chemicals
All compounds (metformin, aminoguanidine, histamine, 5-hydroxytryptamine (5-HT, serotonin), putrescine, dianisidine, Tris, 2-[N-morpholino]ethanesulfonic Acid (MES), acetic acid, porcine diamine oxidase and peroxidase) were purchased from Sigma Aldrich (St. Louis, MO). Cell culture media (DMEM H-21), fetal bovine serum, penicillin/streptomycin, hygromycin B and Hank’s balanced salt solution (HBSS) were obtained from University of California San Francisco Cell Culture Facility. [14C]-Metformin (MC 2043) was purchased from Moravek Biochemicals (Brea, CA), [3H]-histamine (ART 1432) and [3H]-5-hydroxytryptamine (5-HT) (ART 0350) were purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO).
Cell culture
Stably transfected HEK-293 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) H-21 medium supplemented with 10 % fetal bovine serum, 100 units/mL penicillin, 100 units/mL streptomycin, and 150 lg/mL hygromycin B. HEK-293 cells which were used for transient transfection were cultured in the above medium but without the selection antibiotic, hygromycin B. All cell lines were grown at 37 °C in a humidified atmosphere with 5 % CO2.
Cell lines and transfection
Human embryonic kidney (HEK-293) Flp-In-293 cells stably transfected with the full-length reference human OCT1 cDNA (HEK-hOCT1), OCT3 cDNA (HEK-hOCT3), and the empty vector (HEK-EV) were previously established in our laboratory [22, 46, 47]. HEK-293 cells transiently transfected with the full-length reference SERT cDNA (HEK-hSERT) were established by transfecting 500 ng/well of pCMV6-XL4 vector containing the full-length transporter cDNA (NM_001045.2) from Origene (Rockville, MD) into HEK-293 cells using Lipofectamine LTX from Life Technologies (Grand Island, NY) per manufacturer instructions. The expression level of each transporter mRNA in each transfected HEK-293 cell line was verified by quantitative RT-PCR. The transport activity of metformin and serotonin in transfected cell lines were compared to the HEK-EV cell line in every transport assay.
Transporter inhibition studies
Transporter inhibition studies were performed using HEK-293 cells stably expressing the vector only (HEK-EV), SLC22A1 (OCT1), SLC22A3 (OCT3) and SLC6A4 (SERT). Briefly, transfected cell lines were grown on poly-D-lysine coated 24-well plates in DMEM H-21 medium supplemented with 10 % fetal bovine serum to at least 90 % confluence (16–48 h post seeding). For uptake studies, HEK-EV, HEK-hOCT1, HEK-hOCT3 and HEK-hSERT were preincubated in Hank’s balanced salt solution (HBSS; 5.4 mmol/L potassium chloride, 0.44 mmol/L monobasic potassium phosphate, 4.2 mmol/L sodium bicarbonate, 137 mmol/L sodium chloride, 0.34 mmol/L dibasic sodium phosphate, 5.6 mmol/L D-glucose, 1.3 mmol/L calcium chloride, 0.49 mmol/L magnesium chloride, 0.41 mmol/L magnesium sulfate, pH 7.4) for 15–20 min. The buffer was removed and replaced with uptake buffer (HBSS containing 10 nmol/L unlabeled histamine and a trace amount of [3H]-labeled histamine or 10 nmol/L unlabeled serotonin and a trace amount of [3H]-labeled 5-hydroxytryptamine). Uptake was performed at 37 °C for a designated period of time for which linear uptake was observed, and then the cells were washed twice with 1 mL of ice-cold HBSS. To determine the inhibition of the uptake, the HEK-293 cells were simultaneously exposed to the substrate (histamine or serotonin) and metformin at various concentrations (0, 1, 5, 10, 15 and 30 mM). Cells were lysed with 0.1 N NaOH and 0.1 % SDS, and the lysate was subjected to scintillation counting on a LS6500 Scintillation Counter (Beckman Coulter) and normalized by protein content per well as determined with a Pierce BCA Protein Assay Kit (Life Technologies).
Similarity ensemble approach (SEA) (http://sea.bkslab.org/)
SEA predictions were calculated as previously described [45, 48] using RDKit (http://rdkit.org) ECFP4 (Morgan) fingerprints. For the SEA target panel, we used subsets of ChEMBL-14 and ChEMBL-16, extracted as previously described [42], and whose ligand structures were prepared using the ChEMBL standardiser (https://github.com/flat kinson/standardiser). Small SEA p-values (for ChEMBL-14 predictions) and expectation values (E-values; for ChEMBL-16 predictions) denote relationships between drugs and ligand sets that were stronger than would be expected by random chance alone; these values have meanings related to the more familiar E-values from BLAST searches, as the same statistical engine is used by SEA. High maximum Tanimoto coefficients (Max Tc) reflect high pair-wise chemical structural similarities between the drug and its closest ligand neighbor annotated to the predicted target. A maximum Tc of 1.00 means that a drug has already been reported within the ChEMBL database to bind to the predicted target.
Diamine oxidase assay
Metformin was tested for inhibition of porcine kidney diamine oxidase (E.C. No. 1.4.3.6, Sigma-Aldrich). Stocks of the compounds were prepared in purified water or in 50 mM phosphate buffer and subsequently diluted into a three-component, constant ionic strength buffer [49] (final concentration of 50 mM acetic acid, 50 mM MES and 100 mM Tris) to yield a final reaction volume of 1 mL. This buffer helped to alleviate the previously reported ionic strength effects on DAO activity [50]. In addition to the buffer, assays for diamine oxidase contained 0.15 mg/mL O-dianisidine, 100 pyrogallol U/mL peroxidase, 0.024 Units/mL diamine oxidase, 0–100 mM putrescine and 0–100 mM metformin. All reagents and water were added to the cuvette, the reactions were incubated at 37 °C for 5 min, and the reaction was initiated with diamine oxidase. Assays were performed at 37 °C, pH 7.0. Data acquisition was performed by an HP8453a spectrophotometer using kinetic mode in the software UV–Vis ChemStation (Agilent Technologies). The rate of reaction was monitored spectrophotometrically at 440 nm. Activity was measured in triplicate for six different concentrations of metformin. The assay was repeated at least three times over several days. Dose–response curves were plotted, and Ki values were calculated using the enzyme kinetics mixed model inhibition equation in Graphpad Prism 6 (Graphpad, San Diego, CA). During the curve fitting process, the inhibition constant was defined as a global parameter. The Ki values reported for metformin represent the best-fit values for the respective data sets.
