

Abstract
Surface plasmon resonance (SPR) is a powerful analytical technique used to quantitatively examine the interactions between various biomolecules, such as proteins and nucleic acids. The technique has been particularly useful in screening and evaluating binding affinity of novel small molecule and biomolecule-derived therapeutics for various diseases and applications including lupus medications, thrombin inhibitors, HIV protease inhibitors, DNA gyrase inhibitors and many others. Recently, there has been increasing interest in nanotherapeutics (nanoRx), due to their unique properties and potential for controlled release of encapsulated drugs and structure-specific targeting to diseased tissues. Targeted nanoRx offer the potential to solve many drug delivery challenges by enabling, specific interactions between molecules on the surface of the nanoparticle and molecules in the diseased tissue, while minimizing off-target interactions toward non-diseased tissues. These properties are largely dependent upon careful control and balance of nanoRx interactions and binding properties with tissues in vivo. Given the great promise of nanoRx with regard to engineering specific molecular interactions, SPR can rapidly quantify small aliquots of nanoRx formulations for desired and undesired molecular interactions. Moving forward, we believe that utilization of SPR in the screening and design of nanoRx has the potential to greatly improve the development of targeted nanoRx formulations and eventually lead to improved therapeutic efficacy. In this review, we discuss (1) the fundamental principles of SPR and basic quantitative analysis of SPR data, (2) previous applications of SPR in the study of non-particulate therapeutics and nanoRx, and (3) future opportunities for the use of SPR in the evaluation of nanoRx.
Keywords: surface plasmon resonance (SPR), biacore, molecular interactions, nanotechnology, targeted therapy
Graphical abstract
Surface plasmon resonance (SPR) evaluation of specific and nonspecific interactions of a targeted nanotherapeutics. (A) SPR study of specific binding of PEG-coated polystyrene nanoparticles (NP) with no ITEM4 antibody (CNP, black), a low density of ITEM4 (CNP-ITEM4 (low), red), and a high density of ITEM4 (CNP-ITEM4 (high), blue) to a CM5 chip with immobilized extracellular domain of Fn14 protein. (B) SPR study of non-specific binding of uncoated NP (no PEG, UNP, black), CNP, CNP-ITEM4 (low), and CNP-ITEM4 (high) to a CM5 chip with immobilized mouse brain extracellular matrix proteins. (C) Zoomed view of dotted box in (B).
I. Introduction to surface plasmon resonance (SPR)
Surface plasmon resonance (SPR) is a powerful analytical technique that relies on changes in the refractive index at the surface of a gold chip to detect the binding of a ligand that is generally covalently fixed to the chip surface with an analyte molecule that is constantly flowing over the chip surface. Current SPR systems can detect very small (~pg/mm2) changes in the mass of the analyte bound to the chip at a temporal resolution of approximately 0.1 s [1]. Thus, SPR is an extremely useful technique for accurately determining both equilibrium and kinetic rate constants of ligand/analyte binding events. SPR does not require any special labeling of analyte molecules, as does traditional fluorescent or radio-labeled ligand binding assays, and can be used to obtain quantitative binding parameters for analytes ranging from small molecules (~100–200 Da) to entire cells. Indeed, SPR techniques have been used to investigate binding and interactions of proteins and protein conjugates, antibodies and antibody conjugates, nucleic acids, lipid micelles, viruses, nanoparticles (NP), and cells. Further, SPR provides data in real time and, in many cases, quantitative binding data can be generated in a matter of minutes. In this section, we provide a brief overview of SPR methodology and, where appropriate, we highlight some important concepts when considering the use of SPR for nanotherapeutics (nanoRx) binding analysis.
I.A. SPR basics – device overview
A schematic of a typical SPR unit is shown in Figure 1. Current systems generally consist of two pumps, an autosampler, a microfluidic system, a detector unit, and a flow cell that is formed partially by the introduction of an interchangeable sensor chip. Generally, a sensor chip that has been covalently-modified with a ligand of interest (see Section I.B. below for details) is docked to the machine and makes up one of the walls of the flow cell, such that buffer flow through the flow cell constantly runs across the surface of the docked chip. Analytes can then be injected into the flow cell either manually or automatically via the autosampler and a second pump system and subsequent binding events at the surface of the chip are measured by the detector as a change in refractive index at the surface of the chip. The change in refractive index is typically reported in “resonance units” or RU and this number is proportional to the mass concentration of the analyte bound at the surface of the chip. After a binding experiment, bound analyte is removed from the chip by a process called regeneration (see section I.D.). Typically, a single flow channel on a chip can be used for 50–100 runs with repeatable results, so long as a proper regeneration protocol and sensor chip storage procedures are in place.
Figure 1. Schematic of a typical SPR instrument.

Arrows indicate the direction of liquid flow through the system. IFC represents the integrated microfluidic cartridge.
I.B. The SPR sensor chip and chip preparation
A typical SPR chip consists of a glass slide that is coated with a thin layer of gold. The gold layer, in turn, is modified with various types of linker molecules and dextran depending on the type of chip/application and the desired surface chemistry required for ligand immobilization (Figure 2). A variety of chips are commercially available for specific SPR applications, with carboxymethyl-dextran chips (specifically CM5) being most popular. Addition of ligands to the surface of the chip is achieved via either (1) covalent modification or (2) high affinity capture. As covalent modification is the most commonly used method for ligand immobilization, we will focus on this method of ligand mobilization here. Information on high affinity capture as a means for ligand immobilization can be found elsewhere [1–2].
Figure 2. Schematic of an SPR sensor chip and chip surface.

Schematic shows a typical CM5 sensor chip that is surface modified with a carboxymethyl-dextran layer.
Covalent modification of the SPR chip surfaces is performed via irreversible chemical reaction of ligand with amine, thiol, or aldehyde groups found in linker layers. Of the various functional groups, amine groups are the most commonly exploited for covalent conjugation of carboxyl-containing ligands to the chip surface via simple EDC/NHS chemistry. However, care must be taken to ensure the selected immobilization chemistry preserves the biochemical activity of the immobilized ligand; therefore, if vetted immobilization strategies are not available for the ligand of interest, it may be appropriate to test several immobilization chemistries to ensure maximum preservation of ligand activity.
Aside from immobilization chemistry considerations, chip selection may be particularly important for nanoRx SPR studies due to the relatively larger size of nanoRx compared to more conventional SPR analytes. For instance, linker layers on SPR chips can be 10’s of nm thick, giving the surface of the chip a 3-D slab type geometry. Ligand can be immobilized throughout this 3-D slab with some immobilized ligands being 10’s of nm from the outermost part of the linker layer. Small conventional SPR analytes (e.g. proteins) are able to diffuse into the entangled carboxymethyl-dextran linker layer to reach the immobilized ligand in deeper layers, but larger analytes, like nanoRx, may be sterically limited from accessing all immobilized ligand. Tassa et al. observed much lower nanoRx binding (in RU) than expected from the Analyte Binding Capacity, when using a CM5 chip that utilized a carboxymethyl-dextran linker layer and hypothesized that this was due to a failure of nanoRx to penetrate fully into the linker layer and access all immobilized ligand [3]. To test this hypothesis, the authors examined the binding of nanoRx to two different chips with the same immobilized ligand. The chips were a CM5 chip and a C1 chip that lacks a dextran layer, thus providing a flatter, more 2-D like surface to eliminate issues with nanoRx penetration. The authors found that there was 15-fold more binding of nanoRx to the C1 chip than the CM5 chip, even though there was slightly less ligand immobilized to the C1 chip, supporting the hypothesis that more immobilized ligand was available to nanoRx with the C1 chip compared to the CM5 chip. However, the authors noted that more non-specific binding of nanoRx to the C1 chip was observed compared to the CM5 chip, likely due to the lack of dextran, which serves to passivate the CM5 chip surface from non-specific interactions.
In addition to ligand immobilization chemistry, the density of immobilized ligand on the chip surface is an important parameter that should be considered for experiments. In particular, it is important to immobilize sufficient ligand to ensure analyte binding results in measurable changes in RU. This is most important for low molecular weight analytes that contribute a relatively small mass increase at the chip surface for each binding event. Current SPR machines allow the user to set the level of immobilized ligand in RU during immobilization reaction procedures. Methods for selecting appropriate ligand immobilization densities can be found elsewhere [2]. An additional consideration in the case of nanoRx, the focus of this review, is the selection of ligand densities (e.g. in pmol/nm2) that correspond to physiologic densities on target cells and tissues. Using relevant ligand densities can give SPR experiments physiologic meaning in addition to providing a quantitative measure to assess nanoRx binding characteristics. This point is discussed further in Section III.C.
Another important consideration regarding ligand immobilization for nanoRx applications of SPR is related to mass transfer-limited SPR measurements. As nanoRx are relatively large molecules compared to traditional SPR analytes, they have a correspondingly small diffusion coefficient. If diffusion of nanoRx from the bulk fluid phase flowing through the flow cell across the stagnant fluid layer at the chip surface is slow, but the binding of the nanoRx once it reaches the chip surface is very fast, the kinetics measured by SPR will not be kinetic measurements related to binding/dissociation rate constants, but will be a measure of the diffusion of the nanoRx to the surface of the chip. However, this effect is limited to kinetic experiments, as mass transfer limitations do not have an effect on equilibrium constants. To reduce mass transfer limitations in SPR, low ligand immobilizations and higher flow rates of analyte through the flow cell should be used, as they serve to reduce the rate of binding and decrease the boundary layer for diffusion to the chip surface, respectively.
I.C. SPR experiments and sensorgrams, the optical measurement, and conversion to mass concentrations
With a properly designed SPR chip, SPR experiments may begin by flowing varying concentrations of the analyte of interest into the flow cell and across the surface of the sensor chip, where interactions with the immobilized ligand can occur. The analyte is typically delivered at a constant flow rate for a set period of time depending on the goal of the experiment. As analyte molecules diffuse to the surface of the chip and bind to immobilized ligand, the mass at the surface of the sensor chip will begin to increase. During the experiment, a monochromatic light source that illuminates the gold surface at various incident light angles and a CCD detector that measures reflected light are used to determine the incident angle at which a minimal light reflection occurs (θmin). The minimal light reflection occurs due to absorption of photons resulting in the creation of surface plasmons at the gold surface in direct contact with the flow cell. Surface plasmon properties are strongly influenced by the refractive index at the flow cell surface of the gold, such that as analyte binds, the refractive index at the surface changes, and θmin changes (surface plasmon creation).
SPR systems are exquisitely sensitive to changes in θmin and typically changes on the order of 0.1° are observed in SPR experiments. The “resonance units” or RU reported during SPR experiments represents this change in angle, with 1000 RU being equal to a 0.1° change in θmin. Changes in θmin can also be converted to mass densities of the ligand or analyte, though this conversion is dependent on the refractive index of the particular molecule [4–5]. For proteins, 1000 RU represents binding of ~1 ng/mm2 of protein onto the surface of the sensor chip [6]. During SPR experiments, data is displayed as “sensorgrams”, which show the RU as a function of time. A typical sensorgram for a single chain variable fragment of an anti-Fn14 monoclonal antibody binding to a chip coated with an immobilized Fn14 receptor extracellular fragment is shown in Figure 3 (unpublished data). The portion of the curve labeled “A” shows the baseline RU of the chip when bathed in running buffer. At point B, the analyte is injected into the flow cell and the flow rate held for 3 minutes during the portion of the curve labeled “C”. As can be seen in the figure, the RU increases due to binding of the single chain variable fragment monoclonal antibody (mAb) analyte to the chip surface. This is called the association phase of the sensorgram. At point D, the flow of analyte is stopped and running buffer is again constantly delivered through the flow cell for two minutes to allow observation of dissociation behavior. This is called the dissociation phase of the sensorgram (labeled “E”). During analysis, both the association and dissociation phases of the curve are used to extract binding parameters from the data (see Section I.E.1).
