

Abstract
Background
Cardiac myosin heavy chain-α (Myhc), an intracellular protein expressed in the cardiomyocytes, has been identified as a major autoantigen in cardiac autoimmunity. In our studies with Myhc334-352-induced experimental autoimmune myocarditis in A/J mice (H-2a), we discovered that Myhc334-352, supposedly a CD4 T cell epitope, also induced antigen-specific CD8 T cells that transfer disease to naïve animals.
Methods and Results
In our efforts to identify the CD8 T cell determinants, we localized Myhc338-348 within the full length-Myhc334-352, leading to four key findings. (1) By acting as a dual epitope, Myhc338-348 induces both CD4 and CD8 T cell responses. (2) In a major histocompatibility complex (MHC) class I-stabilization assay, Myhc338-348 was found to bind H-2Dd – but not H-2Kk or H-2Ld – alleles. (3) The CD8 T cell response induced by Myhc338-348 was antigen-specific, as evaluated by MHC class I/H-2Dd dextramer staining. The antigen-sensitized T cells predominantly produced interferon-γ, the critical cytokine of effector cytotoxic T lymphocytes. (4) Myhc338-348 was found to induce myocarditis in immunized animals as determined by histology and magnetic resonance microscopy imaging.
Conclusions
Our data provide new insights as to how different immune cells can recognize the same antigen and inflict damage through different mechanisms.
Keywords: cardiac myosin, CD8 T cells, autoimmunity, myocarditis, MHC class I dextramers
1. Introduction
Autoimmunity ensues when immune responses are directed against self-antigens, resulting from the mediation of either autoantibodies or autoreactive T cells. However, when autoreactive T cells are involved, CD4 T helper (Th) cells are commonly implicated because they play a central role in the generation of adaptive immune responses. For example, cytokines produced by CD4 T cells are critical for B cells to produce antibodies to protein antigens, and Th cytokines like interferon (IFN)-γ mediate macrophage activation. On the other hand, interleukin (IL)-2 helps in the differentiation and expansion of CD8 T cells. Because CD4 T cells are needed for B cells to make antibodies, both cell types are expected to recognize the same antigens, but their epitopes could be different. Thus, an argument can be made that therapeutically targeting the pathogenic autoreactive B cells that produce autoantibodies, as well as the CD4 T cells that help the B cells do so, can suppress autoimmune responses in diseases driven by antibodies, such as systemic lupus erythematosus, Hashimoto's thyroiditis, Sjogren's syndrome, and rheumatoid arthritis [1, 2].
CD4 T cells and CD8 T cells are distinct in their recognition of peptides, in that they recognize antigens presented by two different molecules of the major histocompatibility complex (MHC) locus. While MHC class II molecules expressed by professional antigen-presenting cells (APCs) present peptides to CD4 T cells, MHC class I molecules expressed by almost all nucleated cells can present peptides to CD8 T cells. The ends of peptide-binding grooves are open in the class II MHC molecules, enabling them to present long peptides (up to 20 amino acids), as opposed to short peptides of ~8-11 amino acids presented by class I molecules, in which the ends of the MHC-clefts are closed [3, 4]. Therefore, the immunodominant epitopes of protein antigens can be processed and presented in the context of class I or class II molecules through endogenous and exogenous pathways, respectively, leading to the induction of corresponding CD8 and CD4 T cell responses. If such an event happens for self-proteins, then the tissue destruction in the target organs is mediated by CD4 T cells via delayed-type hypersensitivity reaction, whereas CD8 T cells inflict damage through cytolysis [2, 5].
To study the immune events of cardiac autoimmunity, rodent models of experimental autoimmune myocarditis (EAM) are commonly employed. These include cardiac myosin heavy chain-α (Myhc)334-352-induced EAM in A/J (H-2a), CBA/J (H-2k) and B10.A (H-2k) mice; Myhc614-643-induced EAM in Balb/c (H-2d) mice; and Myhc1304-1320-induced EAM in Lewis rats (RT1) [6-11]. The disease is induced by immunizing the animals with the immunodominant epitopes of cardiac antigens in complete Freund’s adjuvant (CFA). In all the EAM models described above, the disease has been shown to be mediated by CD4 T cells, [8, 10, 11] with a complex disease pathogenesis [12-14]. In our studies with EAM induced by Myhc334-352 in A/J mice, however, we noted an unexpected finding that the full-length Myhc334-352 (FL-Myhc334-352) epitope was also found to contain Myhc338-348, which is a determinant for CD8 T cells. By acting as a dual epitope, Myhc338-348 induces both CD4 and CD8 T cell responses that are antigen-specific, and induces myocarditis in immunized animals through the predominant production of IFN-γ, the critical cytokine of effector cytotoxic lymphocytes.
2. Materials and Methods
2.1 Mice
Our studies involved the use of 6- to 8-week-old female A/J mice (H-2a), obtained from the Jackson Laboratory (Bar Harbor, ME) and maintained in accordance with the guidelines of the University of Nebraska-Lincoln. All animals used in the study received humane care, and the Institutional Animal Care and Use Committee approved experimental permit and protocol numbers are A3459-01 and 973, respectively.
2.2 Peptide synthesis and immunization procedures
The indicated peptides, including the controls (bovine ribonuclease [RNase]43-56, VNTFVHESLADVQA; human immunodeficiency virus glycoprotein 120 [HIV]P18-I10, RGPGRAFVTI), were synthesized on 9-fluorenylmethyloxycarbonyl chemistry (Neopeptide, Cambridge, MA). All peptides were HPLC-purified (>90%), identity-confirmed by mass spectroscopy, and dissolved in 1x PBS prior to use. For immunizations, peptides were emulsified in CFA supplemented with Mycobacterium tuberculosis H37RA extract to a final concentration of 5 mg/ml, and administered subcutaneously in multiple sites in the flank and sternal regions (100 μg per mouse). For EAM induction, animals were immunized twice as above at 7 day intervals, and pertussis toxin (100 ng/mouse) was administered on day 0 and day 2 after the first immunization [11, 15].
2.3 Measurement of recall responses and derivation of primary T cell cultures
Fourteen days after immunization, animals were euthanized and single cell suspensions were prepared using the draining lymph nodes (maxillary, mandibular, axillary, inguinal and popliteal) and spleens [11, 15]. After lysing the erythrocytes with 1x ammonium chloride potassium buffer (Lonza, Walkersville, MD) and washing, cells were resuspended in 1x IMAG buffer (BD Biosciences, San Diego, CA). CD4 or CD8 T cells were then enriched to a purity of >95 % by negative selection based on magnetic separation using IMAG (BD Biosciences) [15]. Cells were suspended in growth medium containing RPMI medium supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 4 mM L-glutamine, 1x each of non-essential amino acids and vitamin mixture, and 100 U/ml penicillin-streptomycin (Lonza). To measure the recall responses, cells were stimulated for 2 days at a density of 10 × 106 cells/ml in the presence of syngeneic irradiated APCs at a ratio of 1:1, as well as the indicated peptides (0-100 μg/ml). After pulsing with 1 μCi of tritiated 3[H]-thymidine (MP Biomedicals, Santa Ana, CA) for 16 hours, proliferative responses were measured as counts per minute (cpm) using a Wallac liquid scintillation counter (Perkin Elmer, Waltham, MA) [11]. To derive primary T cell cultures, whole cell populations were stimulated with the peptides (50 μg/ml), and supplemented after two days with IL-2 medium (eBioscience, San Diego, CA; Advanced Biotechnologies, Columbia, MD) [11, 15].
2.4 Disease induction by adoptive transfer
A/J mice were immunized with FL-Myhc334-352 in CFA, and at termination on day 14, spleens and lymph nodes were harvested to prepare single cell suspensions. CD8 T cells were then enriched by negative selection based on magnetic separation as described in section 2.3, then stimulated with concanavalin-A (Con-A, 2.5 μg/ml; Sigma Aldrich, St. Louis, MO) for two days. Viable cells were harvested and administered i.p., (14 × 106 cells per animal) into whole body-irradiated (400 rads/mouse) naïve animals [16-19]. Irradiated naïve mice that received 1 × PBS or Con-A activated CD8 T cells obtained from naïve mice, served as controls. Additionally, each mouse received pertussis toxin (100 ng/mouse) i.p., on day 0 and day 2 posttransfer, and the animals were euthanized on day 18 to harvest hearts for histology.
2.5 Staining for markers of effector CD8 T cells
Groups of A/J mice were immunized with Myhc334-352, and after 10 days, lymph nodes were obtained and lymph node cells (LNCs) were prepared. Cells were stimulated with the peptide (50 μg/ml) for 2 days, and the cultures were maintained in IL-2 medium. On days 4, 6, 8 and 10 post-stimulation, cells were stained with CD8 and CD107a/b or Rat-IgG2a (isotype control) antibodies (eBioscience), and 7-amino-actinomycin-D (7-AAD; Invitrogen, Carlsbad, CA). After washing, cells were acquired by flow cytometry (FACS Calibur, BD Biosciences, San Diego, CA), and the frequencies of CD107a/b+ cells were enumerated in the live (7-AAD−) CD8 T cell subset using Flow Jo software (Tree Star, Ashland, OR) [20, 21]. Similarly, on the indicated days, after staining with CD8 antibodies and 7-AAD, cells were fixed and permeabilized, then stained with granzyme B or perforin antibodies and Rat-IgG2a (isotype control) [22-24]. After washing, cells were acquired by flow cytometry and the frequencies of granzyme B+ and perforin+ cells were enumerated in the live (7-AAD−) population as above.