Results
Metformin-mediated inhibition of histamine and serotonin uptake by OCT1, OCT3 and SERT
Various concentrations of metformin were used to determine its potency in inhibiting OCT1 (SLC22A1), OCT3 (SLC22A3) and SERT (SLC6A4) in overexpressing cell lines. Figure 2 shows the effects of various concentrations of metformin on inhibition of transporter-mediated serotonin uptake (Fig. 2a, c, e) and histamine uptake (Fig. 2b, d). Among the three transporters tested, metformin inhibited OCT1 most potently, with IC50 of 1.46 ± 0.14 mM and 1.46 ± 0.06 mM for serotonin and histamine, respectively (Fig. 2f). In contrast, metformin had a lower potency in inhibiting monoamine uptake by OCT3 and SERT (<50 % inhibition at 10 mM, Fig. 2c–e).
Fig. 2.
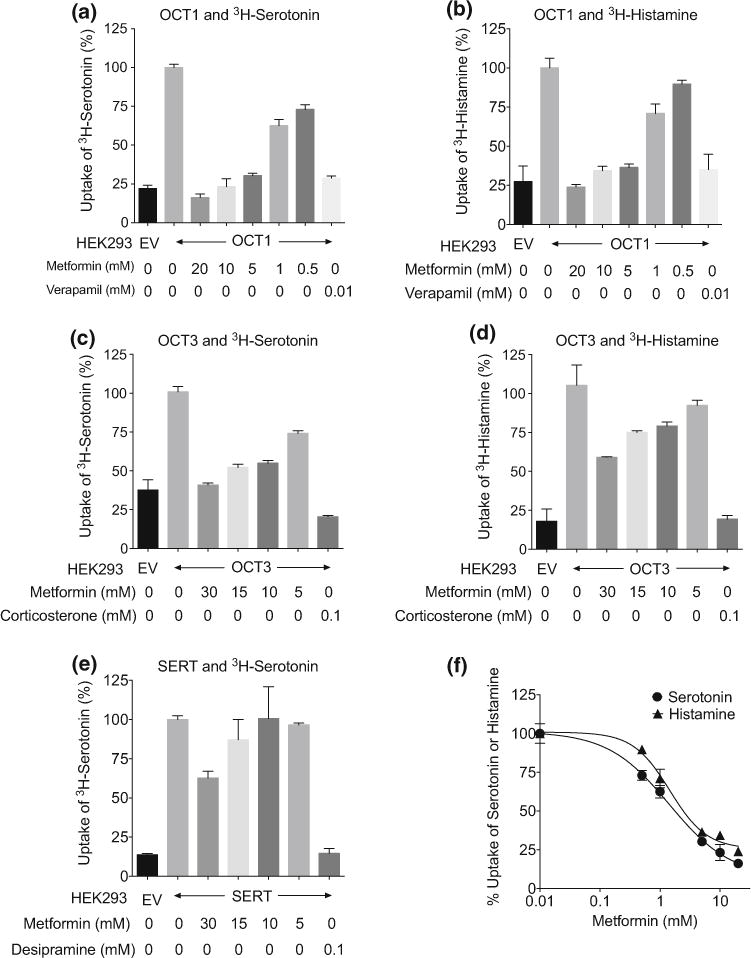
Inhibition of serotonin- and histamine-mediated uptake by metformin in cells recombinantly expressing OCT1 (SLC22A1), OCT3 (SLC22A3), and SERT (SLC6A4). HEK-293 cells stably expressing SLC22A1 (a, b, f) or SLC22A3 (c, d) were used for serotonin and histamine uptake. HEK-293 cells transiently expressing SLC6A4 (e) was used for serotonin uptake. Various concentrations of metformin were used for the inhibition studies and a canonical inhibitor of the transporter was used in the study
Using SEA to predict metformin off-targets
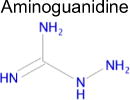
We used SEA to predict whether metformin might bind to any unreported molecular targets. Of the approximately 2,100 protein targets present in ChEMBL-16, SEA predicted that metformin would bind to only one: diamine oxidase (DAO, also known as ABP1). Notably, aminoguanidine has a similar structure to metformin and is a potent inhibitor of diamine oxidase (Table 1), but inhibition of DAO activity by metformin has not been previously reported. Diamine oxidase is highly expressed in the gut [51, 52] and metabolizes polyamines such as putrescine and histamine; for the latter, it is one of only two enzymes to do so. Diamine oxidase plays an important role in degrading exogenous polyamines, and reduced levels of DAO have been associated with histamine intolerance and allergy [53–56].
Table 1.