Figure 3. Example SPR sensorgram data displaying the main parts of the sensorgram.

Data shown was obtained by running serial dilutions of a single chain variable fragment anti-Fn14 antibody over the surface of a CM5 chip with immobilized recombinant Fn14 extracellular domain.. “A” marks the pre-injection baseline, “B” represents the beginning of the injection, “C” represents the association phase of the sensorgram, “D” represents the end of the injection, and “E” represents the dissociation phase of the sensorgram.
As the change in refractive index at the chip surface is the presumed reason for changes in RU in SPR experiments, other alterations to the experimental system that change surface refractive indices can cause difficulty in data analysis and extraction of incorrect kinetic or equilibrium parameters. The most common example is the use of a different buffer (with different refractive index from the running buffer) for injection of analyte into the flow cells. As the buffer is injected with analyte, it will replace the running buffer at the surface of the chip, causing a change in refractive index that is not due to analyte binding events. These buffer effects can be minimized by using a buffer that is as close to the running buffer as possible (ideally the same) and by running control experiments where buffer alone is injected into the flow cell and sensorgram data is collected and used as a “blank”.
Due to the complex physics involved in SPR measurements, we have limited our discussion in this section to the basic points to give the reader a general understanding of how SPR measurements are made. A more detailed explanation of the physics behind SPR measurements can be found elsewhere [7].
I.D. Regeneration of the SPR chip
After an SPR binding experiment, bound analyte must be removed from the chip to regenerate the immobilized ligand binding sites for future experiments. This regeneration step is typically performed by injection of an acidic solution into the flow cell. The change in pH disrupts the interactions between the analyte and immobilized ligand, promoting dissociation of the analyte from the chip surface. Some typical regeneration solutions include 10 mM glycine pH 1.5 – 3.0, and may include surfactants such as SDS or Tween 20. The regeneration method is highly dependent upon the analyte-ligand pair in question and for new analyte-ligand pairs, several regeneration conditions may need to be tested to ensure repeatable binding experiments following regeneration. For nanoRx SPR experiments, selection of an appropriate regeneration method is critically important. For classical, non-nanoRx analytes (e.g. a protein) used in SPR experiments, a single site on the analyte is able to bind the immobilized ligand. Thus, for dissociation of an individual analyte molecule from the chip surface, a single binding site interaction must dissociate. However, in the case of nanoRx that typically have many binding sites per nanoRx (e.g. a nanoRx decorated with 50 mAb against the immobilized ligand), multiple binding interactions must be disrupted simultaneously to release nanoRx from the chip surface to regenerate the chip for further use. Thus, chip regeneration for nanoRx analytes may require more extreme or harsh regeneration conditions to completely dissociate bound nanoRx analyte. Indeed, Stella et al. reported an inability to regenerate SPR chips with folate binding protein as the immobilized ligand and folate on the surface of poly(cyanoacrylate) NP [8]. The binding interactions were likely so strong and multivalent that strong regeneration solutions (3 M KSCN) were not able to adequately regenerate the chip. However, this is not the case for all nanoRx SPR experiments, as we recently showed effective regeneration of a CM5 SPR chip with the extracellular domain of the membrane protein Fn14 as the immobilized ligand and ITEM4 (an Fn14 mAb)-decorated densely PEG-coated polystyrene (PS) NP as the analyte using 100 mM phosphoric acid or 10 mM glycine, pH 2.5 as the regeneration solution [9].
I.E. Kinetic models and extractable parameters for SPR experiments
There are a variety of models available for extraction of kinetic and equilibrium parameters from SPR data and it is not the intent of this review to cover them all. In this subsection, we will provide a brief overview of a simple 1:1 binding model to give the reader an idea of how SPR sensorgram data can be used to determine relevant kinetic and equilibrium binding parameters. For a more complete review of SPR data analysis and models, please consult the following resources [1–2, 10–11]. Further, as mentioned above, in most cases nanoRx are capable of multivalent interactions, as each nanoRx has several binding sites on the surface that can each individually interact with a single immobilized ligand. Unfortunately, most models provided for SPR data analysis assume 1:1 or at most 2:1 (ligand:analyte) binding. Thus, parameters extracted from these fits, while still useful in making qualitative comparisons between nanoRx, should not be taken to be the true, quantitative parameters.
I.E.1. Kinetic analysis
Interaction kinetics are evaluated via SPR by running a series of experiments at varying analyte concentrations. Sensorgrams are generated on the same chip for various analyte concentrations and the RU vs. time data is then fitted to an appropriate mathematical model to globally extract kinetic parameters. The particular model used will depend upon the analyte-ligand in question and specific interaction mechanisms. It is important to note that just because data may exhibit a good “closeness of fit” for a particular model does not necessarily mean the binding obeys the mechanism defined by the model.
For illustrative purposes, kinetic analysis of SPR data assuming a 1:1 binding model is shown below for an analyte (A) and immobilized ligand (B) generating a bound complex (AB). For 1:1 binding, the following association/dissociation relationship would be observed.
With simple mass-action kinetics and appropriate derivations described elsewhere [2], one can express the change in the concentration of analyte bound to the chip for the association phase and the dissociation phase of the sensorgram completely in terms of RU and constants, where RU is the resonance units, t is time, ka is the association rate constant, kd is the dissociation rate constant, and “C” is simply a lumped constant.
| (1) |
| (2) |
With this equation, global numerical fitting of the RU vs time data from sensorgrams to extract ka and kd parameters is then performed.
I.E.2. Affinity analysis
There are several ways affinity constants (e.g. KA, KD) can be obtained for an analyte-ligand pair via SPR. First, if the binding mechanism is known and the data fit the corresponding mathematical model, the affinity constants can be derived directly from the kinetic constants as defined below:
| (3) |
| (4) |
Second, affinity constants can be derived from steady state data, where at each analyte concentration, the analyte is flowed over the chip for sufficient time to allow an equilibrium RU value (equilibrium binding) to be obtained (RUeq). From there, the RUeq vs Canalyte data can be fitted to an appropriate model equation to determine the KA and KD values. For example, for the case of 1:1 binding kinetics and assuming equilibrium binding at a particular analyte concentration, [A], is reached (at equilibrium ), the following equation results (see [2] for details of derivation):
| (5) |
From equation (5), using the RUeq vs. Canalyte data obtained from SPR experiments, numerical fitting can be performed to determine KA and then KD (simply as the inverse of KA).
As mentioned previously, most interaction models assume 1:1 or 2:1 binding and, thus, as far as we know, there is no defined method to get true, quantitative binding constants for nanoRx interaction with receptors. However, the interaction models and equations described above can be used to generate parameter values that can be useful in making qualitative comparisons between sets of nanoRx and this has been the approach of groups thus far utilizing SPR for analysis of targeted nanoRx. For example, Tassa et al. used a 1:1 kinetic binding model to determine KD values for a wide range of iron oxide NP with varying targeting ligands tethered to the surface [3]. Piletska et al. used calculated KD values to compare binding of silica NP with varied NP diameter and varied densities of surface biotin [12]. Recently, our group compared KD values of polystyrene NP decorated with varying amounts of mAb against Fn14 (ITEM-4) in comparison to ITEM-4 alone, using single class of sites model [9].
One other important note regarding analysis of nanoRx binding affinity analysis regards mass transfer limitations. As described above, the relatively large size of nanoRx compared to more classical SPR analytes means that nanoRx measurements may be affected by mass transfer limitations. Accordingly, there are mathematical methods to account for mass transfer limitations in fitting of SPR data, including some modules built directly into SPR software. Discussion of theses mathematical methods is beyond the scope of this review, but can be found in the literature [13–16].
II. SPR for design and characterization of non-particulate therapeutics
Although the goal of this review is to highlight the utility of SPR in studying nanoRx interactions, we feel a brief overview of non-nanoRx applications is appropriate, as many of the applications of SPR for non-nanoRx may be directly transferrable to nanoRx. As this review is focused primarily on the use of SPR in screening of nanoRx, we have focused this section on SPR screening of traditional drug candidates. However, we should note that SPR has been used extensively in studying interactions of various drug classes (small molecule, mAb, protein-drug conjugates, etc.) beyond screening experiments. While we provide a brief summary of some of these other drug classes (see Section II.4), additional information regarding these other applications can be found elsewhere [7, 17–23].
II.1. Primary screening of drug candidates for receptor binding
Due to advances in SPR technology over the past 15–20 years, the lower limit for the size of analytes for which binding can be reliably detected is now between 100 and 200 Da. These advances have opened the door for the use of SPR as a primary screening tool in identifying small molecule drug candidates, whereas in the past SPR was typically only used as a secondary test to confirm binding detected from a primary screen. The major advantages of using SPR as a primary screening methodology are that SPR (i) does not require any chemical modifications of the compounds to be screened, (ii) requires only a small amount of the compound, unlike more traditional assays that rely on fluorescence or radio-labeling, and (iii) the process can be fully automated. This could potentially save time, money, and reduce the possibility of altering the binding properties of screened compounds from labeling reactions. There are now SPR systems that have been designed specifically to facilitate high-throughput drug screening [21]. For example, the Biacore™ 4000 allows up to 16 targets to be screened at once, allowing 4800 compounds to be screened over a 24 hour period without user input after starting the protocol. Primary SPR screening has been used to screen a variety of small molecule compounds for many therapeutic applications including thrombin [24], HIV protease [25–26], and DNA gyrase [27] inhibitors.
II.2. Characterization of promising compounds from primary screen
While primary SPR screens are currently being utilized for drug discovery, the main role of SPR in drug screening and discovery to date has been as a secondary method to confirm the results of an alternate primary screen and to provide quantitative information about binding behaviors of positive binders. For example, Lan et al. used SPR to confirm the binding of a colon cancer candidate drug, (−)-glossypol, that was originally discovered from a fluorescence polarization assay designed to identify molecules that prevented binding of an RNA-binding protein (Murashi-1 or MSI-1) involved in colon cancer to its target RNA sequence [28]. For SPR experiments, the RNA binding domain of the MSI-1 protein was first immobilized to a CM5 chip and the binding of varying concentrations of (−)-glossypol was tested. The authors confirmed that (−)-glossypol did indeed bind to the RNA-binding domain of MSI-1 and that binding was dose dependent. Thus, SPR was used to confirm the original screening result and provide proof of a direct interaction between the candidate drug (−)-glossypol and the MSI-1 protein’s RNA binding domain. Navratilova et al. used SPR as a secondary screen to determine binding properties of drug candidates that were identified in a primary high-throughput enzymatic screen of a tyrosine kinase (IL-2 inducible T-cell kinase or ITK). The authors used SPR to validate binding of drug candidates to ITK (immobilized to SPR chip), to determine quantitative binding parameters (both kinetic and affinity), and to extract information about binding mechanisms for each drug candidate [29].
The quantitative binding data from secondary SPR testing can give insights into binding mechanisms and provide the links between these interactions and the chemical structure of the candidate small molecules drugs (i.e. structure-activity relationships [25, 30]). This information can then be used to optimize the chemistry of small molecule drug candidates to generate desired binding characteristics. Markgren et al. used SPR to characterize binding of 58 previously identified transition state analogue inhibitors of HIV protease 1 [30]. Both affinity (KD) and kinetic (ka and kd) binding parameters were determined by flowing candidate inhibitors over an SPR chip with immobilized HIV protease 1. Comparison of binding parameters with chemical structures of the inhibitors revealed structure-binding relationships between distinct structural classes of inhibitors, whereby specific structural features were associated with either an increase or decrease in ka or kd. This suggests that intelligent modification of drug candidates may be guided by the use of structure-binding relationship data from SPR to generate optimized drug candidates with specific binding characteristics.