2.6 MHC class I stabilization assay
To identify the epitopes that bind MHC molecules, we used a MHC class I stabilization assay [25], which involved the use of a transporter associated with antigen-processing 2-deficient RMA-S cell lines that were stably transfected with these MHC class I genes: H-2Dd (kind gift from David H. Raulet, University of California, Berkeley), H-2Kk (kind gift from Peter Cresswell, Yale School of Medicine, New Haven), and H-2Ld (kind gift from Ted Hansen, Washington University, St. Louis). RMA-S-Dd cells were cultured in Kappler-Marrack complete tumor medium [26]. RMA-S-Kk and RMA-S-Ld cells were cultured in RPMI medium supplemented with 10% FBS, 10 mM HEPES, 2 mM L-glutamine and 100 U/ml penicillin-streptomycin. Briefly, cells were plated in 96-well plates at a density of 2 × 106/ml and were incubated with or without Trunk A-4 and Trunk A-5 peptides (0 to 400 μM) at 26°C/5% CO2 for 18 hours. The plates were then transferred to a 37°C incubator for 1.5 hours, and after washing, cells were stained with fluorophore-labelled antibodies for H-2Dd (BD Pharmingen, San Diego, CA), H-2Kk (BD Pharmingen), and H-2Ld (Cedarlane, Burlington, NC). After washing, cells were acquired by flow cytometry, and the mean fluorescence intensities (MFI) were calculated using CellQuest software (BD Biosciences). Finally, MFI ratios were derived using the formula: MFI ratio = (MFI observed in the presence of peptide at 37°C/MFI observed in the absence of peptide at 26°C) – (MFI observed in the absence of peptide at 37°C/MFI observed in the absence of peptide at 26°C) × 100 [25].
2.7 Derivation of MHC dextramers and determination of antigen-specificity of Myhc-reactive T cells by dextramer staining
The antigen specificity of Myhc-reactive CD4 T cells was determined by MHC class II/IAk dextramer staining. MHC class II/IAk dextramers (Myhc334-352 and RNase43-56), were prepared as we have described previously [27]. CD4 T cells obtained from mice immunized with Myhc334-352 were stimulated with the peptide (50 μg/ml) for 2 days, and the cells were maintained in IL-2 medium. On day 8, cells were stained with dextramers for two hours at room temperature followed by staining with anti-CD4 and 7-AAD [27]. After washing, cells were acquired by flow cytometry, and the percentages of dextramer+ cells were determined in the live (7-AAD−) cell populations using Flow Jo software.
The antigen specificity of Myhc-reactive CD8 T cells was determined by MHC class I/H-2Dd dextramer staining. The process followed three stages.
2.7.1 Expression of MHC class I heavy chains and β2-microglobulin (β2m)
Derivation of MHC class I dextramers required the expression of the class I heavy chain for H-2Dd and the light chain β2m. The expression vector for the mouse MHC class I heavy chain of H-2Dd was constructed as previously described [28]. Briefly, the full-length extracellular domains of the H-2Dd allele were amplified from the cDNA synthesized from the splenocytes of naïve A/J mice using the sequence-specific forward and reverse primers 5'GGGAATTCATATGGGCTCACACTCGCTGAGG3' and 5'AGAGGATCCGGATGAAG GAGGCTCCTCCTTGCC3'. The forward and reverse primers were designed to include the in-frame Nde1 site/start codon and in-frame BamH1 sites, respectively. The amplified fragments were digested with the respective enzymes and cloned into a pET22b+ vector (Novagen, Madison, WI). Finally, the sequence for the biotinylation site (5’GATCCCTGCATCATATTC TGGATGCACAGAAAATGGTGTGGAATCATCGTTAAA3’) was cloned immediately after the codon for the 280th amino acid of the MHC class I heavy chain [28].
The construct for human β2m (pHN1-β2m) was received as a kind gift from Dr. David Garboczi [29]. MHC class I monomers and β2m were expressed in BL21(DE3)pLysS competent cells (Promega, Madison, WI) induced by isopropyl β-D-1-thiogalactopyranoside (ThermoScientific, Waltham, MA) at a final concentration of 1 mM and 0.4 mM, respectively [28]. The proteins expressed as insoluble inclusion bodies were purified, solubilized in urea and stored snap-frozen in aliquots at −80°C until use [28].
2.7.2 Preparation of H-2Dd dextramers
The heavy chain of H-2Dd was refolded with β2m and Trunk A-4, Trunk A-5 or Trunk A-6 peptides in individual reactions as described previously [28]. To prepare control dextramers, we used HIVP18-I10, which binds the H-2Dd allele, as reported by others [30]. Briefly, peptides (1.5 mg), β2m (2000 nM), and MHC heavy chain (1000 nM) dissolved in injection buffer (3 mM guanidine HCl, 10 mM sodium acetate, 10 mM EDTA, pH 4.2) were injected sequentially into a beaker containing folding buffer (100 mM Tris base, 0.4 M L-arginine, 2 mM EDTA, pH 8.0, supplemented with 5 mM reduced glutathione, 0.5 mM oxidized glutathione and 100 mM phenylmethylsulfonyl fluoride) at 4°C with continuous stirring. At 12 hours and 18 hours post incubation, the reaction mixtures were supplemented with additional amounts of heavy chain protein (1000 nM), and the incubation was continued for a total of 36 hours. The precipitate containing unfolded protein complexes was removed by centrifuging the samples at 13,000 rpm for 20 minutes, and the MHC heavy chain/β2m/peptide complexes in the supernatant were concentrated using ultracentrifugal filters (Millipore, Billerica, MA). These complexes were then desalted using gel-filtration columns (GE Healthcare Bio-Sciences, Pittsburgh, PA) and biotinylated using the biotin ligase, BirA ([25 μg or 10 nM; Avidity, Denver, CO]/ 2.5 mg/ml of the MHC/peptide complexes) at 30°C overnight. After desalting, biotinylated MHC/peptide monomers were assembled with dextran-streptavidin-phycoerythrin molecules (kindly provided by Immudex Aps, Copenhagen, Denmark) at a molar ratio of 20:1 by incubating the reaction mixtures at room temperature (RT) for 30 minutes in the dark, followed by reconstitution in the dextramer buffer (50 mM Tris Hcl, pH 7.2). The reagents were stored at 4°C until use.
2.7.3 Dextramer staining
To ascertain the antigen specificity of the CD8 T cells, we used H-2Dd dextramers, which included specific (H-2Dd/Trunk A-4, H-2Dd/Trunk A-5 and H-2Dd/Trunk A-6) and control (H-2Dd/ HIVP18-I10) dextramers [27]. Briefly, LNCs obtained from the animals immunized with FL-Myhc334-352 were stimulated with the immunizing peptide (50 μg/ml), and on day 5 post stimulation, viable cells were harvested and maintained in IL-2 medium. Cells harvested on day 8 were washed and chilled on ice for 20 minutes, followed by staining with the MHC class I dextramers using FBS (2.5%)-supplemented IL-2 medium, pH 7.63, at RT for 1 hour. Cells were washed and stained with anti-CD8, anti-CD4 and 7-AAD. After washing, cells were acquired by flow cytometry, and the percentages of dextramer+ CD8+ cells were determined in the live (7-AAD−) population using Flow Jo software.
2.8 Intracellular cytokine staining
Single cell suspensions were prepared from mice immunized with FL-Myhc334-352, Trunk A-4 peptide or Trunk A-5 peptide on day 14 post immunization. Cells (5x106/ml) were stimulated with the immunizing peptides (50 μg/ml) for two days, and IL-2 medium was then added. On day 6, viable cells were stimulated briefly with phorbol-12-myristate 13-acetate (PMA; 20 ng/ml) and ionomycin (300 ng/ml; Sigma Aldrich) in the presence of monensin (2 mM; Golgi stop; BD Pharmingen) for 5 hours. Cells were washed and stained with antibodies for CD4 and CD8 (eBioscience) and 7-AAD. After washing and fixing, cells were permeabilized and stained with cytokine antibodies or their respective isotype controls (rat IgG1, rat IgG2a and rat IgG2b). Cells were acquired by flow cytometry, and frequencies of cytokine-producing CD4 or CD8 T cells were analyzed in the live (7-AAD−) population using Flow Jo software by utilizing the gates drawn for isotype controls corresponding to each cytokine [11]. The following clones of cytokine antibodies were used: IL-2 (JES6-5H4); IFN-γ (XMG1.2); IL-4 (11B11); IL-10 (JES5-16E3); IL-17A (eBio 17B7); IL-17 F (eBio 18F10); and tumor necrosis factor (TNF)-α (MP6-XT22) (all from eBioscience) and IL-22 (140301; R&D Systems, Minneapolis, MN).
2.9. Histopathology
Hearts were collected at termination on day 21 post immunization with the peptides, and the tissues were fixed by immersion in 10% phosphate buffered formalin [11, 31]. Three longitudinal sections 5 μm thick were made from each heart and stained with hematoxylin and eosin (H and E). The sections were examined by a board-certified pathologist blinded to treatment. Inflammation and severity scores were obtained based on the total number of inflammatory foci added across the three sections, deriving the percent inflammation as reported previously [9].
2.10 Magnetic resonance microscopy (MRM imaging)
To determine the structural and functional abnormalities of hearts in mice affected with myocarditis, we used MRM imaging as a non-invasive tool [32]. Briefly, mice were anaesthetized using 2% isoflurane; a rectal thermometer was placed in the rectum to monitor body temperature, and a pulse oximeter was connected to monitor the heart rate. An animal monitoring system involving respirometry and pulse oximetry was used to minimize image blurring due to respiratory and cardiac movements. Animals were subjected to MRM imaging using a wide-bore (89 mm) 9.4 T vertical-bore magnet (Varian, Inc. Walnut Creek, CA) equipped with triple axis gradients of 100 G/cm and a 4 cm radio-frequency imaging coil as described previously [32]. Using an echo-based cine pulse sequence, short-axis slices of hearts were captured in eight time frames and stored. The images were analyzed using Segment software (Segment v1.8 R1430, Medviso, Sweden) to determine the changes, if any, in the structural (left ventricular [LV] wall thickness) and functional parameters (end diastolic volume and ejection fraction).
2.11 Statistics
Student's t-test was used to determine differences in T cell proliferative responses, MHC class I stabilization assay, cytokine-producing cells, and degree of inflammation between groups. p ≤ 0.05 values were considered significant.