Metformin off-target prediction
| Drug | Closest ligand for predicted target | Predicted target | SEA E-value | Closest (max) Tc |
|---|---|---|---|---|
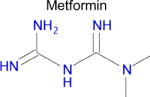
|
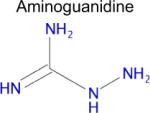
|
DAO (ABP1) | 7.4 × 10−5 | 0.35 Tc |
Tc Tanimoto coefficient. The Tc calculates the number of on bits in common between fingerprints divided by the total number of nonoverlapping on bits between fingerprints
In vitro analysis of diamine oxidase inhibition
Using an enzymatic assay, we found that metformin inhibited porcine DAO with mixed-type inhibition, with a Ki value of 8.6 ± 3.1 mM (Table 2). Figure 3 shows a plot of porcine diamine oxidase inhibition by metformin. The KM value of putrescine in the experiment was 0.33 ± 0.16 mM. Meanwhile, aminoguanidine, a known inhibitor of DAO, inhibited porcine DAO greater than 80 % at 100 μM in the presence of putrescine (100 μM) as the substrate. Previously, Cubria et al. [57] reported that phenformin, a biguanide and a predecessor of metformin, inhibited diamine oxidase with Ki of 4 mM. Furthermore, the reported KM value of putrescine using porcine diamine oxidase is 0.35 mM [58], which is within the error of our measurements. Both this result and the high inhibitory activity of aminoguanidine support the reliability of the assay. Whereas 9 mM is a high concentration, we note that metformin is a small molecule (MW 129.16 g/mol) and a polar drug (i.e. bearing at least one and possibly two positive charges), and concentrations higher than this are thought to be reached in the human intestine upon standard dosing [59, 60].
Table 2.
Top prescription drugs predicted to target DAO
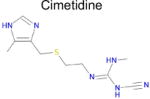
| Drug | DAO SEA E valuea | Affinity binb (μM) | Max Tcc | IC50 from literatured | |
|---|---|---|---|---|---|
| 1 |
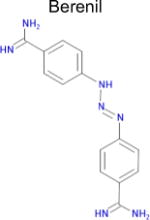
|
4.4 × 10−30 (rat)e | 10 | 0.36 Tc | 13 ± 1f nM |
| 2 |
|
1.8 × 10−19 | 1 | 1.00 Tc (known in ChEMBL) | 10–1508 nM |
| 3 |
|
1.1 × 10−16e | 1 | 0.28 Tc | 970± 50f μM |
| 4 |
|
6.8 × 10−14 (rat) | 10 | 0.38 Tc | 5.1 ± 0.8g μM |
| 5 |
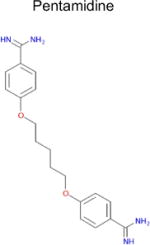
|
2.2 × 10−8 (rat)e | 10 | 0.29 Tc | 0.29–3f μM |
| 6 |
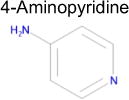
|
4.4 × 10−5 | 1 | 0.36 Tc | Not known in literature |
| 7 |

|
7.4 × 10−5 | 1 | 0.35 Tc | Not known in literature |
| 8 |
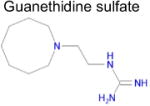
|
3.5 × 10−4 | 1 | 0.32 Tc | Not known in literature |
| 9 |
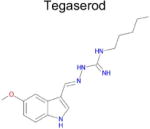
|
4.8 × 10−4 (rat) | 10 | 0.44 Tc | Not known in literature |
| 10 |
|
(Not predicted) | Not predicted and thus no value | Not predicted and thus no value | 90 ± 14f μM |
| 11 |

|
(Not predicted) | Not predicted and thus no value | Not predicted and thus no value | 100 ± 8g μM |
The descriptions of the data shown in this table is described below
E-value is derived from a statistical model which presents the probability of observing this raw score by random chance alone. The model is similar to that underlying BLAST [45]. The smaller the E-value, the stronger it is, and this indicates strong overall chemical structural similarity between two sets of compounds. Typically with small E-values, <0.001, are unlikely to be random chance [74]
Affinity bin The ligands for each protein target that is used as a reference (e.g., DAO) are subdivided (“binned”) by the logs of their affinities, and a SEA calculation is run independently against each bin. Thus, when asking whether a particular drug has a strong SEA E-value for DAO, we first compute the E-value for the compound against DAO ligands at 0.1 μM, DAO ligands at 1 μM,…, DAO ligands at 10 μM, and finally report the affinity bin at which DAO achieves its strongest E-value for the drug of interest. This may be thought of as an approximate prediction of the drug’s binding affinity at the target, but this interpretation has not been tested at scale
Tc Tanimoto coefficient. The Tc calculates the number of “on” bits in common between two fingerprints, divided by the total number of non-overlapping “on” bits between them. It is an overall measure of the similarity between any two molecular fingerprints, on a 0.0 (completely dissimilar) to 1.0 (completely similar) scale
IC50 IC50 value indicates the concentration needed to inhibit a biological function by half (by 50 %). In this study, it will be the concentration needed to inhibit formation of the product by diamine oxidase using putrescine as the substrate
Predicted using ChEMBL-14 (p-value) instead of ChEMBL-16 (E-value)
IC50 values (inhibition concentrations of DAO by 50 %) were obtained from literature [75]
Fig. 3.
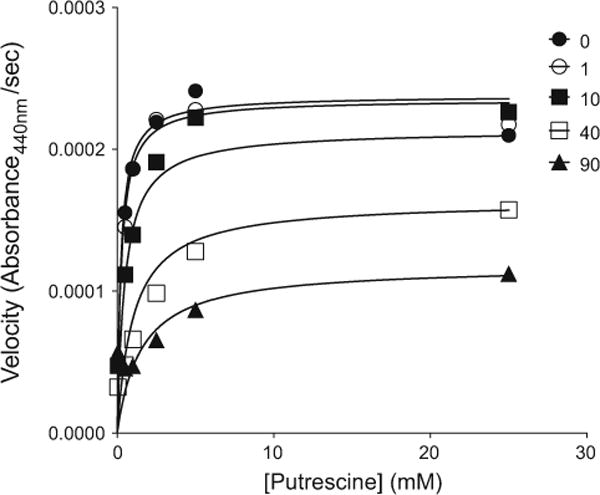
An example of inhibition of porcine diamine oxidase (DAO). Experimental data curve fits were obtained through global model nonlinear regression analysis. Metformin inhibition of porcine diamine oxidase is shown [0 mM, (closed circle), 1 mM (open circle), 10 mM (closed square); 40 mM (open square); 90 mM (triangle)]. The overall fit values obtained from three separate experiments were Ki (metformin) = 8.6 ± 3.1 mM; and KM (putrescine) = 0.33 ± 0.16 mM
Discussion
Metformin is one of the most widely prescribed drugs worldwide due to its efficacy in treating patients with type 2 diabetes. Its major adverse effects are gastrointestinal and include diarrhea, nausea, vomiting and bloating. Several studies have reported that up to 50 % of patients on metformin have one or two of these symptoms [3–6], and in one recent study, Dujic et al. reported that approximately 5 % of patients treated with metformin exhibit intolerance due to gastrointestinal symptoms that leads to discontinuation of the drug [40].