II.3. Screening candidates for serum protein binding and membrane interactions
In addition to evaluating desired binding of analyte to immobilized drug targets, SPR has also been used to look at off-target binding of candidate drugs (including mAb drugs) by serum proteins [31–35]. Binding of drugs to serum proteins, such as albumin and others, is extremely important in determining the pharmacokinetic profile of candidate drugs and thus the in vivo efficacy and side effect profiles [36]. Even positive drug candidates with high specificity and high affinity for targets may be wholly ineffective in vivo if they have unfavorable interactions with serum proteins and poor plasma pharmacokinetics. Using purified plasma proteins immobilized to SPR chips, binding of candidate drug to plasma protein constituents has been evaluated and has been found to correlate well with other established methods that are used for evaluating drug-plasma protein interactions [36]. The advantage of SPR in this regard is that it is relatively high-throughput and requires low sample volume, compared to more traditional assays. Graham et al. used SPR to study the interactions between a small molecule drug, GDC-0449, with plasma proteins in order to explain the unusual pharmacokinetics that were observed during a Phase I clinical trial for patients with refractory solid tumors [35]. The binding affinity of GDC-0449 for human serum albumin (HSA) and alpha-1-acid glycoprotein (AAG) were determined by flowing GDC-0449 at various concentrations over SPR chips with immobilized HSA or AAG. This study revealed that the binding affinity of GDC-0449 was much greater for AAG (KD = 13 μM) than for HSA (120 μM). This was in good agreement with analysis of plasma samples of patients receiving GDC-0449, where GDC-0449 and AAG plasma levels were closely correlated and little free GDC-0449 was observed. Further, a pharmacokinetics simulation using binding data from SPR suggested that the abnormal pharmacokinetics behavior of GDC-0449 was indeed due to relatively strong molecular interactions with AAG.
In addition to screening candidate drugs for binding to serum proteins, SPR has been used for studying candidate drug/membrane interactions, as a surrogate for drug interactions with membranes in the gastrointestinal tract and in endothelial cell membranes at the blood-brain barrier [37–40]. For example, Danelian et al. utilized SPR to study the interaction of 27 small molecule drugs with liposomes (as a membrane surrogate) that were immobilized to the surface of an L1 sensor chip, as a high-throughput surrogate for intestinal absorption after oral administration [39]. Interestingly, the amount of binding of drugs to liposomes correlated well with the known GI absorption fraction in humans. These results suggest that SPR studies of membrane interactions may be useful as a secondary screening method for selecting drugs with high GI absorption and bioavailability.
II.4. Screening of therapeutic antibodies and antibody-toxins
Because of its versatility, SPR technology has proved to be a useful tool in assessing a variety of non-nanoRx drug candidates. Here we highlight a few examples of other therapeutic applications for SPR where it has enabled streamlining and optimization of the therapeutic development process.
The use of various antibody conjugates and therapeutic antibodies is a burgeoning field due to the high specificity and the unique mechanisms of action these types of therapies offer. SPR has played a key role in many studies as a method of validation when designing such constructs. An example comes from the development of antibody-based therapies surrounding the TWEAK-Fn14 cytokine-receptor axis, which has been implicated in various disease processes and has been identified as a potential drug target [41]. Initial studies of the interaction between the TWEAK ligand and the Fn14 cell surface receptor were conducted by Brown et al. using SPR [42]. The TWEAK ligand was immobilized on a sensor chip after which Fn14 wild-type or mutant proteins were flowed across the chip and KD, ka, and kd measurements were obtained to determine the major regions of the protein responsible for ligand binding. More recently, Zhou et al. employed SPR assays to evaluate the binding of various Fn14 mAb-toxin chemical conjugates or fusion proteins to immobilized Fn14 extracellular domain [43–45]. Following confirmation of high affinity binding using the SPR technique, the Fn14-targeted proteins were tested for cellular uptake and cytotoxic activity on cancer cells both in vitro and in vivo after subcutaneous implantation in immunodeficient mice.
Another major type of antibody conjugate therapy involves the conjugation of a small molecule drug to an antibody (aka antibody-drug conjugates or ADC). As with antibody-toxins, SPR has contributed to ADC development. There are many examples of SPR use in the context of these conjugates, which can be found elsewhere [46–49]. Here, we highlight the SPR characterization of the anti-cancer ADC inotuzumab ozogamicin (CMC-544), which consists of a cytotoxic drug, calicheamicin, conjugated to a mAb targeting CD33, a receptor that is aberrantly expressed in malignant blast cells. Boghaert et al. determined the pharmacokinetic values and confirmed the functional properties of CMC-544 using an SPR method known as sandwich plasmon resonance [47]. With this method, the CD33 target ligand was immobilized on a sensor chip, and then, diluted plasma samples of mice treated with CMC-544 were flowed across the chip to measure the amount of antibody-drug conjugate in plasma circulation. Next, calicheamicin-specific mAbs were flowed across the chip, creating a ligand-drug conjugate-antibody “sandwich” to measure the calicheamicin bound on the conjugate and in effect the stability of the conjugate in vivo. From these data, the amount of conjugate present in plasma was determined by interpolation of the RU values from a calibration curve. Such use of SPR technology demonstrates its potential to assess pharmacokinetic properties and antibody-drug conjugate stability as a high-throughput proxy for preclinical therapeutic efficacy and systemic toxicity studies.
Similarly, therapeutic antibodies have been widely evaluated using SPR technology. Maurer et al. employed SPR to aid in the generation and characterization of mAbs that can neutralize the human IL-21 protein to mitigate inflammation and autoimmune disease symptoms [50]. In this study, SPR was used to assess the binding kinetics of the neutralizing antibodies as well as their effectiveness in neutralizing IL-21. First, the anti-IL-21 antibodies were immobilized on a sensor chip using an antibody capture surface, and dilutions of IL-21 were flowed over the SPR chip. Binding affinity was assessed by comparing the antibody association and dissociation constants. In order to characterize the competitive binding interaction between the neutralizing antibodies and IL-21, another SPR assay was performed in which IL-21 was flowed over the neutralizing antibody coated chip. Then, anti-IL-21 antibodies were run over the chip to identify the competitive binding capabilities of the neutralizing antibodies immobilized on the chip. It was observed that the binding kinetics results produced by SPR correlated with the in vivo functional assay of neutralization. In effect, the authors categorized the neutralizing mAbs based on functionality and binding using SPR methodology. These findings underline the utility of SPR-based evaluations of therapeutic interactions and mechanisms of antibodies.
II.5. QA/QC – use of SPR as a quality control mechanism
Quality Assurance (QA) and Quality Control (QC) are important checkpoints that ensure manufactured pharmaceuticals are consistent, safe, and effective from batch to batch. Accordingly, pharmaceutical manufacturers are required to be compliant with laws regarding Good Laboratory Practice (GLP) and Good Manufacturing Practice (GMP). SPR is a suitable technique for QA/QC of pharmaceuticals as it can be used to verify binding, rule out microbial contamination, and verify concentrations of batches of manufactured drugs/therapeutics quickly without the need for more cumbersome biological assays [51–57]. SPR poses several advantages over more traditional techniques such as ELISA and liquid chromatography as it provides rapid results in real time, requires little manual sample manipulation, and is highly accurate and precise [51–52]. The utility of SPR in QA/QC applications is evidenced by the fact that there are now specific SPR machines available that are dedicated for use in quality control applications (e.g. Biacore™ C).
III. SPR studies of nanotherapeutics (nanoRx)
III.A. SPR to confirm biological activity of targeting moieties following surface chemistry and nanoRx formulation
One of the most simple and straightforward applications of SPR technology for nanoRx evaluation is the use of SPR as a rapid test to confirm successful chemical coupling of the targeting moiety to the surface of nanoRX. This is particularly important for targeted nanoRX to ensure that the chemical conjugation technique did not ablate the binding activity of the targeting moiety. This strategy has been used in a variety of nanoRx targeting studies to validate nanoRx binding before more time consuming cell culture and animal model studies. For example, Wang et al. [58] used SPR to confirm the biological activity of IgG conjugated to the surface of multi-walled carbon nanotubes (MWCNT). The authors first immobilized streptavidin to a CM5 sensor chip and then introduced the biotinylated antigen (corresponding to the IgG on the MWCNT) to generate a sensor chip with a RU value of 750 for binding experiments. Then, injections of various IgG functionalized MWCNT were used to determine if conjugated IgG retained biological activity, prior to in vivo SPECT imaging study to determine biodistribution following IV injection. SPR revealed that all IgG-conjugated MWCNT bound specifically to antigen-immobilized chips and that binding was concentration-dependent.
Salvati and coworkers used SPR to compare the merits of two different conjugation strategies for preparing transferrin-targeted liposomes [59]. They utilized SPR to evaluate the binding affinity of two different liposomes that were synthesized by conjugating an anti-transferrin antibody (RI7217) via either streptavidin/biotin or maleimide/thiol chemistry. The overall goal was to synthesize optimal transferrin-targeted liposomes to facilitate crossing of the blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. Using a transferrin immobilized sensor chip, the binding of the two different targeted liposomes was evaluated with a single binding site isotherm model and revealed that targeted liposomes made via maleimide/thiol chemistry exhibited more than 4-fold greater binding affinity compared to targeted liposomes made via the streptavidin/biotin method. The results were in close agreement with in vitro experiments where targeted liposomes made via maleimide/thiol chemistry were taken up more readily by transferrin-expressing cells and were able to penetrate more efficiently across an in vitro BBB model.
Patil et al. used SPR to confirm the successful surface partitioning and activity of two separate targeting moieties, folic acid and biotin, on the surface of poly(lactic-co-glycolic acid)-co-polyethylene glycol (PLGA-PEG) NP [60]. Prior to NP formation, folic acid or biotin were chemically conjugated to the hydrophilic PEG portion of a PLGA-PEG diblock copolymer. NP were then formulated by a single emulsion/evaporation method where the hydrophobic PLGA block partitions to the core of the NP and the hydrophilic PEG block partitions to the surface of the NP (in contact with aqueous phase). After particle formation, SPR binding experiments were performed with either a streptavidin coated sensor chip or a sensor chip with immobilized anti-folate antibody to determine whether (1) biotin and folate partitioned to the surface (and not the core) of the NP and (2) whether biotin and folate molecules remained biologically active after the NP preparation process (which involved exposure to organic solvent and high intensity sonication). A single concentration of various NP was used to qualitatively compare NP binding and the authors found that specific binding was observed for both biotin-modified and folate-modified NP. Thus, SPR validated that the polymer chemistry and particle formulation strategies preserved the activity of targeting moieties on the surface of the NP, which provide a versatile method to incorporate multiple targeting ligands into a single PLGA-PEG NP.