3. Results
3.1 A/J mice immunized with FL-Myhc334-352 show generation of antigen-specific CD4 and CD8 T cell responses
EAM induced with FL-Myhc334-352 has been used to study the cellular mechanisms of cardiac autoimmunity that involve the mediation of Myhc-reactive CD4 T cells [6, 11]. However, by characterizing the infiltrates of the hearts, we noted that CD8 T cells contributed a proportion of T cells (CD3+ CD8+: 3.50 ± 1.40%; CD3+ CD4+:11.84 ± 5.09%), leading us to conduct tests to determine their antigen specificity. Groups of A/J mice were immunized with Myhc334-352, and after 14 days, CD4 and CD8 T cells were enriched by negative selection with a purity of more than 95% (Fig. 1, top panel). Cells were stimulated with irradiated APCs pulsed with Myhc334-352 to measure the proliferative responses based on 3[H]-thymidine-incorporation assay. Predictably, CD4 T cells specifically and significantly responded to Myhc334-352 (p<0.0053), since the responses to a control peptide, RNase43-56, were absent (Fig. 1, bottom left panel). Their antigen specificity was later confirmed by staining the cells with MHC class II/IAk dextramers. In brief, LNCs from mice immunized with Myhc334-352 were stimulated with the peptide and the cells were rested in IL-2 medium. Viable cells harvested on day 8 were stained with IAk/Myhc334-352 and RNase43-56 (control) dextramers, leading to the detection of Myhc-specific CD4 T cells (2.59±0.32%) since the staining for control dextramers was negligible (0.24±0.073%) (Supplemental fig. 1; p=0.00016). However, unexpectedly, CD8 T cells obtained from immunized animals also responded to Myhc334-352, dose- and antigen-dependently, and the responses were significant when compared with control (p<0.00058; Fig. 1, bottom right panel). Importantly, the magnitude of CD8 T cell responses was comparable to that of CD4 T cells, as the responses between the two did not differ significantly.
Figure 1. FL-Myhc334-352 induces both CD4 and CD8 T cell responses.

A/J mice were immunized with FL-Myhc334-352 in CFA; after 14 days animals were euthanized to prepare single cell suspensions from spleens and lymph nodes, from which CD4 and CD8 T cells were enriched by magnetic separation. The purity of CD4 and CD8 T cell preparations were ascertained by staining with antibodies for CD3, CD4 and CD8, and 7-AAD, where the percentages of respective cell populations were determined within the live (7-AAD−) cells (top panel). Cells were stimulated with the irradiated APCs pulsed with the indicated peptides for two days. After pulsing with 3[H]-thymidine (1μCi/well) for 16 hours, proliferative responses were measured as cpm. Mean ± SEM values derived from five experiments, each involving 5 to 8 mice are shown (bottom panels). RNase43-56 (control).
We then asked if the CD8 T cells were to respond to Myhc, whether the antigen-sensitized cells would produce IFN-γ, the signature cytokine of CD8 T cells. To test this possibility, LNCs from immunized mice were stimulated with Myhc334-352, and their cytokine-producing abilities were tested in relation to CD4 and CD8 T cell subsets. Supplemental figure 2 shows that the cells capable of producing Th1 (IFN-γ) and Th17 (IL-17A, IL-17F and IL-22) cytokines were predominant, followed by IL-10-producing Th2 cells. In contrast, under the same conditions, the IFN-γ-producing ability of CD8 T cells was enhanced four-fold (p≤0.0032), but Th17 cytokines were limited to IL-17F and IL-22, and IL-10-producing cells were negligible. The enhanced IFN-γ-producing ability of CD8 T cells further supported our proposal that they might be antigen-specific and play a role in disease mediation.
To determine the pathogenic role of Myhc-reactive CD8 T cells, lymphocytes were obtained from animals immunized with Myhc334-352, and the CD8 T cells were enriched. After stimulating with Con-A for two days, cells were administered into naïve animals. Upon evaluating hearts at termination on day 18, we detected inflammatory foci accompanied by hemorrhagic necrosis and mineralization in the recipients of antigen-primed CD8 T cells (Fig. 2ai and 2aii), but not in the saline group (Fig. 2aiii) or the recipients of naïve CD8 T cells (Fig. 2aiv). This finding suggested that Myhc-reactive CD8 T cells are pathogenic, and the effector mechanisms might involve their cytolytic functions. Consistent with this notion, we verified that Myhc334-352-reactive CD8 T cells express granzyme B (Fig. 2b left panel; p<0.027) and perforin (Fig. 2b middle panel; p<0.026) intracellularly, and express CD107a/CD107b (Fig. 2b right panel; p<0.020), the surface markers of degranulation process of effector cells [20-22, 24, 33, 34].
Figure 2. The effector FL-Myhc334-352-sensitized CD8 T cells transfer disease to naïve recipients.

(a) Histological evaluation of hearts. A/J mice were immunized with FL-Myhc334-352; after 14 days, CD8 T cells were enriched from spleens and lymph nodes. Cells were stimulated with Con-A for two days, and viable lymphoblasts were administered i.p. into naïve irradiated mice; each mouse also received pertussis toxin i.p. Whereas, naïve irradiated mice that received1 × PBS or Con-A stimulated CD8 T cells from unimmunized A/j mice served as controls. The animals were euthanized on day 18 post transfer, and hearts were examined by H and E staining. Panel-a (i and ii; Antigen-primed CD8 T cell-recipients) shows lymphocyte infiltrations, hemorrhagic necrosis and mineralization; multifocal necrotic areas and degenerative leukocytes, respectively; panel-a (iii and iv) shows normal sections from recipients of saline or naïve CD8 T cells respectively. Representative sections are shown from experiments involving four to six mice per group. (b) Evaluation of the markers of effector CD8 T cells. LNC were isolated from mice immunized with Myhc334-352, and after stimulating with peptide for two days, cultures were maintained in IL-2 medium. CD8 T cells were tested for granzyme B and perforin intracellularly, and also for surface expression of CD107a/CD107b by flow cytometry using anti-mouse antibodies, and their frequencies were enumerated in the CD8 subset. Mean ± SEM values derived from seven experiments each involving three to six mice are shown.
3.2 Localization of CD8 T cell epitopes within the FL-Myhc334-352
CD8 T cells generally recognize epitopes with a length of 8 to 11 amino acids, although they can recognize longer peptides [3, 35-37]. However, in our case, the finding that the 19-mer FL-Myhc334-352 induced CD8 T cell responses suggested that the CD8 T cell epitopes must be localized within this peptide sequence, and identification of these determinants required three levels of screening.
In the primary screening, we sought to use the truncated peptides Trunk A (Myhc334-349), Trunk B (Myhc338-352), Trunk C (Myhc334-347) and Trunk D (Myhc340-352), generated from FL-Myhc334-352 in a proliferation assay, in which the truncations resulted from the deletions of 3 to 6 amino acids from the N- or C-terminal ends in each (Table 1). Briefly, CD4 and CD8 T cells were enriched from animals immunized with Myhc334-352, as above, and the cells were tested for their responses to Myhc334-352 (positive control), truncated peptides and RNase43-56 (negative control), leading us to make two observations.
Table 1.
The list of peptides used to evaluate CD4 and CD8 T cell responses in the primary screening
| Peptide | Sequence | ||
|---|---|---|---|
| minor | major | ||
| TCR contacts | 344 | 348 | |
| ↑ | ↑ | ||
| FL-Myhc334-352 | DSAFDVLSFTAEEKAGVYK† | ||
| ↓ | |||
| 338 | MHC-anchor | ||
| Trunk A (Myhc334-349) | DSAFDVLSFTAEEKAG | ||
| Trunk B (Myhc338-352) | DVLSFTAEEKAGVYK | ||
| Trunk C (Myhc334-347) | DSAFDVLSFTAEEK | ||
| Trunk D (Myhc340-352) | LSFTAEEKAGVYK | ||
putative MHC class II (IAk)-binding residue is shown with dotted arrow and two TCR-contact residues are shown with bold arrows
(a) D4 T cells responded vigorously to Myhc334-352 (p=0.03) as well as Trunk A (p=0.02) and Trunk B (p=0.02) peptides, but their responses to Trunk C and Trunk D peptides were progressively reduced (Fig. 3, top panels). The responses to Myhc peptides were specific, since the antigen-sensitized T cells did not respond to the control. These patterns were expected because the putative MHC class II (IAk)-binding residue Asp at position 338 (Asp338), as well as the two CD4 T cell receptor (TCR)-contact residues (Ala348, major; and Ala344, minor), for CD4 T cells are preserved in both Trunk A and Trunk B (Table 1, [6]). As such, the relative reduction in response to Trunk C was expected because the major TCR-contact residue Ala348 is lacking (Table 1). Although the Trunk D peptide contains both TCR-contact residues, the response was low, perhaps because the peptide lacked the putative MHC-binding residue, Asp338.
Figure 3. Evaluation of CD4 and CD8 T cell responses to the truncated peptides of FL-Myhc334-352 (13 to 16-mers) in the primary screening.

A/J mice were immunized with Myhc334-352 in CFA; after 14 days, single cell suspensions were prepared from spleens and lymph nodes, from which CD4 and CD8 T cells were enriched by magnetic separation. Cells were stimulated with Myhc334-352 and its truncated peptides (13 to 16-mers: Trunk A to D) in the presence of irradiated APCs for two days, followed by pulsing with 3[H]-thymidine. After 16 hours, proliferative responses were measured as cpm. RNase43-56 (control). Mean ± SEM values obtained from two individual experiments, each involving 10 to 11 mice are shown.
(b) Like CD4 T cells, CD8 T cells also responded to various truncated peptides of Myhc, but not the control, with similar trends in a decreasing order (Trunk A > Trunk B > Trunk C > Trunk D; Fig. 3, bottom panels). The magnitude of responses to Trunk A and Trunk B peptides, was significantly higher than responses to the control (RNase 43-56, p<0.05). Since, the Trunk A peptide (with deletions of three amino acids [VYK] at the C-terminus) showed a comparable response to that obtained with the Trunk B peptide (with deletions of four amino acids [DSAF] at the N-terminus) (Table 1), we focused on the central core region (12-mer: DVLSFTAEEKAG), which was common to both in order to identify shorter peptides that could potentially induce CD8 T cell responses. In support of this proposal, we noted that responses to the Trunk C peptide, with deletion of two additional residues (AG) at the C-terminus, and to the Trunk D peptide, with deletion of six residues (DSAFDV) at the N-terminus, remained low (Fig. 3, bottom panels).