Our group and others have shown that the organic cation transporters, OCT1 and OCT3, as well as the serotonin transporter, SERT, play a role in the uptake of serotonin and histamine as well as metformin [20, 22, 61]. In the intestine, these transporters appear to be involved in the influx of their substrates from the lumen into the intestinal epithelial cells or transport between the blood and intestinal epithelial cells. Thus, these transporters are thought to be important determinants of the disposition of signaling molecules (e.g., histamine and serotonin) and as such to play a role in gastrointestinal physiology. Notably, both histamine and serotonin have important roles in gastrointestinal physiology and pathophysiology [14, 15]. For example, extrinsic sensory neurons activated by serotonin initiate sensations from the bowel, which may lead to nausea and bloating [16, 17, 62]. Similarly, histamine also plays important roles in gut motility and gastrointestinal symptoms [14]. In this study, we hypothesized that metformin inhibits intestinal transporters and enzymes known to function in the disposition of these important monoamines.
Our inhibition studies of serotonin and histamine uptake via OCT1, OCT3 and SERT showed that metformin inhibits OCT1 more potently than the other two transporters (Fig. 2). Previously, we showed that metformin can competitively inhibit the uptake of thiamine through OCT1 (IC50 ~ 1.4 mM), which may lead to alterations in thiamine disposition [20]. Since the primary mechanisms by which serotonin and histamine are cleared are through cellular uptake and subsequent intracellular degradation, inhibition of these transporters by metformin may result in higher extracellular concentrations, particularly in the intestinal lumen, thereby enhancing or prolonging serotonergic or histaminergic signaling in the intestine (Fig. 1). These effects may result in gastrointestinal side effects. Because metformin doses are high, its predicted concentrations in the intestinal lumen are about 10 to 20 mM; thus the drug would not have to interact potently with intestinal targets to exhibit pharmacologic effects on the intestinal tract in vivo [63].
Our data clearly showed that at concentrations achievable in the GI tract, metformin can inhibit OCT1-mediated transport of both serotonin and histamine (Fig. 2). Thus, though speculative, it is possible that metformin modulates levels of these monoamines in the GI tract, contributing to its adverse effects on the gut. This idea is supported by a recent publication, in which patients with reduced function non-synonymous variants (R61C, G401S, 420del, G465R) in OCT1 were found to be more likely to have metformin-induced gastrointestinal side effects [40]. This recent study of type 2 diabetic patients on metformin found that patients with concomitant use of medications known to inhibit OCT1 activity (e.g. calcium channel blockers, proton pump inhibitors, and alpha-adrenergic blockers) were more likely to be metformin intolerant [40]. Thus, it is possible that OCT1 genetic variants or concomitant use of prescription drugs that are OCT1 inhibitors may exacerbate metformin gastrointestinal side effects through effects on histamine or serotonin. Recent genomewide association studies of the human metabolome suggest an important link between OCT1 and serotonin. In particular, Shin et al. observed a strong association between the reduced-function OCT1 variant, rs683369, with serotonin levels, suggesting that OCT1 is involved in the systemic disposition of serotonin [64]. Serotonin and other 5-HT3 agonists through interactions with 5-HT3 receptors are associated with vomiting and diarrhea in rodents [65]. Thus, through inhibition of OCT1, metformin may modulate intestinal or systemic levels of serotonin, which may contribute to the GI side effects of the drug. Metformin also inhibited serotonin uptake by OCT3 and SERT but at much higher concentrations (>30 mM to inhibit 50 % of serotonin or histamine) (Fig. 2). These transporters may also play a role in serotonin (and histamine) disposition and actions in the body. Other organic cation transporters in intestinal epithelial cells, for example, PMAT (SLC29A4), which transports monoamines [23], and MATE1 (SLC47A1) were not examined in this current research.
We further tested whether metformin may inhibit other proteins involved in histamine disposition. To identify metformin off-targets, we used a chemoinformatic method, SEA. This method relates drugs to proteins based on the chemical similarity of the drug to each target’s set of ligands (http://sea.bkslab.org). It has successfully predicted off-target binding partners of prescription drugs that mediate side-effects [42]; for example, selective serotonin receptor inhibitors (SSRIs), were predicted and found to bind to β1-adrenergic receptors, which may contribute to sexual dysfunction and the SSRI discontinuation syndrome [48].
Using SEA, we predicted one target for metformin with an E-value better than 10−4, the enzyme diamine oxidase (DAO). It is known that DAO, localized inside enterocytes, is involved in metabolism of putrescine and histamine to their inactive metabolites (Fig. 1). Previous studies have demonstrated that genetic polymorphisms in DAO and serum DAO activity are associated with food intolerance and polyamine levels [54, 55, 64]. Inhibition of DAO by metformin could therefore lead to aberrant histamine levels in the intestine and cause metformin-induced GI side effects. Metformin inhibited DAO less potently than it did OCT1 (IC50 = 8.6 mM for DAO and IC50 = 1.5 mM for OCT1), but DAO inhibition may nonetheless occur in vivo based on estimated intestinal concentrations following a therapeutic dose (10–20 mM).