Kocbek et al. used SPR to examine two types of mAb coated immunonanoparticles (immunoNP) for targeting invasive epithelial breast tumor cells [61]. Two different PLGA NP were surface modified by either adsorption or covalent conjugation (via EDC catalysis) of a mAb that targets specific cytokeratins on the surface of breast tumor cells. The authors used a streptavidin coated sensor chip to immobilize biotinylated protein A to the chip surface, which specifically binds to the Fc region of IgG molecules (such as the mAb used in this study). After injection of a single concentration of NP, the authors found that both NPs made by adsorption and covalent methods bound specifically to the chip surface, though the binding of the covalently-conjugated NP was much lower (in RU). The reason for the reduced activity of the covalently-conjugated NP was not known. The authors speculated it may be due to (1) lack of a linker molecule between the NP surface and the mAb (e.g. PEG) leading to reduced availability of the mAb to the target, (2) significant self-polymerization of the mAb via amide linkage in place of amide linkage with the NP (as a separate EDC/NHS activation step was not performed separate from the coupling portion of the reaction), or (3) the presence of an important NH2 group in the antigen binding domain that was consumed during amide bond formation. Thus, the authors concluded that the EDC-mediated covalent linkage was not effective for formulation of the specific mAb-decorated immunoNP.
Debotton et al. conjugated an anti-H-ferritin antibody (AMB8LK) to the surface of biodegradable PEG-PLA NP via maleimide-thiol chemistry to generate immunoNP for site-specific therapy of pancreatic and lung tumors overexpressing H-ferritin [62]. In their experiments, the authors compared the binding affinity of native AMB8LK antibody to the binding affinity of immunoNP, which contained ~76 AMB8LK molecules per NP. However, unlike most other SPR experiments with NP, the authors immobilized either the AMB8LK antibody or the immunoNP onto the surface of a CM5 sensor chip and evaluated the binding of H-ferritin at different concentrations to the sensor chip surface. Both the shapes and the extracted kinetic and affinity parameters were very similar between the AMB8LK chip surface and the immunoNP chip surface experiments, suggesting that the conjugation of the AMB8LK antibody to the surface of the NP did not alter the binding affinity of AMB8LK. (ka = 3.45 × 104 M−1s−1, kd = 1.53 × 10−4 s−1, KD = 6.7 nM and ka = 3.3 × 104 M−1s−1, kd = 1.27 × 10−4 s−1, KD = 8.2 nM, respectively). Therefore, the maleimide-thiol chemistry was an effective coupling mechanism for generating immunoNP with AMB8LK. To confirm that binding was specific to H-ferritin, transferrin was used as the negative control analyte and no binding was observed to either sensor chip surface.
III.B. SPR studies of various nano-sized therapeutics
In addition to using SPR as a screening tool to ensure coupling and activity of targeting moieties, numerous groups have used SPR to evaluate and optimize the binding properties of targeted nanoRx. As a variety of different nanoRx have been studied with SPR, the following subsections give a brief review of the SPR findings from experiments on dendrimers (III.B.1.), micelles, (III.B.2), liposomal and lipid NP (III.B.3), and polymeric NP (III.B.4.).
III.B.1. Dendrimers
Dendrimers are a subset of nanoRx that have been relatively well-studied by SPR due to the availability of monodisperse commercially available dendrimers (e.g. PAMAM dendrimers) and the ease of control over targeting ligand density per dendrimer, which can be controlled by changing the dendrimer generation number. As such, dendrimers have been extensively used to look at the effects of multivalent binding and avidity (discussed in section IV). In this section, we focus briefly on several application based studies utilizing SPR to characterize therapeutic dendrimers.
Wada et al. used SPR to confirm the binding of mannosylated, cyclodextrin-conjugated PAMAM dendrimer gene carriers (Man-α-CDE/pDNA) to lectins [63]. In their experiments, the authors immobilized the lectin concavilin A to the surface of the sensor chip and compared the binding of Man-α-CDE/pDNA to cyclodextrin-conjugated PAMAM dendrimer gene carriers (α-CDE/pDNA) and PAMAM dendrimer gene carrier alone (PAMAM/pDNA). The calculated binding affinity of Man-α-CDE/pDNA was 25- and 470-fold higher than that calculated for α-CDE/pDNA and PAMAM/pDNA, respectively, confirming the lectin-specific binding activity of Man-α-CDE/pDNA.
Thomas et al. used SPR to evaluate the binding activity of a methotrexate (MTX) conjugated polyvalent dendrimers to folic acid receptors (Figure 4A, [64]). The generation 5 dendrimers (G5) with either 5 or 10 MTX molecules conjugated by amide-linkages (G5-MTX5 or G5-MTX10), were tested at varying concentrations via flowing over a folate binding protein (FBP) immobilized CM5 chip and using kinetic analysis to determine ka, kd, and KD. Both G5-MTX5 and G5-MTX10 bound strongly to the chip surface, displaying 857- and 4,360-fold greater binding affinity compared to free MTX (KD of 2.4 × 10−5 M, Figure 4B). No binding to the chip was observed for control dendrimers (no MTX, G5-MTX0, Figure 4C). Specific binding of G5-MTX5 was confirmed in cell uptake studies, where G5-MTX5 was strongly associated with folate receptor expressing KB cells, but not folate receptor negative B16-F10 cells (Figure 4D). The authors found that a lower dissociation rate constant, kd, was the major contributor for the enhanced binding affinity (KD) observed upon increasing MTX valence from 5 to 10. The effect of multivalent interactions on kd and therefore KD has been widely observed and is discussed in more detail in Section IV.A. More recently, Varga et al. used SPR to examine the effectiveness of a set of synthetic carbohydrate dendrimers in blocking the association (and resulting infection) of HIV-1 and Dengue virus with dendritic cells via a specific C-type lectin receptor (DC-SIGN) [65]. In these SPR experiments, mannosylated-bovine serum albumin (man-BSA) was immobilized to the surface of a CM4 sensor chip. As DC-SIGN naturally will bind man-BSA, IC50 values for various carbohydrate dendrimers were determined by co-injecting various concentrations of the dendrimers with a set amount of the extracellular domain of DC-SIGN (ECD-DC-SIGN, 24 μM) and observing the residual activity of ECD-DC-SIGN (in RU) in the presence of these inhibitors. The authors tested a variety of dendrimer scaffold and carbohydrate combinations and generally observed an increase in activity (as a reduced IC50) as the valency of the dendrimer was increased. Based on these SPR experiments, the most effective dendrimer-based inhibitors (on the basis of the lowest IC50) were tested further for the ability to block infection of HIV-1 or Dengue virus in CD4+ T cells from healthy donors in vitro. In excellent agreement with the SPR experiments, the most potent dendrimer-based inhibitor (dendrimer 13.4) was the most efficacious, completely blocking virus infection (0% infected cells) at a concentration of 10 μM, compared to only a 30–40% reduction in infection with previously studied tetramer dendrimers (~60–70% infected cells for dendrimers 1.2 and 11.3).
Figure 4. Use of SPR to optimize formulation chemistry of a dendrimeric nanotherapeutic.

SPR is used to confirm the proposed advantages (A) of a methotrexate-dendrimer, G5-MTXn compared to free methotrexate. SPR binding measurements (B, C) revealed that conjugation of Methotrexate to the G5 dendrimer yielded targeted dendrimers with greatly enhanced binding to a folate binding protein immobilized sensor chip compared to a control dendrimer lacking methotrexate (C). The enhanced binding seen in SPR experiments was confirmed in cell-uptake studies (D). Left panel shows cells not expressing folate receptor and the right panel shows cells expressing the folate receptor. Methotrexate-dendrimer conjugate is localized to only the cells positive for the folate receptor. Figure adapted from ref. [64]
III.B.2. Micelles
Micelles are a promising nanoRx platform that form into nanostructures by a self assembly process and there are a few formulations, such as NC-6004 (nanoplatin) from Nanocarrier Co., Ltd, currently undergoing evaluation in clinical trials. Several studies have used SPR to evaluate the binding behavior of various targeted micelle therapeutics [66–70].
Bae et al. [70] designed a multifunctional polymer-based micelle from an ampiphilic block copolymer, folate–poly(ethylene glycol)–poly(aspartate hydrazone adriamycin) [Fol–PEG–P(Asp-Hyd-ADR)], for targeting the chemotherapeutic drug adriamycin (doxorubicin) to folate binding protein (FBP)-overexpressing tumor cells. The authors used SPR to validate binding of the folate-modified micelles (FMA) compared to unmodified micelles (MA) to FBP immobilized to the sensor chip surface. FMA bound strongly to the FBP coated sensor chip with an RU value of more than 16,000, whereas no binding was observed for MA. When the authors added free folic acid to FMA prior to injection, competitive inhibition was observed, proving the FMA binding was due to a specific interaction between folate functionalized on the surface of FMA and FBP on the sensor chip. The SPR results agreed with subsequent in vitro cell uptake studies where FMA were endocytosed by cancer cells more efficiently than MA.
Schaeffer et al. [68] formulated micelles from mannoside glycolipid conjugates (MGC) in an attempt to increase the inhibitory effect of single molecule MGC in the inhibition of HIV1 transfection of dendritic cells (DC). MGC work by competitive inhibition of the HIV1 interaction with the DC surface protein, DC-specific ICAM3-grabbing nonintegrin (DC-SIGN), thereby inhibiting subsequent transfection of DC by HIV1. The authors immobilized the carbohydrate recognition domain of recombinant DC-SIGN to the surface of a CM5 sensor chip and found that synthesized multivalent micelles of MGC bound more avidly (with lower KD) compared to single molecule MGC or rigid particles made from polymerized MGC. This result was confirmed in in vitro DC infection assays, where micelles were most effective in blocking HIV1 infection of DC. The experiments demonstrated enhanced inhibition of HIV1 by multivalent inhibitors and suggested an important role in maintaining dynamic MGC in micelles and other multivalent inhibitors, as rigid particles derived from polymerized MGC were not efficacious.
Ezzat et al. [66] used SPR to study the binding of antisense oligonucleotides (ASO) micelles to the class A scavenger receptor subtype 1 protein (SCARA1) in order to confirm that SCARA1 was responsible for ASO micelle uptake in cells. The authors studied a variety of modified AOS that spontaneously self-assembled into micelles with average size less than 200 nm. For SPR studies, the authors immobilized SCARA1 with his-tag to a CM5 chip that had previously been covalently modified with an anti-his-tag antibody (via amine chemistry). Then, they flowed various ASO micelles over the surface of the sensor chip and measured the maximum RU max for each ASO micelle at a variety of concentrations to demonstrate direct binding between micelles and SCARA1. The authors confirmed direct binding of all ASO micelles to SCARA1 and found that ASO micelles that more readily form stable nanostructures (100–200 nm) bound more readily to SCARA1 on the SPR chip. Direct binding data from SPR was supported by immunocytochemical analysis of cells that revealed colocalization of anti-SCARA1 antibody with all ASO micelles tested.
III.B.3. Liposomes and lipid based NP
Liposomes and lipid based NP are some of the most widely used and studied nanoRx, with formulations such as Doxil® already in clinical use. However, there have been relatively few SPR studies of these versatile nanoRx. Okamoto et al. used SPR to confirm the binding of siRNA loaded lipid NP (LNP) that were surface functionalized with an Fab′ fragment of an antibody against the protein heparin-binding epidermal growth factor-like growth factor (HB-EGF) to a HB-EGF immobilized sensor chip for the purpose of targeting therapeutic siRNA to solid tumors expressing HB-EGF [71]. The authors compared the binding of NP bearing the anti-HB-EGF Fab′ fragment that was linked to the LNP via a DSPE-PEG linker (LNP-PEG-HB-EGF) to two types of control LNPs, LNP without PEG or anti-HB-EGF Fab′ fragment (cLNP) and LNP with PEG, but without anti-HB-EGF Fab′ fragment (cLNP-PEG). From injection of a single concentration of NP, the authors found extremely strong binding of LNP-PEG-HB-EGF (RU value of nearly 12,000), whereas there was essentially no binding for cLNP-PEG and cLNP. These results correlated well with in vitro experiments that showed greater association and uptake of LNP-PEG-HB-EGF in HB-EGF expressing cells compared to cLNP.