In the secondary screening, within the Trunk A peptide we designed six 10-mer to 12-mer peptides labelled Trunk A-1 to Trunk A-6 (Table 2). We enriched CD4 and CD8 T cells from mice immunized with FL-Myhc334-352 and evaluated their responses to these six peptides.
Table 2.
The list of peptides used to evaluate CD4 and CD8 T cell responses in the secondary screening
| Peptide | Sequence |
|---|---|
| Trunk A (Myhc334-349) | DSAFDVLSFTAEEKAG |
| Trunk A-1 (Myhc340-349) | LSFTAEEKAG |
| Trunk A-2 (Myhc339-349) | VLSFTAEEKAG |
| Trunk A-3 (Myhc338-349) | DVLSFTAEEKAG |
| Trunk A-4 (Myhc338-348) | DVLSFTAEEKA |
| Trunk A-5 (Myhc338-347) | DVLSFTAEEK |
| Trunk A-6 (Myhc339-348) | VLSFTAEEKA |
As shown in figure 4 (top panels), Trunk A-4 and Trunk A-5 peptides induced the responses in CD8 T cells (p≤0.009), and if any, the responses for Trunk A-4 were better than FL-Myhc334-352 (p=0.02), whereas responses for Trunk A-6 were absent. Likewise, responses to three other peptides (Trunk A-1, A-2 and A-3) also were marginal (data not shown).
Figure 4. Evaluation of CD4 and CD8 T cell responses to the short peptides of FL-Myhc334-352 (10 to 11-mers) in the secondary screening.

A/J mice were immunized with Myhc334-352 in CFA; after 14 days, CD4 and CD8 T cells were enriched by magnetic separation using the cell suspensions obtained from spleens and lymph nodes. Cells were stimulated with Myhc334-352 and short peptides (10 to 11-mers: Trunk A-4 to A-6) in the presence of irradiated APCs for two days, followed 16 hours later by pulsing with 3[H]-thymidine, the incorporation of which was measured as cpm. RNase43-56 (control). Mean ± SEM values obtained from two to four individual experiments, each involving 9 to 11 mice are shown.
Furthermore, our attempts to reduce the length of Trunk A-4, and A-5 peptides to fewer than 10 amino acids did not yield better results. To this end, we designed 9-mer peptides, based on the epitope prediction database [38]. Proliferative responses, as indicated by stimulation indices, were marginal compared with responses obtained with the control peptide (RNase43-56) (Supplemental table 1).
In contrast to CD8 T cells however, while all three peptides (Trunk A-4, A-5 and A-6) induced responses in the CD4 T cells comparable to responses induced by Myhc334-352, responses to Trunk A-4 and Trunk A-5 (p≤0.005), but not Trunk A-6 (p=0.061), were significantly higher than responses to the control (Fig. 4, bottom panels). We had expected the responses to Trunk A-5 (with a deletion of major TCR-contact residue Ala348) and Trunk A-6 (with a deletion of putative MHC-contact residue Asp338) to be lower than responses to Trunk A-4, which contained both the MHC-contact residue Asp338 and two TCR-contact residues (Ala344; and Ala348; Table 1). But that was not the case.
We thus identified Trunk A-4 and Trunk A-5 as the putative Myhc peptides that induce both CD4 and CD8 T cell responses. The immunogenicity of these peptides was later confirmed by immunizing groups of mice. As expected, CD4 T cells responded comparably to both Trunk A-4 and A-5 peptides, whereas the response to Trunk A-6 was weak (Supplemental fig. 3, top panels). In contrast, CD8 T cells responded to a greater degree to Trunk A-4 followed by Trunk A-5, and the least response was obtained for Trunk A-6 peptides (Supplemental fig. 3, bottom panels).
3.3 Identification of MHC class I allele that binds CD8 T cell epitopes
It has been previously shown that the FL-Myhc334-352 binds the IAk (MHC class II) allele in A/J mice. Since this mouse strain expresses three MHC class I alleles – H-2Dd, H-2Kk and H-2Ld – it was necessary to determine the specific allele that presents the Myhc peptides to CD8 T cells. This was examined via an MHC class I stabilization assay that involved the use of RMA-S cell lines expressing H-2Dd, H-2Kk and H-2Ld alleles, which stabilize MHC molecules on their surface when the MHC-expressing cells bind with peptides [39]. We used fluorophore-conjugated allele-specific antibodies to ascertain MHC stability, determining the difference in the MFI ratios between cells loaded with or without peptides by flow cytometry. By using the peptides that induced the CD8 T cell responses (Trunk A-4 and Trunk A-5), we found that the MFI ratios obtained with RMA-S-Dd cells were significantly higher for Trunk A-5 peptides (p≤0.03) compared to cells incubated with no peptide (Fig. 5). Although MFI ratios obtained for Trunk A-4 were lower than those obtained with Trunk A-5 peptides, the ratios were significantly higher than those obtained with the control peptide, especially at a concentration of 200 μM or more (p≤0.05). Under similar conditions, however, neither of the two peptides bound to RMA-S-Kk- and RMA-S-Ld-expressing cells (data not shown). Thus, we identified Trunk A-4 and Trunk A-5 peptides as the binders of the H-2Dd molecule. These results were further corroborated using T2-Dd cells (kind gift from Ted Hansen, Washington University, St. Louis; and Joyce Solheim, University of Nebraska Medical Center, Omaha), which also showed stronger affinity for Trunk A-5 than Trunk A-4 (data not shown).
Figure 5. Determination of the binding of CD8 T cell epitopes to H-2Dd allele by MHC class I-stabilization assay.
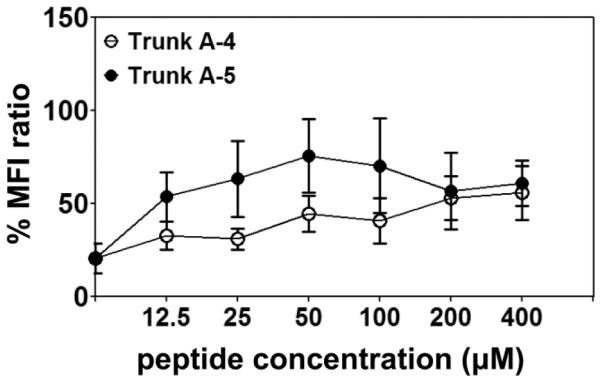
RMA-S-Dd cells were incubated in the presence or absence of the indicated peptides (0 to 400 μM) for 18 hours at 26°C; the plates were then transferred to a 37°C incubator and incubation continued for an additional 1.5 hours. After washing, cells were stained with fluorophore-conjugated H-2Dd antibody and acquired by flow cytometry. MFI values were obtained using CellQuest software. The MFI ratios were then derived using the formula as described in the section 2.6. Mean ± SEM values derived from five to six experiments are shown.
3.4 Antigen-specificity of CD8 T cells sensitized with FL-Myhc334-352
After identifying Trunk A-4 and Trunk A-5 peptides as the putative CD8 T cell epitopes based on peptide screening and MHC-stabilization assay, we sought to create MHC class I dextramers and we recently showed that the MHC dextramers were found to be superior to tetramers in various experimental settings [27, 40, 41]. We generated H-2Dd dextramers by conjugating the peptide-loaded, biotinylated H-2Dd protein assembled with the β2m as described previously [27]. Essentially, we prepared dextramers for Trunk A-4, Trunk A-5 and Trunk A-6 peptides (control) and used them to determine the specificity of CD8 T cells generated in response to Myhc334-352. The expectation was that H-2Dd dextramers of Trunk A-4 and A-5, but not A-6, peptides would bind CD8 T cells, but none would bind CD4 T cells. Briefly, LNC cultures were prepared from A/J mice immunized with Myhc334-352, and the cells were stained with the dextramers and CD4 and CD8 antibodies. As shown in figure 6, H-2Dd/Trunk A-4 (2.74±0.34%; p≤0.0014) and H-2Dd/Trunk A-5 (1.79±0.29%; p≤0.0042) dextramers stained the Myhc334-352-sensitized CD8 T cells, whereas the staining for control (Trunk A-6 dextramers) was negligible (0.05±0.008%). Similarly, dextramers prepared using the irrelevant control antigen HIVP18-I10 also did not stain the cells (0.04±0.0058%), indicating that the staining obtained with H-2Dd /Trunk A-4 and A-5 dextramers was specific. Additionally, none of the H-2Dd dextramers stained the CD4 T cells generated in mice immunized with Myhc334-352, further ensuring the specificity of the MHC class I dextramers (Fig. 6). Taken together, the patterns of staining obtained with H-2Dd dextramers and the MHC-stabilization assay were identical, indicating that the two CD8 T cell epitopes (Trunk A-4 and Trunk A-5) are localized within Myhc334-352.
Figure 6. Evaluation of antigen-specificity of CD8 T cells using MHC class I dextramers.

LNC were obtained from animals immunized with Myhc334-352 and after cells were stimulated with the peptide for two days, IL-2 medium was supplemented. On day 8, cells were stained with the indicated H-2Dd dextramers, followed by anti-CD8, anti-CD4 and 7-AAD. After washing, cells were acquired by flow cytometry, and the percentages of dextramer+ cells were analyzed in relation to CD4 and CD8 T cells by Flow Jo software. H-2Dd/Trunk A-6 and H-2Dd/HIVP18-I10 dextramers, control. Representative flow cytometric plots from one of the six experiments are shown (left panel). Mean ± SEM values derived from six experiments are shown (right panel).