Information from the literature [29–34] and various databases [51, 66, 67] found that OCT1, OCT3, SERT and DAO mRNA and protein are expressed at different levels and in different sections of the intestine (Supplemental Table 1). DAO transcript levels are the highest among the four genes in the stomach, duodenum, small intestine and colon, followed by SERT, which has greater mRNA expression levels in the small intestine, as compared to OCT1 and OCT3 (Supplemental Table 1). Although transcript levels of both OCT1 and OCT3 are low in intestinal tissue, the protein expression of OCT1, OCT3 and DAO are medium to high, depending on the location in the intestine (Supplemental Table 1) [28, 52, 61]. Based on the localization of these transporters (Fig. 1) and our results from this study (Fig. 2), metformin may accumulate inside the intestinal cells through OCT3 [22, 26], inhibit DAO inside the cells, and inhibit OCT1 on the basolateral membrane of the intestine. As a result, histamine and/or serotonin levels will increase inside the enterocytes. Additionally, serotonin and histamine could be released or effluxed into the blood by OCT1 and SERT (Fig. 1). After oral absorption of metformin through the intestine, metformin could inhibit the reabsorption of serotonin or histamine from the blood into the intestinal cells by OCT1. If this occurs, histamine and serotonin levels may increase in the blood. However, it is not known whether metformin-induced gastrointestinal side effects occur as a result of increased histamine or serotonin levels surrounding the basolateral membrane of the intestine, inside the intestinal cells, surrounding the intestinal lumen or by other unrelated mechanisms. Further studies are needed to assess the in vivo effects of metformin on proteins involved in the disposition of histamine and serotonin, as well as the mechanisms of metformin-induced gastrointestinal side-effects.
Conclusions
Our results indicate that metformin inhibits transporters and enzymes in the gut that are associated with the disposition of serotonin and histamine and suggest that clinically, metformin concentrations in the gut may be sufficient to inhibit these proteins. Future studies are necessary to determine whether metformin may modulate the levels of histamine or serotonin in the plasma (pharmacokinetic analysis) or gastrointestinal tract through inhibition effects on intestinal transporters or DAO, and whether such modulation may contribute to its adverse effects on the GI tract (pharmacologic effect).
Postscript
This manuscript embodies two principles that I learned as a graduate student in Gary’s laboratory. First, research should focus on scientific mechanisms, and second, pharmacokinetics remains important, but only in the context of pharmacodynamics. The studies described in this manuscript focus on potential mechanisms by which metformin produces gastrointestinal side effects. The high concentrations that metformin achieves in the intestine, and its interaction kinetics with intestinal transporters and enzymes suggest a potential pharmacokinetic-pharmaco-dynamic relationship that may be responsible for the dose-limiting gastrointestinal side effects of metformin. Gary, your classical work on tubocurarine, beginning in 1964 [68] showing the differences in the temporal relationships of drug concentrations and drug effects, formed the basis of the field of pharmacodynamics. Your published works over many years in which you and your research team characterized the effects of disease on pharmacodynamics were both exploratory and elegant [69–71], and built upon earlier work that we had done together focused on disease effects on pharmacokinetics (e.g., propoxyphene in renal and liver disease) [72, 73]. And there is one final principle I learned from you that truly has influenced my career. That is, “the half-life of your students’ careers far exceeds that of your research citations.” Your impact as a teacher and mentor to me has been long, sustained and profound. I have striven to pass your teachings on to my own students and postdocs. Thank you, Gary, for all you have done for me and my students and postdocs, and indeed for the entire field of pharmacokinetics and pharmacodynamics.
Supplementary Material
Acknowledgments
This project was supported by NIH Grant U19-GM61390 (SWY, KMG), GM71896 (BKS), NIH training grant T32 GM007175 (LL), the Li Ka Shing Foundation (KMG, BKS), the Burroughs Wellcome Fund Innovation in Regulatory Science Awards (Grant BWF ID 1012485) (KMG, BKS), R44GM093456 (MJK), and a Glenn Foundation Award for Research in Biological Mechanisms of Aging (MJK).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10928-015-9436-y) contains supplementary material, which is available to authorized users.