In more mechanistic studies, Guo et al. formulated RGD decorated liposomes with varying surface spacing of RGD by using various linker strategies to investigate the optimal RGD spacing for avid binding to αVβ3 integrins [72]. Monomeric RGD (cRGD), dimeric RGD (diRGD), and flexible dimeric RGD (P-diRGD) decorated liposomes were synthesized and the binding to B16 melanoma cells or MCF-7 breast cancer cells was studied via SPR. In this case, cells were immobilized to the sensor chip via their extracellular membrane proteins using EDC/NHS chemistry (to 300 RU). The authors found that in the B16 cells, which express high levels of αVβ3 integrins, P-diRGD decorated liposomes bound most strongly to the B16 immobilized chip. In contrast, there was no significant difference in binding of the various RGD decorated liposomes to MCF-7 cells, which express very low levels of αVβ3 integrins, as all three liposomes bound poorly to the MCF-7 chip. Similar results were observed in in vitro cell uptake studies in B16 cells and in in vivo tumor targeting studies. The authors rationalized through computational models that the intermediate spacing of RGD molecules in the P-diRGD liposomes (compared to more tight spacing in diRGD and more spread out spacing in cRGD) allows the maximal number of RGD molecules to interact with αVβ3 integrins during receptor clustering. For example, the spacing between RGD molecules on the P-diRGD liposomes (~43Ǻ) and clustered αVβ3 integrin receptors on B16 cells (~41Ǻ to 66Ǻ) were most similar. Consideration of the relative spacing between targeting moieties on the particle surface and corresponding receptors on the target cell is an emerging topic in the field and is discussed further in Section IV.C.
III.B.4. Polymeric NP
Biodegradable polymeric NP such as PLGA NP have been studied extensively in relation to sustained drug release and enhanced therapeutic to toxicity ratio. Despite numerous papers dedicated to targeting studies of therapeutic polymeric NP, there have been only a few studies that utilized SPR to investigate target binding. Hrkach et al. used SPR to confirm the binding of a small molecule PSMA inhibitor (ACUPA) on the surface of self-assembled PLA-PEG NP to PSMA for targeted prostate cancer therapy [73]. A NTA (nitrilotriacetic acid) sensor chip was used to immobilize his-tag labeled PSMA by sequential addition of a nickel solution (NTA on the sensor chip binds Ni via chelation) and then His-tag PSMA to the sensor chip surface (Ni/NTA on sensor chip binds His-tag). After confirmation of 1000 RU of immobilized PSMA, the authors injected self-assembled NP made from either 100% PLA-PEG polymers (PLA-PEG NP) or a mixture of PLA-PEG and PLA-PEG-ACUPA polymers (PLA-PEG-ACUPA NP). The authors confirmed that PLA-PEG-ACUPA NP bound strongly to the PSMA immobilized chip surface, whereas PLA-PEG NP exhibited no binding. The experiments served to confirm that ACUPA molecules localized to the surface, not the core, of self-assembled NP and that binding activity was preserved following conjugation to PLA-PEG polymer and NP assembly. These PLA-PEG-ACUPA NP are currently in Phase II clinical trials for non-small cell lung cancer and prostate cancer therapy (ClinicalTrials.gov identifier: NCT01792479 and NCT01812746).
Stella et al. examined the binding affinity of folic acid conjugated poly[aminopoly(ethylene glycol) cyanoacrylate-co-hexadecyl cyanoacrylate] (poly(H2NPEGCA-co-HDCA)) NP to folate binding protein (FBP) to evaluate the potential for targeting tumors that overexpress folate receptors [8]. Using an FBP immobilized sensor chip, the authors compared the binding characteristics of folic acid-conjugated poly(H2NPEGCA-co-HDCA) NP to non-conjugated control NP and to free folic acid. Using the dissociation phase of the sensorgram data, dissociation constants (kd) were determined for the various analytes tested. As expected, folic acid-conjugated poly(H2NPEGCA-co-HDCA) NP bound more strongly to the FBP-immobilized sensor chip compared to non-conjugated control NP (although significant binding of control NP was observed). In an attempt to evaluate specific vs. non-specific binding of folic acid-conjugated poly(H2NPEGCA-co-HDCA) NP, the authors performed competitive binding experiments in the presence of 50 μM free folic acid (as a competitive binder). Interestingly, no significant difference in NP binding was observed between 0 μM and 50 μM free folic acid. The authors rationalized this result by citing the measured kd values of folic acid-conjugated poly(H2NPEGCA-co-HDCA) NP, obtained from fitting the dissociation phases of the various sensorgrams, which was 108-fold lower than that of free folic acid.
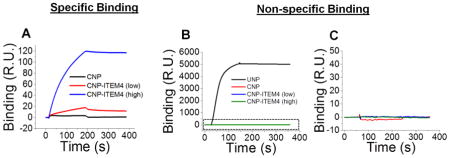
More recently, Schneider et al. used SPR to design PEG-coated, mAb-decorated polystyrene NP that were capable of specifically binding to a membrane protein (Fn14) that is overexpressed on glioblastoma cells (Figure 5A, [9]). The authors used an CM5 SPR chip coated with the Fn14 extracellular domain to assess and quantify the binding of NP with various surface densities of an antibody (ITEM4) known to bind this domain. Using a single class of sites model, the authors found that PEG-coated, ITEM4 functionalized polystyrene NP (CNP-ITEM) exhibited stronger binding affinity compared to free ITEM alone and that binding affinity increased with increasing surface density of ITEM4. Further, these binding data were in agreement with in vitro experiments using Fn14-positive U87 glioblastoma cells that showed increased uptake for NP with higher ITEM4 surface densities [9].
Figure 5. SPR evaluation of specific and nonspecific interactions of a targeted nanoparticles.

(A) SPR study of specific binding of PEG-coated polystyrene NP with no ITEM4 antibody (CNP, black), a low density of ITEM4 (CNP-ITEM4 (low), red), and a high density of ITEM4 (CNP-ITEM4 (high), blue) to a CM5 chip with immobilized extracellular domain of Fn14 protein. (B) SPR study of non-specific binding of uncoated NP (no PEG, UNP, black), CNP, CNP-ITEM4 (low), and CNP-ITEM4 (high) to a CM5 chip with immobilized mouse brain extracellular matrix proteins. (C) Zoomed view of dotted box in (B). Figure adapted from ref. [9]
IV. Fundamental understanding of nanoRx binding and surface interactions
As shown in Section III above, SPR is extremely useful in confirming activity of targeted nanoRx, evaluating particle formulation strategies, and screening and optimizing targeted nanoRx. However, SPR has also been used to investigate fundamental aspects of interactions between nanoRx and biologic surfaces. McGurk and colleagues were one of the first groups to propose the use of SPR for the screening and optimization of nanoRx in place of more complicated and cumbersome in vitro cell studies or in vivo animal experiments [74]. Despite less sophisticated instrumentation compared to current SPR experiments, the authors demonstrated the utility of SPR in studying both specific (Section IV.A.) and non-specific (Section IV.B.) interactions of nanoRx with biologically modified surfaces. In these early studies, the authors coated the surface of model PS NP with F127 surfactant or BSA and used SPR to evaluate specific and non-specific binding events to sensor chips coated with various proteins. F127, a PEO-PPO-PEO triblock copolymer, was used to prevent non-specific interactions between the PS NP surface and a protein coated chip, as the authors observed a high level of non-specific binding with uncoated PS NP (with a very hydrophobic surface) to a BSA-coated chip. Non-specific binding was greatly reduced by adsorption of F127 to the surface of PS NP. Specific binding of BSA-coated NP was then evaluated by looking at the binding of BSA-coated NP to a BSA-coated chip or a chip coated with a mAb to BSA. Binding of BSA-coated NP was only observed on the mAb coated chip, confirming the specificity of the interaction. In the following sections, we highlight more fundamental studies of nanoRx interactions with surfaces, including specific nanoRx interaction and the effect of multivalent interactions (Section IV.A.), non-specific nanoRx interactions (Section IV.B.), and the effects of intermolecular distance in multivalent binding (Section IV.C.).
IV. A. SPR evaluation of the effects of multivalent interactions on binding behavior
The vast majority of mechanistic SPR studies of nanoRx binding have been focused on the effects of multivalent interactions and the concept of avidity. Many studies, including some cited above, discovered that the calculated binding affinity of targeted nanoRx tends to increase (lower KD) with increased nanoRx valency (# of targeting molecules per nanoRx). To better understand this phenomenon, many groups have used SPR to study the change in kinetic and affinity parameters of various nanostructures, including dendrimers and polymeric NP, with varied valency.
Hong et al. studied the effect of increasing folate valency on G5-acetamide terminated dendrimers on the binding affinity to folate binding protein [75]. Folate-conjugated dendrimers exhibited enhanced binding affinity compared to free folate, with small increases in valency resulting in large changes in binding affinity compared to free folic acid. For example, G5 dendrimers with a valency of 2.6 had a calculated binding affinity (KD) of 2 nM, which was 2500-fold lower compared to the binding affinity calculated for free folic acid, 5 μM. Further increasing valency from 2.6 result in further increases in binding affinity (lower KD); however, the authors observed a plateau of binding affinity (KD) at ~0.07 nM once a valency of ~5 was achieved. In vitro cell binding studies in folate receptor expressing KB cells verified these results as the calculated KD values from SPR correlated with mean intensity of FL dendrimers on KB cells. Cell binding data exhibited the same plateau type behavior at valency above 5, as was seen in SPR experiments. Kinetic analysis of SPR data was performed to determine ka and kd values for the various folate-functionalized dendrimers. The authors found that the association rate constant, ka, increased linearly with folate valency, whereas the dissociation rate constant, kd, was found to decrease exponentially with folate valency. Thus, the effect of multivalency on dissociation rate constant was the main driving force behind the observed plateau behavior of the binding affinity with increasing valency.
In a series of comprehensive experiments, Tassa et al. used SPR to mechanistically study the effect of multivalent targeting ligands on the surface of NP on NP avidity and binding affinity to a single protein target, FK506-binding protein 12 (FKBP12), that was immobilized to the surface of a CM5 chip [3]. FKBP12 was chosen for their study due to the well characterized binding to natural small molecule FK506, and the availability of a variety of FK506 variants with binding affinities that spanned more than 3 orders of magnitude, from 24 nM to 110 μM. Thus, this allows for testing the effect of varying targeting ligand binding affinity (KD, sm) on NP binding affinity (KD, NP). The authors chemically conjugated a variety of FK506 variants to the surface of dextranated magnetic NP (cross linked iron oxide, d = 38 nm) at either low (3–5 molecules per NP) or high (13–20 per NP) density. In early experiments, FKBP12-targeted NP bound so strongly and avidly to the surface of the sensor chip that no dissociation was observed even with a dissociation phase of 12 minutes. In order to be able to measure dissociation kinetics, the authors reduced the immobilization density of FKBP12 (to ~100 RU), so that dissociation events could be observed. For each FK506 variant targeting ligand and corresponding targeted NP, the kinetic constants (ka, kd) and equilibrium constant (KD) were calculated from SPR data using a 1:1 binding model. As would be expected for multivalent interactions, the kd values calculated for all NP were greatly reduced (most by 2–3 orders of magnitude or more) compared to the corresponding free FK506 variant small molecules. However, the ka values for NP relative to free small molecules displayed no consistent trend. For example, one of the high density NP (with a valency of 17) exhibited 400-fold slower association kinetics to the free small molecule, whereas another high density NP (with a valency of 20) exhibited ~40-fold faster association compared to the free small molecule. Overall, the equilibrium binding affinity (KD) of all NP were smaller than the corresponding free small molecules, indicating stronger binding via avidity from the multivalent interactions. To get a relative idea of the changes in binding affinity for the various NP compared to corresponding free small molecules, the authors calculated an enhancement factor (β), defined as the KD, NP divided by the KD, sm. The values for β varied greatly, between 4 and 9,500, for the various NP formulations. Interestingly, NP modified with small molecule ligands that had intrinsically weaker binding (lower KD, sm), exhibited the largest β values. This finding demonstrates that multivalent binding of NP can transform a relatively weak single small molecule interaction into a strong, high affinity interaction due to avidity. It is also interesting to note that despite the wide range of binding affinity of the free small molecules (24 nM to 110 μM), all resulting targeting ligand conjugated NP exhibited similar binding affinities in the lower nM range (ranging from 2.8 to 9.1 nM).