3.5 Determination of the pathogenic potential of Myhc-sensitized CD8 T cells
To determine the pathogenic potential of antigen-sensitized CD8 T cells, we analyzed their cytokine-producing abilities and compared the responses with CD4 T cells [11]. In brief, LNCs from mice immunized with Myhc334-352 were restimulated with the Trunk peptides (A-4, and A-5) and Myhc334-352, and the frequencies of cytokine-producing cells were evaluated for Th1, Th2 and Th17 cytokines and TNF-α. As shown in figure 7 (left panels), cells capable of producing Th1 (IFN-γ) cytokines and TNF-α were predominantly present in both CD4 and CD8 T cell cultures, followed by Th17 and Th2 cytokines. Importantly, when the cytokine profiles of CD4 and CD8 T cell subsets were compared, it was evident that the frequencies of IFN-γ-secreting cells were consistently greater in the CD8 T cell cultures, regardless of peptides used (Myhc334-352: 11.19 ± 1.08 vs 1.98 ± 0.43, p = 0.02E-02; Trunk A-4: 12.87 ± 0.46 vs 2.85 ± 0.54, p = 8.08E-06; Trunk A-5: 14.40 ± 1.62 vs 2.96 ± 0.48, p = 4.91E-05). Conversely, percentages of cells producing Th2 cytokines tended to be greater in CD4 T cells stimulated with Trunk A-4 and A-5 peptides (Fig. 7, right panels). No other major differences were noted for other cytokines except that CD8 T cell cultures produced fewer IL-17F-producing cells than did CD4 T cells (p≤0.009). The findings that the cytokine patterns obtained in CD8 T cell cultures with the Trunk peptides A-4 and A-5 were comparable to those produced with Myhc334-352, and that their responses were largely similar to those produced in CD4 T cells suggest that the A-4 and A-5 peptides might be able to induce disease.
Figure 7. Cytokine responses to dual epitopes of FL-Myhc334-352 in CD4 and CD8 T cells.

LNCs obtained from A/J mice immunized with Myhc334-352, Trunk A-4, Trunk A-5 or Trunk A-6 were stimulated with the immunizing peptides for two days, and the cultures were maintained in IL-2 medium. Cells were briefly stimulated on day 6 with PMA and ionomycin for 5 hours in the presence of monensin. After staining with anti-CD4, anti-CD8 and 7-AAD, cells were fixed and permeabilized, and then stained with cytokine antibodies or their respective isotype controls. Cells were acquired by flow cytometry and the percentages of cytokine-producing cells in the live (7-AAD−) CD4 or CD8 T cell subsets were determined using Flow Jo software in relation to the gates drawn for isotype controls corresponding to each cytokine. Mean ± SEM values from four to six independent experiments are shown. In the left panel, cells positive for individual cytokines are shown (left, y-axis: IL-2, IFN-γ, IL-4, IL-10, IL-17A, IL-17F and IL-22; right, y-axis: TNF-α). The right panel shows the cytokine+ cells with respect to Th1 (IFN-γ), Th2 (IL-4 + IL-10) and Th17 (IL-17A + IL-17F + IL-22) subsets.
3.6 Evaluation of disease induced with Trunk A-4 and Trunk A-5 peptides
We evaluated the disease-inducing abilities of peptides by immunizing groups of mice with Trunk A-4 and Trunk A-5 and compared the disease severity, as assessed by histology, with that of Myhc334-352, which was used as a positive control (Fig. 8). While all three peptides induced myocarditis showing infiltrations of mononuclear cells, the disease severity induced with Trunk A-4 and A-5 peptides was lower than that induced by Myhc334-352 (Fig. 8; Myhc334-352: incidence, 5/5; percent area inflamed, 12.8 ± 5.54). However, among the two truncated peptides, the disease incidence (Trunk A-4, 6/13; Trunk A-5, 3/9) and the homogeneity of histological changes (percent area inflamed: Trunk A-4, 3 ± 0.75; Trunk A-5, 10.7 ± 7.22) were more consistent with Trunk A-4 than Trunk A-5. We also made efforts to determine whether the disease induced with the truncated peptides could also be captured non-invasively. To provide a proof-of-principle for this analysis, we subjected the animals immunized with Trunk A-4 peptides to MRM imaging to evaluate structural and functional abnormalities. The data revealed that the LV wall thickness in the Trunk A-4-immunized mice was increased by ~1.35-fold (p≤2.25E-05) in relation to age-matched healthy mice (Supplemental fig. 4a). Likewise, measurement of cardiac output parameters indicated that both end diastolic volumes (p≤3.45E-05) and ejection fractions (p≤5.67E-07) were low in the myocarditic mice compared to healthy controls (Supplemental fig. 4b). Taken together, the data suggest that the Trunk A-4 and Trunk A-5 peptides can induce myocarditis, and the histological features are similar to those of myocarditis induced by Myhc334-352.
Figure 8. Histological evaluation of hearts obtained from mice immunized with FL-Myhc334-352, and its truncated-derivatives, Trunk A-4 and Trunk A-5.

Groups of A/J mice were immunized with Myhc334-352, Trunk A-4 or Trunk A-5 in CFA on day 0 and day 7, and the animals received pertussis toxin on day 0 and day 2 after the first immunization. Animals were euthanized 21 days later, and hearts were examined for inflammation by H and E staining. Arrows indicate lymphocytic infiltrations. Left panels (Myhc334-352-immunized group): epicardial infiltrates of lymphocytes and macrophages (i); subendocardial infiltrates of lymphocytes and macrophages extend into myocardium (ii); and interstitial myocardial infiltrates of macrophages and lymphocytes (iii). Middle panels (Trunk A-4-immunized group): subendocardial plasma cells, lymphocytes and macrophages extending into the myocardium (i); perivascular and interstitial lymphocytic and histiocytic infiltrates in the myocardium (ii); and subendocardial lymphocytes and macrophages (iii). Right panels (Trunk A-5-immunized group): interstitial lymphocytes and macrophages in myocardium (i); subendocardial lymphocytes and a few macrophages and rare plasma cells infiltrate the atria (ii); and small foci of lymphocytes in subendocardium (iii).
4. Discussion
We report here the localization of the CD8 T cell determinant, Myhc338-348 that induces EAM in A/J mice. Previously, it has been shown that Myhc334-352, an immunodominant epitope that binds the MHC class II allele IAk, induces myocarditis in IAk-expressing A/J mice through the generation of Myhc-reactive CD4 T cells [6, 10, 11, 15]. We recently created MHC class II/IAk tetramers and dextramers and demonstrated that Myhc334-352-specific CD4 T cells are generated in the peripheral repertoire and infiltrate the hearts of immunized animals [15, 27]. However, by examining the heart infiltrates, we noted that CD8 T cells also formed a component of the T cell fraction (~35% of T cells), leading us to determine their potential role in the mediation of cardiac autoimmunity.
We first verified that the responses of CD8 T cells from the animals immunized with Myhc334-352 were comparable with responses of CD4 T cells (Fig. 1). Importantly, we confirmed that Myhc-sensitized CD8 T cells produce IFN-γ (Supplemental fig. 2) and express markers CD107a/CD107b, granzyme and perforin, the markers of their effector functions, and transfer disease to naïve animals (Fig. 2). Based on these observations, we decided to localize the CD8 T cell epitopes within FL-Myhc334-352 (DSAFDVLSFTAEEKAGVYK). In this peptide, three amino acid residues were previously identified to be critical (Asp338, MHC class II/IAk-anchor; and Ala344-minor; and Ala348-major TCR-contacts; Table 1), where deletion of major TCR contact residue Ala348 was found to reduce CD4 T cell responses [6]. Hence, during the primary screening, we took an approach to ablate the CD4 T cell responses by designing peptides to retain or to delete the MHC (Asp338)- or the major TCR (Ala348)-contact residues. These include trunk A and trunk B (both contain MHC- and TCR-contact residues); trunk C (lacks the TCR-contact residue); and trunk D (lacks the MHC-contact residue) (Table 1). Predictably, we noted the responses for Trunk A and Trunk B in CD4 T cells, whereas the responses for Trunk C and Trunk D were low (Fig. 3). In contrast, the two truncated peptides (Trunk A and Trunk B), containing all three residues essential for the induction of CD4 T cell responses, also induced comparable responses in CD8 T cells (Fig. 3). These data pointed to a possibility that the central core region (DVLSFTAEEKAG) may encompass the CD8 T cell epitope/s, with the further possibility that CD4 T cell responses could still be preserved, since the MHC class II anchor and the TCR-contact residues for the generation of CD4 T cells were still conserved (Table 1).
Reports indicate that the peptides binding to MHC class I molecules can be of variable lengths (8 to 11 amino acids), but long peptides up to 15 amino acids can also bind class I molecules under the conditions of conformational change of their peptide-binding sites. Therefore, we designed a total of six peptides with a length of 10 to 12 amino acids, and found that two peptides (Trunk A-4: DVLSFTAEEKA, and Trunk A-5: DVLSFTAEEK) induced responses in CD8 T cells comparable to those obtained with FL-Myhc334-352, whereas response for Trunk A-6 was absent (Fig. 4). But surprisingly, all three peptides, including Trunk A-5, which was missing the major TCR-contact residue Ala348 for CD4 T cells, and Trunk A-6, which was missing the class II MHC-anchor residue Asp338 at the N-terminus, stimulated the CD4 T cells, and their responses were comparable to those stimulated by Myhc334-352. These data support the notion that shorter peptides of 10-mers can also be presented by MHC class II molecules to CD4 T cells as reported by others [42-44]. While our data also suggest the possibility of the existence of an alternative MHC class II-anchor residue, the display of major or minor TCR-contact residues in the short peptide sequences may still be similar to FL-Myhc334-352. Alternatively, the minor TCR-contact residue Ala344 may get displayed differently with an altered binding register in the absence of the major TCR-contact residue as in Trunk A-5 (Table 2), and such a compensatory change may be adequate to facilitate peptide-recognition by TCR. Overall, since Trunk A-4 and Trunk A-5 peptides showed comparable responses in CD8 T cells, we decided to use these for further experimentation. Finally, after confirming that the MHC class I allele H-2Dd presents both Trunk A-4 and Trunk A-5 peptides, we created their dextramers and confirmed that the CD8 T cells reacting to Myhc were antigen-specific, suggesting they play a role in the development of myocarditis.