References
- 1.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122(6):253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viollet B, Foretz M. Revisiting the mechanisms of metformin action in the liver. Ann Endocrinol. 2013;74(2):123–129. doi: 10.1016/j.ando.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Dandona P, Fonseca V, Mier A, Beckett AG. Diarrhea and metformin in a diabetic clinic. Diabetes Care. 1983;6(5):472–474. doi: 10.2337/diacare.6.5.472. [DOI] [PubMed] [Google Scholar]
- 4.Krentz AJ, Ferner RE, Bailey CJ. Comparative tolerability profiles of oral antidiabetic agents. Drug Saf. 1994;11(4):223–241. doi: 10.2165/00002018-199411040-00002. [DOI] [PubMed] [Google Scholar]
- 5.Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the diabetes prevention program outcomes study. Diabetes Care. 2012;35(4):731–737. doi: 10.2337/dc11-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouchoucha M, Uzzan B, Cohen R. Metformin and digestive disorders. Diabetes Metab. 2011;37(2):90–96. doi: 10.1016/j.diabet.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Carter D, Howlett HC, Wiernsperger NF, Bailey C. Effects of metformin on bile salt transport by monolayers of human intestinal Caco-2 cells. Diabetes Obes Metab. 2002;4(6):424–427. doi: 10.1046/j.1463-1326.2002.00223.x. [DOI] [PubMed] [Google Scholar]
- 8.Cubeddu LX, Bonisch H, Gothert M, Molderings G, Racke K, Ramadori G, Miller KJ, Schworer H. Effects of metformin on intestinal 5-hydroxytryptamine (5-HT) release and on 5-HT3 receptors. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;361(1):85–91. doi: 10.1007/s002109900152. [DOI] [PubMed] [Google Scholar]
- 9.Molloy AM, Ardill J, Tomkin GH. The effect of metformin treatment on gastric acid secretion and gastrointestinal hormone levels in normal subjects. Diabetologia. 1980;19(2):93–96. doi: 10.1007/BF00421851. [DOI] [PubMed] [Google Scholar]
- 10.Bertaccini G, Coruzzi G. An update on histamine H3 receptors and gastrointestinal functions. Dig Dis Sci. 1995;40(9):2052–2063. doi: 10.1007/BF02208678. [DOI] [PubMed] [Google Scholar]
- 11.Deiteren A, De Man JG, Pelckmans PA, De Winter BY. Histamine H(4) receptors in the gastrointestinal tract. Br J Pharmacol. 2015;172(5):1165–1178. doi: 10.1111/bph.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leurs R, Brozius MM, Smit MJ, Bast A, Timmerman H. Effects of histamine H1-, H2- and H3-receptor selective drugs on the mechanical activity of guinea-pig small and large intestine. Br J Pharmacol. 1991;102(1):179–185. doi: 10.1111/j.1476-5381.1991.tb12150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smolinska S, Jutel M, Crameri R, O’Mahony L. Histamine and gut mucosal immune regulation. Allergy. 2014;69(3):273–281. doi: 10.1111/all.12330. [DOI] [PubMed] [Google Scholar]
- 14.Maintz L, Novak N. Histamine and histamine intolerance. Am J Clin Nutr. 2007;85(5):1185–1196. doi: 10.1093/ajcn/85.5.1185. [DOI] [PubMed] [Google Scholar]
- 15.Mawe GM, Hoffman JM. Serotonin signalling in the gut—functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2013;10(8):473–486. doi: 10.1038/nrgastro.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gershon MD. Review article: serotonin receptors and transporters—roles in normal and abnormal gastrointestinal motility. Aliment Pharmacol Ther. 2004;20(Suppl 7):3–14. doi: 10.1111/j.1365-2036.2004.02180.x. [DOI] [PubMed] [Google Scholar]
- 17.Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132(1):397–414. doi: 10.1053/j.gastro.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Chen JJ, Li Z, Pan H, Murphy DL, Tamir H, Koepsell H, Gershon MD. Maintenance of serotonin in the intestinal mucosa and ganglia of mice that lack the high-affinity serotonin transporter: Abnormal intestinal motility and the expression of cation transporters. J Neurosci. 2001;21(16):6348–6361. doi: 10.1523/JNEUROSCI.21-16-06348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beasley CM, Jr, Koke SC, Nilsson ME, Gonzales JS. Adverse events and treatment discontinuations in clinical trials of fluoxetine in major depressive disorder: an updated meta-analysis. Clin Ther. 2000;22(11):1319–1330. doi: 10.1016/s0149-2918(00)83028-3. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Shu Y, Liang X, Chen EC, Yee SW, Zur AA, Li S, Xu L, Keshari KR, Lin MJ, Chien HC, Zhang Y, Morrissey KM, Liu J, Ostrem J, Younger NS, Kurhanewicz J, Shokat KM, Ashrafi K, Giacomini KM. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc Natl Acad Sci USA. 2014;111(27):9983–9988. doi: 10.1073/pnas.1314939111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boxberger KH, Hagenbuch B, Lampe JN. Common drugs inhibit human organic cation transporter 1 (OCT1)-mediated neurotransmitter uptake. Drug Metab Dispos. 2014;42(6):990–995. doi: 10.1124/dmd.113.055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L, Pawlikowski B, Schlessinger A, More SS, Stryke D, Johns SJ, Portman MA, Chen E, Ferrin TE, Sali A, Giacomini KM. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet Genomics. 2010;20(11):687–699. doi: 10.1097/FPC.0b013e32833fe789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan H, Wang J. Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther. 2010;335(3):743–753. doi: 10.1124/jpet.110.170142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakata T, Anzai N, Kimura T, Miura D, Fukutomi T, Takeda M, Sakurai H, Endou H. Functional analysis of human organic cation transporter OCT3 (SLC22A3) polymorphisms. J Pharmacol Sci. 2010;113(3):263–266. doi: 10.1254/jphs.09331sc. [DOI] [PubMed] [Google Scholar]
- 25.Jonker JW, Schinkel AH. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3) J Pharmacol Exp Ther. 2004;308(1):2–9. doi: 10.1124/jpet.103.053298. [DOI] [PubMed] [Google Scholar]