Choi et al. demonstrated the power of multivalent interactions of a 5th generation PAMAM dendrimer (G5) functionalized with the cell wall binding antibiotic vancomycin (VMC) that was able to attain sufficient binding affinity via avidity to target the cell wall of VMC-resistant bacteria [76]. In VMC-resistant bacterial strains, VMC have a very low binding affinity (KD ≈ 1 mM) to the modified cell wall, resulting in ineffective targeting of these resistant cells. The authors synthesized a variety of G5 dendrimers with varying valency of VMC (G5-Vn where n is the valency of the dendrimer) and studied binding to CM5 sensor chips with immobilized peptide sequences specific to either VMC-susceptible or VMC-resistant bacterial cell walls. Kinetic analysis of each G5-Vn was determined with a 1:1 kinetic model and kinetic parameters were used to calculate the binding affinities. The authors found that despite a 1000-fold difference in the binding affinity of free VMC between the two chip surfaces (9.5 × 10−7 M and 1.5 × 10−3 M for VMC-susceptible and VMC-resistant cell wall peptides, respectively), multivalent G5-Vn (with valency between 2 and 6) exhibited a KD value that was only 2-fold to 7-fold greater for VMC-susceptible versus VMC-resistant cell wall peptides, with all binding affinities falling in the 0.25 nM to 8.1 nM range.
IV. B. Evaluation of non-specific binding of nanoRx via SPR
Despite extensive studies of non-specific interactions of small molecule drug candidates with plasma proteins and lipid membrane fragments (Section I.F.3.), there have been few studies of non-specific (and undesirable) interactions of nanoRx with biologic molecules via SPR. In an early study, Moghimi and colleagues modified a silver-coated SPR sensor chip with a thin layer of polystyrene (PS) to generate a surface that mimics the surface of PS NP [77]. The authors then performed SPR experiments to investigate the adsorption of serum proteins and poloxamine surfactant onto the PS sensor chip to explain the results of blood circulation and clearance experiments of PS NP with and without poloxamine surfactant coatings. Single injections of either serum or poloxamine 908 showed clear binding to the PS-coated sensor chip. Further, serum pre-treated with poloxamine 908 showed similar binding to the PS sensor chip as poloxamine 908 alone, but not serum alone. The authors correlated this to observations that, in blood clearance experiments, PS NP that were pre-coated with poloxamine 908 or poloxamine 908 pre-treated serum exhibited prolonged residence in the blood and less accumulation in the liver and spleen compared to unmodified PS NP 3 hours after intravenous injection in rats. The results suggested that pre-coating the PS surface with poloxamine 908 prevents binding of specific serum components (such as IgG) that would normally lead to recognition and clearance by the reticuloendothelial system, thereby reducing clearance of PS NP from the blood. Interestingly, when the authors pre-coated the PS sensor chip with an injection of serum and then followed with an injection of poloxamine 908, the poloxamine 908 did not appreciably displace serum proteins from the chip surface. This finding suggests that poloxamine 908 works to stabilize PS NP in the bloodstream by preventing serum protein binding, rather than removing previously bound proteins from the surface of the PS NP.
Schneider et al. evaluated a variety of PEG-coated polystyrene NP for non-specific binding interactions within brain tissue by testing NP binding to a CM5 sensor chip immobilized with extracted mouse brain ECM proteins [9]. The authors found that PEG-coated, ITEM4 mAb-decorated polystyrene NP (CNP-ITEM4) and PEG-coated polystyrene NP (CNP) did not bind to the chip; however, polystyrene NP that lacked a PEG coating (uncoated NP or UNP) bound avidly to the ECM chip (Figure 5B, C). The binding of UNP was so strong that the ECM sensor chip could not be regenerated effectively. These data correlated well with in vivo imaging of NP spread in the brain, where UNP were unable to spread through brain tissue following intracranial injection, whereas CNP-ITEM4 diffused effectively and associated with Fn14-expressing U87 cells [9].
IV.C. Effects of intermolecular distance of immobilized SPR ligands and targeting molecules on nanoRx on nanoRx binding affinities measured by SPR
As mentioned above (in Section I.B.), the immobilization ligand density is an important parameter for measuring the binding of SPR analytes, including nanoRx. Although the effect of SPR ligand immobilization on mass transfer is well known, more recent studies have begun to examine the effects on the relative intermolecular distance between targeting molecules on the nanoRx surface and of immobilized ligands on the surface of the sensor chip. These studies have suggested that this relationship can significantly affect the measured binding affinity of multivalent nano-sized analytes.
Munoz et al. studied the interaction of three different mannosylated gallic acid-triethylene glycol dendrimers with varying generation/branching (with 3–27 mannose residues per dendrimer) with the lectin concavlin A. The authors studied this interaction via both SPR and a solution based technique (competition assay) to examine the effects of lectin clustering/arrangement on binding kinetics and binding equilibrium [78]. Compared to solution phase experiments, SPR surface experiments measured greater differences in binding affinities between dendrimers of varying generation, which was likely due to lectin clustering at the chip surface during SPR experiments. Based on this, the authors suggested that SPR experiments are better determinants of biological membrane receptor-ligand binding compared to solution based measurements.
The authors also examined the same dendrimers binding at two different concavalin A immobilization levels (concavalin A surface densities), which resulted in significantly lower calculated binding affinities for lower concavalin A immobilization (3-fold lower for G1 and G2 dendrimers and 12-fold lower for G3 dendrimers). The authors attributed this result to the fact that at low concavalin A immobilization levels, all three dendrimers have a particle diameter that is less than the average distance between concavalin A molecules (inter-concavalin A distance). On the other hand, at higher immobilization of concavalin A, the inter-concavalin A distance gets smaller and rebinding of dissociating dendrimer becomes more likely. This is because, the dendrimers are, on average, closer to adjacent lectins, increasing probability of binding to an adjacent lectin before diffusing away from the surface of the chip. The 12-fold increase in G3-dendrimer binding affinity, compared to 3-fold for the G1 and G2 dendrimers, was explained by the fact that at higher immobilization densities only the G3 dendrimer has a particle diameter that exceeds the inter-lectin distance, allowing a single G3 dendrimer to bind to multiple concavalin A molecules. This multivalent binding would be expected to significantly decrease the dissociation rate constant (KD) and therefore increase binding affinity, which was observed at the higher concavalin A immobilization density. These results suggest that, when possible, SPR experiments with nanoRx that are capable of multivalent interactions should be carried out with ligand immobilization densities that are as close to physiologic target receptor densities on the cell surface as possible. This will ensure that binding characteristics observed by SPR are more likely to translate to in vitro and in vivo behavior. The results also suggest that average spacing between targeting moieties on nanoRx can be adjusted relative to the distance between target receptors on the cell surface to modulate dendrimer/NP binding properties.
Reynolds et al. used mannose functionalized gold NP (glyco-NP or GNP) to study the multivalent binding properties to an immobilized, divalent lectin (BC2L-A) on a CM5 sensor chip, as well as in solution via isothermal titration calorimetry (ITC) [79]. Similar to studies by Munoz et al., the authors found that binding affinities of multivalent GNP in solution (measured via ITC) were lower than surface binding measurements via SPR. They also observed an enhanced binding affinity for a particular NP, GNP9, which had a mannose surface density that was structurally compatible with the divalent lectin. Specifically, the distance between neighboring mannose molecules on GNP9 was most similar to the distance between the two mannose binding sites on the BC2L-A molecule. This result further illustrates the potential importance of matching targeting moiety surface density to the density of receptor binding sites on the cell or protein of interest.
Altogether, these studies highlight (1) the importance of using physiological immobilization densities in SPR experiments to ensure that measured binding phenomenon translate to in vivo systems and (2) the importance of considering nanoRx size and intermolecular distance in the design of targeted nanoRx.
IV.D. Novel protocol for evaluating the binding of multivalent nanoRx via SPR
In addition to their observations above, Munoz and colleagues noted that the dissociation phase of multivalent dendrimer sensorgrams has two distinct subphases: an early, rapid dissociation subphase and a later, slower dissociation subphase [78]. The authors hypothesized that these subphases may be the result of competition between dendrimers for the same concavalin A molecules on the surface of the chip. For instance, during the beginning of the dissociation phase the maximum amount of dendrimer is bound at the surface of the chip. Thus as a dendrimer dissociates from interaction with a concavalin A molecule, it is less likely to rebind to another adjacent concavlin A molecule due to a high local concentration of other dendrimers at the chip surface. During the later phase, after many dendrimers have dissociated and been washed away from the chip surface via running buffer, dendrimers that dissociate have less competition from other dendrimers locally and are therefore more likely to rebind to the chip. This would explain the early, rapid and late, slow phases of dissociation observed in their kinetic SPR experiments.
Based on these observations, the same group went on to design an analytical SPR experimental protocol to better characterize binding of multivalent analytes (e.g. dendrimers) [80]. The protocol relies on evaluation of kinetic data during the early parts of the association phase and the late parts of the dissociation phase, when there are relatively low amounts of the dendrimer at the surface of the chip. The purpose of focusing in on these areas of the data is that, in these regions, there is less likely to be competition between two dendrimers for the same immobilized ligand, which would serve to either prevent multivalent crosslinking (if the dendrimer is large enough) or rebinding of dendrimers to adjacent immobilized ligand after dissociation. Thus, analyzing these portions of the curve exclusively is more likely to give a better representation of the binding affinity of the multivalent analyte. In the paper, the authors analyzed data from mannosylated dendrimers using this method, and showed that an excellent fit is obtained to pseudo-first order kinetics with less binding heterogeneity compared to analysis of the entire association and dissociation phases. The authors further found that analysis of SPR data from lower analyte concentrations with this method was ideal for measuring the multivalent binding behavior of the tested dendrimers (due to less competition between dendrimers for concavalin A). For those that wish to measure nanoRx binding affinities with SPR, we recommend a comprehensive reading and consideration of these papers [78, 80].
V. Future applications of SPR for nanoRx studies
Despite numerous studies highlighted in this review, SPR remains an underutilized technique in the field of nanoRx development. In the future, we foresee SPR being used routinely for high-throughput screening of nanoRx as well as continued fundamental studies of nanoRx interactions. In this final section, we suggest some applications of SPR that we believe are primed for exploration and that will aid in the development of effective nanoRx.
V.A. SPR for rapid screening and validation of nanoRx
One of the major strengths of the SPR technology is the ability to generate rapid, real-time kinetic binding data on the order of ~10–15 minutes per sample. Coupled with autosampler units that are standard on most SPR instruments, screening of 10’s to 100’s of nanoRx is feasible within a 24 hour period of instrument use. While SPR has been previously used for screening targeted nanoRx, we believe SPR will take on a much bigger role in evaluating libraries of non-targeted and targeted nanoRx for binding to receptor targets and in performing rapid quality control checks prior to in vitro and in vivo testing once an optimal nanoRx has been identified.