To evaluate the pathogenic potential of CD8 T cell epitopes, we first analyzed the cytokine-producing ability of CD8 T cells based on intracellular staining using panels of Th1, Th2 and Th17 cytokines and TNF-α. Essentially, the Myhc-reactive CD8 T cell cultures contained cells capable of producing all cytokines tested, and their profiles were comparable to those of CD4 T cells. Importantly, the frequencies of IFN-γ-producing cells were significantly greater in CD8 than in CD4 cultures, except that IL-17F-producing cells were low in the cultures responsive to Trunk A-4 and A-5 peptides. However, regardless of cell type, Th2 cytokines were consistently low. However, it is to be noted that conflicting reports were available as to the role of cytokines in the mediation of autoimmune myocarditis, in part, due to the variation in the genetic backgrounds of the mouse strains used [12, 13]. For example, IL-13-deficient and IL-12p40-decificient Balb/c mice showed opposing disease phenotypes, in that the disease severity was enhanced in IL-13-deficient mice, whereas in IL-12p40-deficient mice, the severity was relatively low [13, 45-47]. Conversely, while, Balb/c mice deficient for IFN-γ showed more severe disease, the IL-4-deficient A/J mice showed an attenuated myocarditis [31, 47]. It is currently held that the cytokines represented by both Th1 (IFN-γ) and Th17 (IL-17 family cytokines and IL-22) are critical to the mediation of autoimmune myocarditis, since cytokines other than IL-12 produced by APCs, such as IL-1, IL-6, IL-23 and transforming growth factor-β, favor the development of proinflammatory Th17 cytokines [13, 46]. However, it is to be noted that IL-17A appears not critical for the development of myocarditis as IL-17A-deficient mice can develop severe disease [48]. Adding to this complexity is the possibility of plasticity for the interconversion of Th1 to Th17 and vice versa [49, 50]. Finally, by verifying the disease-inducing potential, we noted that the histological changes observed with myocarditis resulting from Trunk A-4 and Trunk A-5 peptides were comparable to those resulting from FL-Myhc334-352, but the disease severity induced by the Trunk peptides remained mild to moderate (Fig. 8). The relative lower ability of CD8 T cells to produce IL-17F (Fig. 7), which otherwise contributes to the aggravation of inflammation by promoting neutrophil/monocyte migration [51, 52], might explain the lower disease severity induced with the determinants for CD8 T cells.
4.1 Limitations
In this study, we noted the following limitations: (i) for intracellular cytokine staining, we could not compare the cytokine profiles obtained with Myhc peptide-stimulations, with the cells cultured in medium-alone, or the cells stimulated with the irrelevant peptide, since the cells in the latter two conditions were not expected to be viable in the cultures for six days. Although, we evaluated the cytokine-secretion based on cytokine bead array analysis in the whole cell cultures specific to Myhc peptides (data not shown), we could not analyze the cytokines in the individual CD4 and CD8 subsets enriched by magnetic separation; (ii) we performed the MHC class I stabilization assay based on published protocols, but these protocols do not involve the use of a positive control peptide; its use otherwise would have been helpful to compare the affinity of Myhc peptides; (iii) the animals immunized with Trunk A-5 peptide were not subjected for MRM imaging since the incidence of disease induced with this peptide was low; and (iv) we made no efforts to identify the amino acid residues within Myhc338-348 that are critical for binding MHC class I (H-2Dd) and the TCR of CD8 T cells.
5. Conclusion
We have demonstrated that Myhc338-348 acts as a dual epitope for both CD4 and CD8 T cells, and the CD8 T cell epitopes induce myocarditis through the generation of Myhc-specific IFN-γ-producing cells. Interestingly, the shorter peptides, Trunk A-5 and A-6, lacking the major TCR-contact residue and MHC class II-anchor, respectively, also induced CD4 T cell responses comparable to those produced by FL-Myhc334-352. These observations may lead to the conclusion that a proportion of antigen-specific CD4 T cells that arise in response to Myhc334-352 may be the result of recognition of shorter peptides possibly generated by the processing of FL-Myhc334-352 by APCs. In this scenario, the presence of the single TCR-contact residue Ala344 in FL-Myhc334-352, supposedly a minor TCR-contact residue in Trunk A-5, may be sufficient to induce activation of CD4 T cells. During this process, the cardiac-reactive CD8 T cells can also be produced and participate in the disease pathogenesis. Thus, the hierarchical generation of cardiac-reactive CD4 and CD8 T cells may be possible, but they may collectively contribute to the tissue destruction. Although the disease induced by CD4 cells is typically mediated by Th1 and Th17 cytokines, in addition to their cytolytic functions, cytokines produced by CD8 T cells similar to Th1 and Th17 phenotypes [53, 54] also have been implicated. Furthermore, Myhc, being an intracellular protein, is expressed by cardiac myocytes, and the resident APCs in A/J mice have also been shown to display fragments of Myhc [6]. Yet, the animals do not develop myocarditis spontaneously, possibly because of self-tolerance due to the lack of costimulatory signals. In inflammatory conditions, however, upregulation of costimulatory molecules may favor the presentation of peptides, leading to a break in self-tolerance, and cardiac autoimmunity may ensue, possibly involving the generation of Myhc-reactive CD4 and CD8 T cells. Of note, the generation of pathogenic, dual T cell-specific responses through the recognition of a single epitope similar to Myhc334-352 has been described for at least three other self-antigens, myelin oligodendrocyte glycoprotein 35-55, cancer testes antigen and interphotoreceptor retinoid-binding protein 201-216 [42, 55-57]. However, reports also indicate that induction of antigen-specific CD8 T cells can be potentially beneficial to controlling autoimmune responses, because CD8 T cells that lack the expression of CD28 can behave as regulatory T (Treg) cells, as shown in systemic lupus erythematosus and rheumatoid arthritis [58-60]. This was not the case, however, with Myhc-specific CD8 T cell responses, because the Myhc-reactive cells were positive for CD28, and did not express other markers of Treg cells like forkhead box P3 (data not shown). Nonetheless, the finding that a solitary peptide fragment bearing the epitopes for two different T cell types induced myocarditis provides new insights as to how different immune cells can recognize the same antigen and mediate cardiac damage by different mechanisms. In autoimmune settings, identification of the disease-inducing epitopes and their cellular mechanisms may possibly create opportunities to target antigen-specific T cells for therapy. As to human relevance, IgG autoantibodies for various cardiac antigens like cardiac myosin, β1-adrenergic receptor, cardiac troponin I, Na-K-ATPase and heart-specific mitochondrial inner membrane proteins M7 type A and cardiac adenine-nucleotide transporter have been demonstrated in patients with myocarditis/dilated cardiomyopathy, and experimentally, in animal models of adjuvant and viral myocarditis, but their pathological significance continues to be uncertain [61-65]. However, the requirement that T cell help is critical for B cells to produce IgG antibodies suggests that cardiac-reactive T cells might play a pathogenic role. Our data may point to this possibility.
Supplementary Material
Supplemental figure 1: Evaluation of the antigen-specificity of Myhc-sensitized T cells by IAk/Myhc334-352 dextramer staining. CD4 T cells enriched from the animals immunized with Myhc334-352 were stimulated with peptide-loaded APCs for two days, and the cultures were maintained in IL-2 medium. On day 8, cells were stained with dextramers, CD4 antibodies, and 7-AAD. After acquiring by flow cytometry, dextramer+ live (7-AAD−) cells were enumerated in the CD4 T cell subsets. RNase43-56, control. Representative flow cytometric plots from one of the five experiments is shown (top panel). Mean ± SEM values from five experiments are shown (bottom panel).
Supplemental figure 2: Cytokine responses in CD4 and CD8 T cells derived from animals immunized with FL-Myhc334-352. CD4 and CD8 T cells from the animals immunized with Myhc334-352 were enriched and after stimulation with the peptide-loaded APCs for two days, IL-2 medium was added. On day 6, cells were stimulated briefly with PMA and ionomycin in the presence of monensin for 5 hours and the cells were stained with anti-CD4 or anti-CD8, and 7-AAD. After fixation and permeabilization, cells were stained with the indicated cytokine antibodies or their respective isotype controls and acquired by flow cytometry. Percentages of cytokine producing CD4+ or CD8+ T cells were then analyzed in the live (7-AAD−) population using Flow Jo software in relation to the gates drawn for isotype controls corresponding to each cytokine. Top panel: Representative flow cytometric plots are shown. Bottom panels: Mean ± SEM values derived from five to eight experiments are shown (left panel, CD4 T cells; right panel, CD8 T cells). Left, y-axis: IL-2 and IFN-γ; right, y-axis: IL-4, IL-10, IL-17A, IL-17F and IL-22. (*, p=0.0068; **, p=0.0032).
Supplemental figure 3: Evaluation of the immunogenicity of Trunk A-4, Trunk A-5 and Trunk A-6. Groups of A/J mice were immunized with Trunk A-4, Trunk A-5 or Trunk A-6, and on day 14 postimmunization, CD4 and CD8 T cells were enriched from the lymphocytes based on magnetic separation. Cells were stimulated with APCs loaded with the immunizing peptides or RNase43-56 (control) for two days. After pulsing with 3[H]-thymidine, mean proliferative responses were measured as cpm 16 hours later. Mean ± SEM values from three independent experiments each involving 5 to 8 mice per group is shown.
Supplemental figure 4: MRM-imaging of myocarditic mice immunized with Trunk A-4. Mice were immunized with Trunk A-4 in CFA on day 0 and day 7, and pertussis toxin was administered on day 0 and day 2 after the first immunization. On day 21, animals were subjected to MRM imaging to evaluate cardiac abnormalities. (a) LV wall thickness. In the healthy and myocarditic mice, short-axis slices of hearts were captured in eight frames using echo-based cine pulse sequence, and LV wall thickness was calculated using segment software (arrows: LV wall thickness). (b) Cardiac output. Cardiac outputs represented by LV end-diastolic volume (i) and ejection fraction (ii) in the above groups were calculated using quantitative medical image analysis with Segment software. Mean ± SEM values for a group of mice are shown (n = 5 to 6 per group).