- 26.Chen EC, Liang X, Yee SW, Geier EG, Stocker SL, Chen L, Giacomini KM. Targeted disruption of organic cation transporter 3 attenuates the pharmacologic response to met-formin. Mol Pharmacol. 2015;88(1):75–83. doi: 10.1124/mol.114.096776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee N, Duan H, Hebert MF, Liang CJ, Rice KM, Wang J. Taste of a pill: organic cation transporter-3 (OCT3) mediates metformin accumulation and secretion in salivary glands. J Biol Chem. 2014;289(39):27055–27064. doi: 10.1074/jbc.M114.570564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han TK, Everett RS, Proctor WR, Ng CM, Costales CL, Brouwer KL, Thakker DR. Organic cation transporter 1 (OCT1/mOct1) is localized in the apical membrane of Caco-2 cell monolayers and enterocytes. Mol Pharmacol. 2013;84(2):182–189. doi: 10.1124/mol.112.084517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT) Biochem Pharmacol. 2005;70(12):1851–1860. doi: 10.1016/j.bcp.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe K, Sawano T, Endo T, Sakata M, Sato J. Studies on intestinal absorption of sulpiride (2): transepithelial transport of sulpiride across the human intestinal cell line Caco-2. Biol Pharm Bull. 2002;25(10):1345–1350. doi: 10.1248/bpb.25.1345. [DOI] [PubMed] [Google Scholar]
- 31.Jonker JW, Wagenaar E, Mol CA, Buitelaar M, Koepsell H, Smit JW, Schinkel AH. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (Oct1 [Slc22a1]) gene. Mol Cell Biol. 2001;21(16):5471–5477. doi: 10.1128/MCB.21.16.5471-5477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302(2):510–515. doi: 10.1124/jpet.102.034140. [DOI] [PubMed] [Google Scholar]
- 33.International Transporter C. Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and bio-pharmaceutical implications. Pharm Res. 2007;24(7):1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 35.Nies AT, Koepsell H, Winter S, Burk O, Klein K, Kerb R, Zanger UM, Keppler D, Schwab M, Schaeffeler E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009;50(4):1227–1240. doi: 10.1002/hep.23103. [DOI] [PubMed] [Google Scholar]
- 36.Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol. 2007;74(2):359–371. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Investig. 2007;117(5):1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17(8):2127–2135. doi: 10.1681/ASN.2006030205. [DOI] [PubMed] [Google Scholar]
- 39.Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, Inui K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005;20(5):379–386. doi: 10.2133/dmpk.20.379. [DOI] [PubMed] [Google Scholar]
- 40.Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CN, Pearson ER. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: a GoDARTS study. Diabetes. 2015;64(5):1786–1793. doi: 10.2337/db14-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cameron RT, Coleman RG, Day JP, Yalla KC, Houslay MD, Adams DR, Shoichet BK, Baillie GS. Chemical informatics uncovers a new role for moexipril as a novel inhibitor of cAMP phosphodiesterase-4 (PDE4) Biochem Pharmacol. 2013;85(9):1297–1305. doi: 10.1016/j.bcp.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Cote S, Shoichet BK, Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486(7403):361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gregori-Puigjane E, Setola V, Hert J, Crews BA, Irwin JJ, Lounkine E, Marnett L, Roth BL, Shoichet BK. Identifying mechanism-of-action targets for drugs and probes. Proc Natl Acad Sci USA. 2012;109(28):11178–11183. doi: 10.1073/pnas.1204524109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hert J, Keiser MJ, Irwin JJ, Oprea TI, Shoichet BK. Quantifying the relationships among drug classes. J Chem Inf Model. 2008;48(4):755–765. doi: 10.1021/ci8000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25(2):197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 46.Wittwer MB, Zur AA, Khuri N, Kido Y, Kosaka A, Zhang X, Morrissey KM, Sali A, Huang Y, Giacomini KM. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47A1) inhibitors through prescription drug profiling and computational modeling. J Med Chem. 2013;56(3):781–795. doi: 10.1021/jm301302s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10(3):531–539. doi: 10.1158/1535-7163.MCT-10-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellis KJ, Morrison JF. Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 1982;87:405–426. doi: 10.1016/s0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
- 50.Bardsley WG, Crabbe MJ, Shindler JS. Kinetics of the diamine oxidase reaction. Biochem J. 1973;131(3):459–469. doi: 10.1042/bj1310459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 52.Schwelberger HG, Feurle J, Houen G. New tools for studying old questions: antibodies for human diamine oxidase. J Neural Transm. 2013;120(6):1019–1026. doi: 10.1007/s00702-012-0936-2. [DOI] [PubMed] [Google Scholar]
- 53.Shamji MH, Layhadi JA, Scadding GW, Cheung DK, Calderon MA, Turka LA, Phippard D, Durham SR. Basophil expression of diamine oxidase: a novel biomarker of allergen immunotherapy response. J Allergy Clin Immunol. 2015;135(4):913–921. doi: 10.1016/j.jaci.2014.09.049. e919. [DOI] [PubMed] [Google Scholar]
- 54.Music E, Korosec P, Silar M, Adamic K, Kosnik M, Rijavec M. Serum diamine oxidase activity as a diagnostic test for histamine intolerance. Wien Klin Wochenschr. 2013;125(9–10):239–243. doi: 10.1007/s00508-013-0354-y. [DOI] [PubMed] [Google Scholar]
- 55.Maintz L, Yu CF, Rodriguez E, Baurecht H, Bieber T, Illig T, Weidinger S, Novak N. Association of single nucleotide polymorphisms in the diamine oxidase gene with diamine oxidase serum activities. Allergy. 2011;66(7):893–902. doi: 10.1111/j.1398-9995.2011.02548.x. [DOI] [PubMed] [Google Scholar]
- 56.Wantke F, Gotz M, Jarisch R. Histamine-free diet: treatment of choice for histamine-induced food intolerance and supporting treatment for chronic headaches. Clin Exp Allergy. 1993;23(12):982–985. doi: 10.1111/j.1365-2222.1993.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 57.Cubria JC, Ordonez D, Alvarez-Bujidos ML, Negro A, Ortiz AI. Inhibition of diamine oxidase from porcine kidney by pentamidine and other aminoguanidine compounds. Comp Biochem Physiol B. 1991;100(3):543–546. doi: 10.1016/0305-0491(91)90217-2. [DOI] [PubMed] [Google Scholar]