V.A.1. Screening candidate nanoRx for specific vs. non-specific binding
The most straightforward application of nanoRx via SPR is rapid screening to qualitatively examine binding to sensor chips with immobilized target. A single injection of each nanoRx to be tested (all at the same concentration) can provide an RUmax value that can be used to qualitatively judge the binding of a library of nanoRx, with each sample run taking roughly 10–15 minutes. In general, for nanoRx to be effective, they must avoid non-specific interactions to overcome biological barriers and successfully reach the target tissue. Examples of potential barriers include adsorption of plasma proteins in the blood leading to RES clearance following systemic injection [81], adhesion to sticky glycoprotein components of mucus at mucosal surfaces [82–84], and adhesion to extracellular matrix in tissue following local injection [9, 85–86] and/or extravasation of nanoRx from the blood stream (for instance, after nanoRx accumulation in tumor tissues via the EPR effect) [87]. Therefore, it would also be useful to rapidly screen nanoRx for the ability to overcome biological barriers before time consuming and expensive in vitro and in vivo experiments are performed. Limited studies utilizing SPR for this purpose have been discussed in this review and we believe this is a key area for advancement in the field.
V.A.2. Quality control validation of nanoRx batches before experiments
Once an optimal nanoRx formulation has been identified, it is important that there is little batch-to-batch variability in binding behavior and other important parameters as you move into advanced preclinical and clinical evaluation. Therefore, it is necessary to have a means to rapidly check and validate each batch of nanoRx prior to initiation of experiments. SPR is an ideal technique for this type of quality control validation due to its short run time, minimal sample requirements, and versatility. In the following subsections, we highlight a couple applications of SPR for quality control of batches of targeted nanoRx.
V.A.2.i. Validation of target binding
Similar to nanoRx screening studies, a single injection or a series of injections of a newly synthesized batch of nanoRx can be used to qualitatively or quantitatively validate the binding activity in minutes to a couple of hours, depending on the desired complexity of analysis. This would provide a more rapid and reproducible method for validating nanoRx compared to validation in in vitro cell binding or in vivo assays, increasing the efficiency of the lab and ensuring the veracity of downstream experimental results.
V.A.2.ii. Validation of sterility
In addition to validating target binding, SPR can be used to ensure that newly synthesized batches of nanoRx are free from contaminants that are ubiquitous in biomedical laboratories, such as lipolysaccharide (LPS) fragments from the cell wall of bacteria. When moving on to test the effects of promising nanoRx in vitro and in vivo, it is important that samples are free of LPS, as this peptide is strongly immunogenic and can therefore cause unexpected inflammatory reactions when introduced into cells or animals. One could imagine that an SPR sensor chip coated with immobilized antibodies against common immunogenic proteins and peptides could help to ensure that contaminated nanoRx are excluded from important downstream experiments.
V.B. Additional applications of SPR for studying nanoRx
Due to the versatility of the SPR technology, there are essentially endless possibilities for novel applications involving testing of non-targeted or targeted nanoRx. Below, we highlight a couple areas we believe where SPR might be useful in advancing the field.
V.B.1. Studying protein corona formation on nanoRx
Various studies have illustrated the importance of protein corona formation on the in vivo behavior and fate of NP [88–91]. In the future, SPR could be used to study interactions between nanoRx and protein components of biological fluids to better understand the formation of protein coronas. The results of such studies may provide insights to control/modify protein corona formation and therefore in vivo fate and effectiveness of designed nanoRx.
V.B.2 Studying nanoRx binding under flow
As SPR experiments are performed under constant flow conditions with a flow rate that can be easily controlled with the associated software, SPR is an ideal technique to study nanoRx binding under shear stresses induced by flow. This could be useful for the study of a variety of physiologically relevant nanoRx targeting scenarios, such as targeting nanoRx to arterial plaques or clots following systemic administration. However, these studies are limited to the flow rate limitations of the particular SPR instruments and to laminar flow conditions.
VI. Conclusion
SPR is a versatile technology that has been extensively used to characterize and screen both traditional small molecule therapeutics and newer biologics. Despite current limitations in analysis of SPR data of nanoRx, SPR is a useful technique to characterize nanoRx binding, screen and optimize potential nanoRx, and perform quality control checks of batches of nanoRx. SPR is widely applicable to a variety of nanoRx including dendrimers, liposomes, polymeric, and inorganic NP and in the future may play a greater role in high throughput nanoRx development.
Table 1.
Summary of SPR studies of targeted nanotherapeutics
| NanoRx type | Targeting ligand | Immobilized agent | Application | Reference |
|---|---|---|---|---|
| MWCNT | IgG | Streptavidin | Biological activity of IgG | 58 |
| PLGA-PEG NPs | Folic acid; Biotin | Anti-folate antibody; Streptavidin | Surface partitioning and activity of targeting ligands | 60 |
| PLGA NPs | mAb | Biotinylated protein A | Binding affinity of two conjugation techniques | 61 |
| PEG-PLA NPs | AMB8LK | AMB8LK; AMB8LK- PEG-PLA NPs | Binding affinity of native antibody compared to NPs | 62 |
| PEG-PLA NPs | ACUPA | His-tag PSMA | Binding affinity to PSMA for prostate cancer therapy | 73 |
| Poly(H2NPEGCA-co- HDCA) NPs | Folic acid | Folate binding protein | Binding affinity for targeting tumors over-expressing folate receptors | 8 |
| PAMAM dendrimers | Mannose and cyclodextrin | Lectin concavilin A | Lectin-specific binding activity | 63 |
| Generation 5 polyvalent dendrimers | Methotrexate | Folate binding protein | Folate-specific binding activity | 64 |
| Dendrimers | Carbohydrate | Mannosylated-bovine serum albumin | Binding inhibition of HIV-1 and Dengue virus to DC-SIGN | 65 |
| ASO micelles | — | SCARA1 | Cellular uptake/binding affinity of micelles to SCARA1 | 66 |
| MGC micelles | — | Carbohydrate recognition domain of recombinant DC-SIGN | Binding inhibition of HIV-1 to DC- SIGN by multivalent micelles and single molecule micelles | 68 |
| PEG-P(Asp-Hyd- ADR) micelles | Folic acid | Folate binding protein | Binding affinity of folate-modified micelles and unmodified micelles | 70 |
| Liposomes | Anti-transferrin antibody (RI7217) | Transferrin | Binding affinity of two conjugation techniques | 59 |
| siRNA-loaded liposomes | Anti-HB-EGF Fab fragment | HB-EGF | Targeting therapeutic siRNA to solid tumors expressing HB-EGF | 71 |
| Liposomes | RGD | B16 melanoma cells; MCF-7 breast cancer cells | Binding affinity of monomeric, dimeric, flexible dimeric RDG to αvβ3 integrins | 72 |
| Polystyrene NPs | ITEM4 (mAb) | Fn14 extracelluar domain | Binding affinity of NPs with varying | 9 |
Acknowledgments
We thank Dr. Dudley Strickland (University of Maryland School of Medicine) for critical review of the manuscript. We also thank Dr. Yinghua Zhang at the Biosensor Core at the University of Maryland School of Medicine and School of Pharmacy for her expertise. This work was supported in part by the National Institutes of Health (K12NS080223, K25EB018370, K08NS09043), a DOD CDMRP Lung Cancer Research Program IDEA Award (W81XWH-14-1-0324), an Institutional Research Grant (IRG-97-153-10) from the American Cancer Society, a Passano Foundation Physician Scientist Award, a Elsa U. Pardee Foundation Research Grant, a PhRMA Foundation Research Starter Grant in Pharmaceutics, and a AAPS Foundation New Investigator Grant Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
VII. References
- 1.Biacore. BIACORE 2000 Instrument Handbook. Biacore AB; 2001. [Google Scholar]
- 2.Sciences GL. Biacore Assay Handbook. General Electric Company; 2012. [Google Scholar]
- 3.Tassa C, et al. Binding affinity and kinetic analysis of targeted small molecule-modified nanoparticles. Bioconjug Chem. 2010;21(1):14–9. doi: 10.1021/bc900438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis TM, Wilson WD. Surface plasmon resonance biosensor analysis of RNA-small molecule interactions. Methods Enzymol. 2001;340:22–51. doi: 10.1016/s0076-6879(01)40416-2. [DOI] [PubMed] [Google Scholar]
- 5.Davis TM, Wilson WD. Determination of the refractive index increments of small molecules for correction of surface plasmon resonance data. Anal Biochem. 2000;284(2):348–53. doi: 10.1006/abio.2000.4726. [DOI] [PubMed] [Google Scholar]
- 6.Fivash M, Towler EM, Fisher RJ. BIAcore for macromolecular interaction. Curr Opin Biotechnol. 1998;9(1):97–101. doi: 10.1016/s0958-1669(98)80091-8. [DOI] [PubMed] [Google Scholar]
- 7.Pattnaik P. Surface plasmon resonance: applications in understanding receptor-ligand interaction. Appl Biochem Biotechnol. 2005;126(2):79–92. doi: 10.1385/abab:126:2:079. [DOI] [PubMed] [Google Scholar]
- 8.Stella B, et al. Design of folic acid-conjugated nanoparticles for drug targeting. J Pharm Sci. 2000;89(11):1452–64. doi: 10.1002/1520-6017(200011)89:11<1452::aid-jps8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.Schneider CS, et al. Minimizing the non-specific binding of nanoparticles to the brain enables active targeting of Fn14-positive glioblastoma cells. Biomaterials. 2015;42:42–51. doi: 10.1016/j.biomaterials.2014.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol. 2010;161(6):1219–37. doi: 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karlsson R, et al. Analyzing a kinetic titration series using affinity biosensors. Anal Biochem. 2006;349(1):136–47. doi: 10.1016/j.ab.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 12.Piletska EV, Piletsky SA. Size matters: influence of the size of nanoparticles on their interactions with ligands immobilized on the solid surface. Langmuir. 2010;26(6):3783–5. doi: 10.1021/la904834y. [DOI] [PubMed] [Google Scholar]
- 13.Rich RL, Myszka DG. Advances in surface plasmon resonance biosensor analysis. Curr Opin Biotechnol. 2000;11(1):54–61. doi: 10.1016/s0958-1669(99)00054-3. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein B, et al. The influence of transport on the kinetics of binding to surface receptors: application to cells and BIAcore. J Mol Recognit. 1999;12(5):293–9. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<293::AID-JMR472>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Myszka DG, et al. Extending the range of rate constants available from BIACORE: interpreting mass transport-influenced binding data. Biophys J. 1998;75(2):583–94. doi: 10.1016/S0006-3495(98)77549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mason T, et al. Effective rate models for the analysis of transport-dependent biosensor data. Math Biosci. 1999;159(2):123–44. doi: 10.1016/s0025-5564(99)00023-1. [DOI] [PubMed] [Google Scholar]
- 17.Mullett WM, Lai EP, Yeung JM. Surface plasmon resonance-based immunoassays. Methods. 2000;22(1):77–91. doi: 10.1006/meth.2000.1039. [DOI] [PubMed] [Google Scholar]