Acknowledgements
This work was supported by the National Institutes of Health (HL114669). CM is a recipient of a postdoctoral research fellowship grant awarded by the Myocarditis Foundation, NJ.
List of Abbreviations
- Myhc
cardiac myosin heavy chain-α
- MHC
major histocompatibility complex
- Th
T helper
- IFN
interferon
- IL
interleukin
- APCs
antigen-presenting cells
- EAM
experimental autoimmune myocarditis
- CFA
complete Freund’s adjuvant
- FL-Myhc
full-length Myhc
- RNase
bovine ribonuclease
- HIV
human immunodeficiency virus envelope glycoprotein
- 3[H]-thymidine
tritiated-thymidine
- cpm
counts per minute
- Con-A
concanavalin-A
- LNCs
lymph node cells
- 7-AAD
7-amino-actinomycin-D
- MFI
mean fluorescence intensities
- β2m
β2microglobulin
- PMA
phorbol-12-myristate 13-acetate
- Bir A
biotin ligase
- RT
room temperature
- TNF
tumor necrosis factor
- H and E
hematoxylin and eosin
- MRM
magnetic resonance microscopy
- LV
left ventricular
- TCR
T cell receptor
- Treg
regulatory T cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors report no relationships that could be construed as a conflict of interest.
References
- [1].Hampe CS. B Cell in Autoimmune Diseases. Scientifica. 2012:2012. doi: 10.6064/2012/215308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ken Murphy PT, Mark Walport. Immunobiology: The Immune System in Health and Disease. Taylor & Francis; New York: 2007. [Google Scholar]
- [3].Guo HC, Jardetzky TS, Garrett TP, Lane WS, Strominger JL, Wiley DC. Different length peptides bind to HLA-Aw68 similarly at their ends but bulge out in the middle. Nature. 1992;360:364–6. doi: 10.1038/360364a0. [DOI] [PubMed] [Google Scholar]
- [4].O'Brien C, Flower DR, Feighery C. Peptide length significantly influences in vitro affinity for MHC class II molecules. Immunome Res. 2008;4:6. doi: 10.1186/1745-7580-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. 6th Saunders Elsevier; Philadelphia: 2010. [Google Scholar]
- [6].Donermeyer DL, Beisel KW, Allen PM, Smith SC. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J Exp Med. 1995;182:1291–300. doi: 10.1084/jem.182.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kodama M, Matsumoto Y, Fujiwara M, Masani F, Izumi T, Shibata A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1990;57:250–62. doi: 10.1016/0090-1229(90)90039-s. [DOI] [PubMed] [Google Scholar]
- [8].Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–6. [PubMed] [Google Scholar]
- [9].Pummerer CL, Luze K, Grassl G, Bachmaier K, Offner F, Burrell SK, et al. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J Clin Invest. 1996;97:2057–62. doi: 10.1172/JCI118642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141–7. [PubMed] [Google Scholar]
- [11].Massilamany C, Gangaplara A, Steffen D, Reddy J. Identification of novel mimicry epitopes for cardiac myosin heavy chain-alpha that induce autoimmune myocarditis in A/J mice. Cell Immunol. 2011;271:438–49. doi: 10.1016/j.cellimm.2011.08.013. [DOI] [PubMed] [Google Scholar]
- [12].Christoph Berger UE. Autoimmune Murine Myocarditis and Immunomodulatory Interventions. Inflammatory Cardiomyopathy (DCMi) – Pathogenesis and Therapy: Birkhäuser Basel. 2010:37–49. [Google Scholar]
- [13].Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. doi: 10.1016/S0065-2776(08)00604-4. [DOI] [PubMed] [Google Scholar]
- [14].Penninger JM, Pummerer C, Liu P, Neu N, Bachmaier K. Cellular and molecular mechanisms of murine autoimmune myocarditis. APMIS. 1997;105:1–13. doi: 10.1111/j.1699-0463.1997.tb00532.x. [DOI] [PubMed] [Google Scholar]
- [15].Massilamany C, Gangaplara A, Chapman N, Rose N, Reddy J. Detection of cardiac myosin heavy chain-alpha-specific CD4 cells by using MHC class II/IA(k) tetramers in A/J mice. J Immunol Methods. 2011;372:107–18. doi: 10.1016/j.jim.2011.07.004. [DOI] [PubMed] [Google Scholar]
- [16].Gangaplara A, Massilamany C, Brown DM, Delhon G, Pattnaik AK, Chapman N, et al. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-alpha-reactive CD4 T cells in A/J mice. Clin Immunol. 2012;144:237–49. doi: 10.1016/j.clim.2012.07.003. [DOI] [PubMed] [Google Scholar]
- [17].Barin JG, Talor MV, Baldeviano GC, Kimura M, Rose NR, Cihakova D. Mechanisms of IFNgamma regulation of autoimmune myocarditis. Experimental and molecular pathology. 2010;89:83–91. doi: 10.1016/j.yexmp.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Elliott JF, Liu J, Yuan ZN, Bautista-Lopez N, Wallbank SL, Suzuki K, et al. Autoimmune cardiomyopathy and heart block develop spontaneously in HLA-DQ8 transgenic IAbeta knockout NOD mice. Proc Natl Acad Sci U S A. 2003;100:13447–52. doi: 10.1073/pnas.2235552100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Frohlich A, Marsland BJ, Sonderegger I, Kurrer M, Hodge MR, Harris NL, et al. IL-21 receptor signaling is integral to the development of Th2 effector responses in vivo. Blood. 2007;109:2023–31. doi: 10.1182/blood-2006-05-021600. [DOI] [PubMed] [Google Scholar]
- [20].Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- [21].Kannan K, Stewart RM, Bounds W, Carlsson SR, Fukuda M, Betzing KW, et al. Lysosome-associated membrane proteins h-LAMP1 (CD107a) and h-LAMP2 (CD107b) are activation-dependent cell surface glycoproteins in human peripheral blood mononuclear cells which mediate cell adhesion to vascular endothelium. Cell Immunol. 1996;171:10–9. doi: 10.1006/cimm.1996.0167. [DOI] [PubMed] [Google Scholar]
- [22].Brown DM, Lee S, Garcia-Hernandez Mde L, Swain SL. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol. 2012;86:6792–803. doi: 10.1128/JVI.07172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Workman AM, Jacobs AK, Vogel AJ, Condon S, Brown DM. Inflammation enhances IL-2 driven differentiation of cytolytic CD4 T cells. PLoS One. 2014;9:e89010. doi: 10.1371/journal.pone.0089010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kaiserman D, Bird CH, Sun J, Matthews A, Ung K, Whisstock JC, et al. The major human and mouse granzymes are structurally and functionally divergent. The Journal of cell biology. 2006;175:619–30. doi: 10.1083/jcb.200606073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Suzuki M, Aoshi T, Nagata T, Koide Y. Identification of murine H2-Dd- and H2-Ab-restricted T-cell epitopes on a novel protective antigen, MPT51, of Mycobacterium tuberculosis. Infect Immun. 2004;72:3829–37. doi: 10.1128/IAI.72.7.3829-3837.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kappler JW, Skidmore B, White J, Marrack P. Antigen-inducible, H-2-restricted, interleukin-2-producing T cell hybridomas. Lack of independent antigen and H-2 recognition. J Exp Med. 1981;153:1198–214. doi: 10.1084/jem.153.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Massilamany C, Upadhyaya B, Gangaplara A, Kuszynski C, Reddy J. Detection of autoreactive CD4 T cells using major histocompatibility complex class II dextramers. BMC Immunol. 2011;12:40. doi: 10.1186/1471-2172-12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Altman JD, Davis MM. MHC-peptide tetramers to visualize antigen-specific T cells. Curr Protoc Immunol. 2003:3. doi: 10.1002/0471142735.im1703s53. Chapter 17:Unit 17. [DOI] [PubMed] [Google Scholar]
- [29].Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A. 1992;89:3429–33. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu XS, Liu WJ, Zhao KN, Liu YH, Leggatt G, Frazer IH. Route of administration of chimeric BPV1 VLP determines the character of the induced immune responses. Immunol Cell Biol. 2002;80:21–9. doi: 10.1046/j.1440-1711.2002.01051.x. [DOI] [PubMed] [Google Scholar]
- [31].Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, et al. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159:193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Massilamany C, Khalilzad-Sharghi V, Gangaplara A, Steffen D, Othman SF, Reddy J. Noninvasive assessment of cardiac abnormalities in experimental autoimmune myocarditis by magnetic resonance microscopy imaging in the mouse. Journal of visualized experiments : JoVE. 2014:e51654. doi: 10.3791/51654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC. Granzyme B: a natural born killer. Immunol Rev. 2003;193:31–8. doi: 10.1034/j.1600-065x.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- [34].Packard BZ, Telford WG, Komoriya A, Henkart PA. Granzyme B activity in target cells detects attack by cytotoxic lymphocytes. J Immunol. 2007;179:3812–20. doi: 10.4049/jimmunol.179.6.3812. [DOI] [PubMed] [Google Scholar]
- [35].Speir JA, Stevens J, Joly E, Butcher GW, Wilson IA. Two different, highly exposed, bulged structures for an unusually long peptide bound to rat MHC class I RT1-Aa. Immunity. 2001;14:81–92. doi: 10.1016/s1074-7613(01)00091-7. [DOI] [PubMed] [Google Scholar]
- [36].Stevens J, Wiesmuller KH, Walden P, Joly E. Peptide length preferences for rat and mouse MHC class I molecules using random peptide libraries. Eur J Immunol. 1998;28:1272–9. doi: 10.1002/(SICI)1521-4141(199804)28:04<1272::AID-IMMU1272>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- [37].Tynan FE, Borg NA, Miles JJ, Beddoe T, El-Hassen D, Silins SL, et al. High resolution structures of highly bulged viral epitopes bound to major histocompatibility complex class I. Implications for T-cell receptor engagement and T-cell immunodominance. J Biol Chem. 2005;280:23900–9. doi: 10.1074/jbc.M503060200. [DOI] [PubMed] [Google Scholar]