- 58.Schwelberger HG, Bodner E. Purification and characterization of diamine oxidase from porcine kidney and intestine. Biochim Biophys Acta. 1997;1340(1):152–164. doi: 10.1016/s0167-4838(97)00039-3. [DOI] [PubMed] [Google Scholar]
- 59.Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24(1):49–57. doi: 10.3109/00498259409043220. [DOI] [PubMed] [Google Scholar]
- 60.Bailey CJ, Wilcock C, Day C. Effect of metformin on glucose metabolism in the splanchnic bed. Br J Pharmacol. 1992;105(4):1009–1013. doi: 10.1111/j.1476-5381.1992.tb09093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han TK, Proctor WR, Costales CL, Cai H, Everett RS, Thakker DR. Four cation-selective transporters contribute to apical uptake and accumulation of metformin in Caco-2 cell monolayers. J Pharmacol Exp Ther. 2015;352(3):519–528. doi: 10.1124/jpet.114.220350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gershon MD. Review article: roles played by 5-hydroxytryptamine in the physiology of the bowel. Aliment Pharmacol Ther. 1999;13(Suppl 2):15–30. [PubMed] [Google Scholar]
- 63.Proctor WR, Bourdet DL, Thakker DR. Mechanisms underlying saturable intestinal absorption of metformin. Drug Metab Dispos. 2008;36(8):1650–1658. doi: 10.1124/dmd.107.020180. [DOI] [PubMed] [Google Scholar]
- 64.Shin SY, Fauman EB, Petersen AK, Krumsiek J, Santos R, Huang J, Arnold M, Erte I, Forgetta V, Yang TP, Walter K, Menni C, Chen L, Vasquez L, Valdes AM, Hyde CL, Wang V, Ziemek D, Roberts P, Xi L, Grundberg E, Waldenberger M, Richards JB, Mohney RP, Milburn MV, John SL, Trimmer J, Theis FJ, Overington JP, Suhre K, Brosnan MJ, Gieger C, Kastenmuller G, Spector TD, Soranzo N. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46(6):543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller AD, Nonaka S. Mechanisms of vomiting induced by serotonin-3 receptor agonists in the cat: effect of vagotomy, splanchnicectomy or area postrema lesion. J Pharmacol Exp Ther. 1992;260(2):509–517. [PubMed] [Google Scholar]
- 66.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segre AV, Djebali S, Niarchou A, Consortium GT, Wright FA, Lappalainen T, Calvo M, Getz G, Dermitzakis ET, Ardlie KG, Guigo R. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348(6235):660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F. Towards a knowledge-based human protein atlas. Nat Biotechnol. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 68.Levy G. Relationship between rate of elimination of tubocurarine and rate of decline of its pharmacological activity. Br J Anaesth. 1964;36:694–695. doi: 10.1093/bja/36.11.694. [DOI] [PubMed] [Google Scholar]
- 69.Danhof M, Hisaoka M, Levy G. Kinetics of drug action in disease states. VI: effect of experimental diabetes on phenobarbital concentrations in rats at onset of loss of righting reflex. J Pharmacol Exp Ther. 1985;232(2):435–438. [PubMed] [Google Scholar]
- 70.Hisaoka M, Danhof M, Levy G. Kinetics of drug action in disease states. VII: effect of experimental renal dysfunction on the pharmacodynamics of ethanol in rats. J Pharmacol Exp Ther. 1985;232(3):717–721. [PubMed] [Google Scholar]
- 71.Stapley EO, Birnbaum J, Miller AK, Wallick H, Hendlin D, Woodruff HB. Cefoxitin and cephamycins: microbiological studies. Rev Infect Dis. 1979;1(1):73–89. doi: 10.1093/clinids/1.1.73. [DOI] [PubMed] [Google Scholar]
- 72.Giacomini KM, Giacomini JC, Gibson TP, Levy G. Propoxyphene and norpropoxyphene plasma concentrations after oral propoxyphene in cirrhotic patients with and without surgically constructed portacaval shunt. Clin Pharmacol Ther. 1980;28(3):417–424. doi: 10.1038/clpt.1980.182. [DOI] [PubMed] [Google Scholar]
- 73.Gibson TP, Giacomini KM, Briggs WA, Whitman W, Levy G. Propoxyphene and norpropoxyphene plasma concentrations in the anephric patient. Clin Pharmacol Ther. 1980;27(5):665–670. doi: 10.1038/clpt.1980.94. [DOI] [PubMed] [Google Scholar]
- 74.Pearson WR. An introduction to sequence similarity (homology) searching. Current protocols in bioinformatics/editoral board, Andreas D Baxevanis [et al] 2013 doi: 10.1002/0471250953.bi0301s42. Chapter 3:Unit3 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGrath AP, Hilmer KM, Collyer CA, Shepard EM, Elmore BO, Brown DE, Dooley DM, Guss JM. Structure and inhibition of human diamine oxidase. Biochemistry. 2009;48(41):9810–9822. doi: 10.1021/bi9014192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.High A, Prior T, Bell RA, Rangachari PK. Probing the “active site” of diamine oxidase: structure-activity relations for histamine potentiation by O-alkylhydroxylamines on colonic epithelium. J Pharmacol Exp Ther. 1999;288(2):490–501. [PubMed] [Google Scholar]
- 77.Holt A, Baker GB. Metabolism of agmatine (clonidine-displacing substance) by diamine oxidase and the possible implications for studies of imidazoline receptors. Prog Brain Res. 1995;106:187–197. doi: 10.1016/s0079-6123(08)61215-7. [DOI] [PubMed] [Google Scholar]
- 78.Banchelli G, Bertocci B, Raimondi L, Soldani G, Del Tacca M, Buffoni F. Guanabenz as inhibitor of copper-containing amine oxidases. Agents Actions. 1986;18(1–2):46–48. doi: 10.1007/BF01987979. [DOI] [PubMed] [Google Scholar]
- 79.Bieganski T, Kusche J, Lorenz W, Hesterberg R, Stahlknecht CD, Feussner KD. Distribution and properties of human intestinal diamine oxidase and its relevance for the histamine catabolism. Biochim Biophys Acta. 1983;756(2):196–203. doi: 10.1016/0304-4165(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 80.Finazzi-Agro A, Floris G, Fadda MB, Crifo C. Inhibition of diamine oxidase by antihistaminic agents and related drugs. Agents Actions. 1979;9(3):244–247. doi: 10.1007/BF01966695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.