- [38].Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–9. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- [39].Correa I, Raulet DH. Binding of diverse peptides to MHC class I molecules inhibits target cell lysis by activated natural killer cells. Immunity. 1995;2:61–71. doi: 10.1016/1074-7613(95)90079-9. [DOI] [PubMed] [Google Scholar]
- [40].Massilamany C, Gangaplara A, Jia T, Elowsky C, Kang G, Riethoven JJ, et al. Direct staining with major histocompatibility complex class II dextramers permits detection of antigen-specific, autoreactive CD4 T cells in situ. PLoS One. 2014;9:e87519. doi: 10.1371/journal.pone.0087519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Massilamany C, Gangaplara A, Jia T, Elowsky C, Li Q, Zhou Y, et al. In situ detection of autoreactive CD4 T cells in brain and heart using major histocompatibility complex class II dextramers. Journal of visualized experiments : JoVE. 2014:e51679. doi: 10.3791/51679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shao H, Peng Y, Liao T, Wang M, Song M, Kaplan HJ, et al. A shared epitope of the interphotoreceptor retinoid-binding protein recognized by the CD4+ and CD8+ autoreactive T cells. J Immunol. 2005;175:1851–7. doi: 10.4049/jimmunol.175.3.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Buus S, Sette A, Colon SM, Miles C, Grey HM. The relation between major histocompatibility complex (MHC) restriction and the capacity of Ia to bind immunogenic peptides. Science. 1987;235:1353–8. doi: 10.1126/science.2435001. [DOI] [PubMed] [Google Scholar]
- [44].Sercarz EE, Maverakis E. Mhc-guided processing: binding of large antigen fragments. Nat Rev Immunol. 2003;3:621–9. doi: 10.1038/nri1149. [DOI] [PubMed] [Google Scholar]
- [45].Cihakova D, Barin JG, Afanasyeva M, Kimura M, Fairweather D, Berg M, et al. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol. 2008;172:1195–208. doi: 10.2353/ajpath.2008.070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Eriksson U. The role of IL-17 in Experimental Autoimmune Myocarditis. IL-17, IL-22 and their producing cells: role in inflammation and autoimmunity: Springer Basel. 2013:165–75. [Google Scholar]
- [47].Eriksson U, Kurrer MO, Sebald W, Brombacher F, Kopf M. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J Immunol. 2001;167:5464–9. doi: 10.4049/jimmunol.167.9.5464. [DOI] [PubMed] [Google Scholar]
- [48].Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- [49].Cosmi L, Maggi L, Santarlasci V, Liotta F, Annunziato F. T helper cells plasticity in inflammation. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2014;85:36–42. doi: 10.1002/cyto.a.22348. [DOI] [PubMed] [Google Scholar]
- [50].Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. 2013;121:2402–14. doi: 10.1182/blood-2012-09-378653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ge S, Hertel B, Susnik N, Rong S, Dittrich AM, Schmitt R, et al. Interleukin 17 receptor A modulates monocyte subsets and macrophage generation in vivo. PLoS One. 2014;9:e85461. doi: 10.1371/journal.pone.0085461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kimizuka Y, Kimura S, Saga T, Ishii M, Hasegawa N, Betsuyaku T, et al. Roles of interleukin-17 in an experimental Legionella pneumophila pneumonia model. Infect Immun. 2012;80:1121–7. doi: 10.1128/IAI.05544-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mosmann TR, Li L, Sad S. Functions of CD8 T-cell subsets secreting different cytokine patterns. Seminars in immunology. 1997;9:87–92. doi: 10.1006/smim.1997.0065. [DOI] [PubMed] [Google Scholar]
- [54].Yen HR, Harris TJ, Wada S, Grosso JF, Getnet D, Goldberg MV, et al. Tc17 CD8 T cells: functional plasticity and subset diversity. J Immunol. 2009;183:7161–8. doi: 10.4049/jimmunol.0900368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur J Immunol. 2005;35:76–85. doi: 10.1002/eji.200425660. [DOI] [PubMed] [Google Scholar]
- [56].Neumann F, Kubuschok B, Ertan K, Schormann C, Stevanovic S, Preuss KD, et al. A peptide epitope derived from the cancer testis antigen HOM-MEL-40/SSX2 capable of inducing CD4(+) and CD8(+) T-cell as well as B-cell responses. Cancer immunology, immunotherapy : CII. 2011;60:1333–46. doi: 10.1007/s00262-011-1030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sun D, Zhang Y, Wei B, Peiper SC, Shao H, Kaplan HJ. Encephalitogenic activity of truncated myelin oligodendrocyte glycoprotein (MOG) peptides and their recognition by CD8+ MOG-specific T cells on oligomeric MHC class I molecules. Int Immunol. 2003;15:261–8. doi: 10.1093/intimm/dxg023. [DOI] [PubMed] [Google Scholar]
- [58].Davila E, Kang YM, Park YW, Sawai H, He X, Pryshchep S, et al. Cell-based immunotherapy with suppressor CD8+ T cells in rheumatoid arthritis. J Immunol. 2005;174:7292–301. doi: 10.4049/jimmunol.174.11.7292. [DOI] [PubMed] [Google Scholar]
- [59].Dinesh RK, Skaggs BJ, La Cava A, Hahn BH, Singh RP. CD8+ Tregs in lupus, autoimmunity, and beyond. Autoimmun Rev. 2010;9:560–8. doi: 10.1016/j.autrev.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011;134:17–32. doi: 10.1111/j.1365-2567.2011.03470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Caforio AL, Goldman JH, Baig MK, Haven AJ, Dalla Libera L, Keeling PJ, et al. Cardiac autoantibodies in dilated cardiomyopathy become undetectable with disease progression. Heart. 1997;77:62–7. doi: 10.1136/hrt.77.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Caforio AL, Goldman JH, Haven AJ, Baig KM, Libera LD, McKenna WJ. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur Heart J. 1997;18:270–5. doi: 10.1093/oxfordjournals.eurheartj.a015230. [DOI] [PubMed] [Google Scholar]
- [63].Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail. 2002;4:411–7. doi: 10.1016/s1388-9842(02)00010-7. [DOI] [PubMed] [Google Scholar]
- [64].Kaya Z, Leib C, Katus HA. Autoantibodies in heart failure and cardiac dysfunction. Circ Res. 2012;110:145–58. doi: 10.1161/CIRCRESAHA.111.243360. [DOI] [PubMed] [Google Scholar]
- [65].Neu N, Beisel KW, Traystman MD, Rose NR, Craig SW. Autoantibodies specific for the cardiac myosin isoform are found in mice susceptible to Coxsackievirus B3-induced myocarditis. J Immunol. 1987;138:2488–92. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1: Evaluation of the antigen-specificity of Myhc-sensitized T cells by IAk/Myhc334-352 dextramer staining. CD4 T cells enriched from the animals immunized with Myhc334-352 were stimulated with peptide-loaded APCs for two days, and the cultures were maintained in IL-2 medium. On day 8, cells were stained with dextramers, CD4 antibodies, and 7-AAD. After acquiring by flow cytometry, dextramer+ live (7-AAD−) cells were enumerated in the CD4 T cell subsets. RNase43-56, control. Representative flow cytometric plots from one of the five experiments is shown (top panel). Mean ± SEM values from five experiments are shown (bottom panel).
Supplemental figure 2: Cytokine responses in CD4 and CD8 T cells derived from animals immunized with FL-Myhc334-352. CD4 and CD8 T cells from the animals immunized with Myhc334-352 were enriched and after stimulation with the peptide-loaded APCs for two days, IL-2 medium was added. On day 6, cells were stimulated briefly with PMA and ionomycin in the presence of monensin for 5 hours and the cells were stained with anti-CD4 or anti-CD8, and 7-AAD. After fixation and permeabilization, cells were stained with the indicated cytokine antibodies or their respective isotype controls and acquired by flow cytometry. Percentages of cytokine producing CD4+ or CD8+ T cells were then analyzed in the live (7-AAD−) population using Flow Jo software in relation to the gates drawn for isotype controls corresponding to each cytokine. Top panel: Representative flow cytometric plots are shown. Bottom panels: Mean ± SEM values derived from five to eight experiments are shown (left panel, CD4 T cells; right panel, CD8 T cells). Left, y-axis: IL-2 and IFN-γ; right, y-axis: IL-4, IL-10, IL-17A, IL-17F and IL-22. (*, p=0.0068; **, p=0.0032).
Supplemental figure 3: Evaluation of the immunogenicity of Trunk A-4, Trunk A-5 and Trunk A-6. Groups of A/J mice were immunized with Trunk A-4, Trunk A-5 or Trunk A-6, and on day 14 postimmunization, CD4 and CD8 T cells were enriched from the lymphocytes based on magnetic separation. Cells were stimulated with APCs loaded with the immunizing peptides or RNase43-56 (control) for two days. After pulsing with 3[H]-thymidine, mean proliferative responses were measured as cpm 16 hours later. Mean ± SEM values from three independent experiments each involving 5 to 8 mice per group is shown.
Supplemental figure 4: MRM-imaging of myocarditic mice immunized with Trunk A-4. Mice were immunized with Trunk A-4 in CFA on day 0 and day 7, and pertussis toxin was administered on day 0 and day 2 after the first immunization. On day 21, animals were subjected to MRM imaging to evaluate cardiac abnormalities. (a) LV wall thickness. In the healthy and myocarditic mice, short-axis slices of hearts were captured in eight frames using echo-based cine pulse sequence, and LV wall thickness was calculated using segment software (arrows: LV wall thickness). (b) Cardiac output. Cardiac outputs represented by LV end-diastolic volume (i) and ejection fraction (ii) in the above groups were calculated using quantitative medical image analysis with Segment software. Mean ± SEM values for a group of mice are shown (n = 5 to 6 per group).