- 18.Homola J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem Rev. 2008;108(2):462–93. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 19.Schuck P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Annu Rev Biophys Biomol Struct. 1997;26:541–66. doi: 10.1146/annurev.biophys.26.1.541. [DOI] [PubMed] [Google Scholar]
- 20.Green RJ, et al. Surface plasmon resonance analysis of dynamic biological interactions with biomaterials. Biomaterials. 2000;21(18):1823–35. doi: 10.1016/s0142-9612(00)00077-6. [DOI] [PubMed] [Google Scholar]
- 21.Boozer C, et al. Looking towards label-free biomolecular interaction analysis in a high-throughput format: a review of new surface plasmon resonance technologies. Curr Opin Biotechnol. 2006;17(4):400–5. doi: 10.1016/j.copbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Malmqvist M. Surface plasmon resonance for detection and measurement of antibody-antigen affinity and kinetics. Curr Opin Immunol. 1993;5(2):282–6. doi: 10.1016/0952-7915(93)90019-o. [DOI] [PubMed] [Google Scholar]
- 23.Huber W, Mueller F. Biomolecular interaction analysis in drug discovery using surface plasmon resonance technology. Curr Pharm Des. 2006;12(31):3999–4021. doi: 10.2174/138161206778743600. [DOI] [PubMed] [Google Scholar]
- 24.Karlsson R, et al. Biosensor analysis of drug-target interactions: direct and competitive binding assays for investigation of interactions between thrombin and thrombin inhibitors. Anal Biochem. 2000;278(1):1–13. doi: 10.1006/abio.1999.4406. [DOI] [PubMed] [Google Scholar]
- 25.Markgren PO, Hamalainen M, Danielson UH. Kinetic analysis of the interaction between HIV-1 protease and inhibitors using optical biosensor technology. Anal Biochem. 2000;279(1):71–8. doi: 10.1006/abio.1999.4467. [DOI] [PubMed] [Google Scholar]
- 26.Hamalainen MD, et al. Characterization of a set of HIV-1 protease inhibitors using binding kinetics data from a biosensor-based screen. J Biomol Screen. 2000;5(5):353–60. doi: 10.1177/108705710000500507. [DOI] [PubMed] [Google Scholar]
- 27.Boehm HJ, et al. Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimization. A promising alternative to random screening. J Med Chem. 2000;43(14):2664–74. doi: 10.1021/jm000017s. [DOI] [PubMed] [Google Scholar]
- 28.Lan L, et al. Natural product (−)-gossypol inhibits colon cancer cell growth by targeting RNA-binding protein Musashi-1. Mol Oncol. 2015 doi: 10.1016/j.molonc.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navratilova I, et al. Biosensor-based approach to the identification of protein kinase ligands with dual-site modes of action. J Biomol Screen. 2012;17(2):183–93. doi: 10.1177/1087057111422746. [DOI] [PubMed] [Google Scholar]
- 30.Markgren PO, et al. Relationships between structure and interaction kinetics for HIV-1 protease inhibitors. J Med Chem. 2002;45(25):5430–9. doi: 10.1021/jm0208370. [DOI] [PubMed] [Google Scholar]
- 31.Rowland A, et al. Characterization of the comparative drug binding to intra- (liver fatty acid binding protein) and extra- (human serum albumin) cellular proteins. Xenobiotica. 2015:1–11. doi: 10.3109/00498254.2015.1021403. [DOI] [PubMed] [Google Scholar]
- 32.Schneider EK, et al. Drug-drug plasma protein binding interactions of ivacaftor. J Mol Recognit. 2015;28(6):339–48. doi: 10.1002/jmr.2447. [DOI] [PubMed] [Google Scholar]
- 33.Shim YY, Reaney MJ. Kinetic Interactions between Cyclolinopeptides and Immobilized Human Serum Albumin by Surface Plasmon Resonance. J Agric Food Chem. 2015 doi: 10.1021/jf504811x. [DOI] [PubMed] [Google Scholar]
- 34.Vuignier K, et al. Global analytical strategy to measure drug-plasma protein interactions: from high-throughput to in-depth analysis. Drug Discov Today. 2013;18(21–22):1030–4. doi: 10.1016/j.drudis.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 35.Graham RA, et al. Pharmacokinetics of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clin Cancer Res. 2011;17(8):2512–20. doi: 10.1158/1078-0432.CCR-10-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frostell-Karlsson A, et al. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J Med Chem. 2000;43(10):1986–92. doi: 10.1021/jm991174y. [DOI] [PubMed] [Google Scholar]
- 37.Baird CL, Courtenay ES, Myszka DG. Surface plasmon resonance characterization of drug/liposome interactions. Anal Biochem. 2002;310(1):93–9. doi: 10.1016/s0003-2697(02)00278-6. [DOI] [PubMed] [Google Scholar]
- 38.Cimitan S, et al. Early absorption and distribution analysis of antitumor and anti-AIDS drugs: lipid membrane and plasma protein interactions. J Med Chem. 2005;48(10):3536–46. doi: 10.1021/jm049343o. [DOI] [PubMed] [Google Scholar]
- 39.Danelian E, et al. SPR biosensor studies of the direct interaction between 27 drugs and a liposome surface: correlation with fraction absorbed in humans. J Med Chem. 2000;43(11):2083–6. doi: 10.1021/jm991156g. [DOI] [PubMed] [Google Scholar]
- 40.Ramsden JJ. Partition coefficients of drugs in bilayer lipid membranes. Experientia. 1993;49(8):688–92. doi: 10.1007/BF01923952. [DOI] [PubMed] [Google Scholar]
- 41.Cheng E, et al. TWEAK/Fn14 Axis-Targeted Therapeutics: Moving Basic Science Discoveries to the Clinic. Front Immunol. 2013;4:473. doi: 10.3389/fimmu.2013.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown SA, et al. TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem J. 2006;397(2):297–304. doi: 10.1042/BJ20051362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou H, et al. Development and characterization of a potent immunoconjugate targeting the Fn14 receptor on solid tumor cells. Mol Cancer Ther. 2011;10(7):1276–88. doi: 10.1158/1535-7163.MCT-11-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou H, et al. The TWEAK receptor Fn14 is a therapeutic target in melanoma: immunotoxins targeting Fn14 receptor for malignant melanoma treatment. J Invest Dermatol. 2013;133(4):1052–62. doi: 10.1038/jid.2012.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou H, et al. Development of human serine protease-based therapeutics targeting Fn14 and identification of Fn14 as a new target overexpressed in TNBC. Mol Cancer Ther. 2014;13(11):2688–705. doi: 10.1158/1535-7163.MCT-14-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breij EC, et al. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014;74(4):1214–26. doi: 10.1158/0008-5472.CAN-13-2440. [DOI] [PubMed] [Google Scholar]
- 47.Boghaert ER, et al. Determination of pharmacokinetic values of calicheamicin-antibody conjugates in mice by plasmon resonance analysis of small (5 microl) blood samples. Cancer Chemother Pharmacol. 2008;61(6):1027–35. doi: 10.1007/s00280-007-0560-2. [DOI] [PubMed] [Google Scholar]
- 48.Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 49.Hong LPT, et al. Cancer-targeting Antibody. Drug Conjugates: Site-specific Conjugation of Doxorubicin to Anti-EGFR 528 Fab′ through a Polyethylene Glycol Linker. Australian Journal of Chemistry. 2011;64(6):779–789. [Google Scholar]
- 50.Maurer MF, et al. Generation and characterization of human anti-human IL-21 neutralizing monoclonal antibodies. MAbs. 2012;4(1):69–83. doi: 10.4161/mabs.4.1.18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thillaivinayagalingam P, et al. Biopharmaceutical production: Applications of surface plasmon resonance biosensors. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(2):149–53. doi: 10.1016/j.jchromb.2009.08.040. [DOI] [PubMed] [Google Scholar]
- 52.Cooper MA. Optical biosensors in drug discovery. Nat Rev Drug Discov. 2002;1(7):515–28. doi: 10.1038/nrd838. [DOI] [PubMed] [Google Scholar]
- 53.Wang H, et al. Development of biosensor-based SPR technology for biological quantification and quality control of pharmaceutical proteins. J Pharm Biomed Anal. 2009;50(5):1026–9. doi: 10.1016/j.jpba.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 54.Dudak FC, I, Boyaci H. Rapid and label-free bacteria detection by surface plasmon resonance (SPR) biosensors. Biotechnol J. 2009;4(7):1003–11. doi: 10.1002/biot.200800316. [DOI] [PubMed] [Google Scholar]
- 55.Bergwerff AA, van Knapen F. Surface plasmon resonance biosensors for detection of pathogenic microorganisms: strategies to secure food and environmental safety. J AOAC Int. 2006;89(3):826–31. [PubMed] [Google Scholar]
- 56.Nilsson CE, et al. A novel assay for influenza virus quantification using surface plasmon resonance. Vaccine. 2010;28(3):759–66. doi: 10.1016/j.vaccine.2009.10.070. [DOI] [PubMed] [Google Scholar]
- 57.Wong RB, et al. A quantitative human monoclonal antibody immunoassay using anti-idiotypic antibody as a membrane antigen surrogate with surface-plasmon-resonance detection. Biotechnol Appl Biochem. 2009;53(Pt 1):51–61. doi: 10.1042/BA20080072. [DOI] [PubMed] [Google Scholar]
- 58.Wang JT, et al. The relationship between the diameter of chemically-functionalized multi-walled carbon nanotubes and their organ biodistribution profiles in vivo. Biomaterials. 2014;35(35):9517–28. doi: 10.1016/j.biomaterials.2014.07.054. [DOI] [PubMed] [Google Scholar]
- 59.Salvati E, et al. Liposomes functionalized to overcome the blood-brain barrier and to target amyloid-beta peptide: the chemical design affects the permeability across an in vitro model. Int J Nanomedicine. 2013;8:1749–58. doi: 10.2147/IJN.S42783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patil YB, et al. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials. 2009;30(5):859–66. doi: 10.1016/j.biomaterials.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kocbek P, et al. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120(1–2):18–26. doi: 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 62.Debotton N, et al. A quantitative evaluation of the molecular binding affinity between a monoclonal antibody conjugated to a nanoparticle and an antigen by surface plasmon resonance. Eur J Pharm Biopharm. 2010;74(2):148–56. doi: 10.1016/j.ejpb.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 63.Wada K, et al. Improvement of gene delivery mediated by mannosylated dendrimer/alpha-cyclodextrin conjugates. J Control Release. 2005;104(2):397–413. doi: 10.1016/j.jconrel.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 64.Thomas TP, et al. Polyvalent dendrimer-methotrexate as a folate receptor-targeted cancer therapeutic. Mol Pharm. 2012;9(9):2669–76. doi: 10.1021/mp3002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varga N, et al. A multivalent inhibitor of the DC-SIGN dependent uptake of HIV-1 and Dengue virus. Biomaterials. 2014;35(13):4175–84. doi: 10.1016/j.biomaterials.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 66.Ezzat K, et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett. 2015;15(7):4364–73. doi: 10.1021/acs.nanolett.5b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lai MH, et al. Bacteria-mimicking nanoparticle surface functionalization with targeting motifs. Nanoscale. 2015;7(15):6737–44. doi: 10.1039/c5nr00736d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaeffer E, et al. Dynamic micelles of mannoside glycolipids are more efficient than polymers for inhibiting HIV-1 trans-infection. Bioconjug Chem. 2013;24(11):1813–23. doi: 10.1021/bc4000806. [DOI] [PubMed] [Google Scholar]
- 69.Reulen SW, et al. Collagen targeting using protein-functionalized micelles: the strength of multiple weak interactions. J Am Chem Soc. 2009;131(21):7304–12. doi: 10.1021/ja807723p. [DOI] [PubMed] [Google Scholar]
- 70.Bae Y, et al. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Mol Biosyst. 2005;1(3):242–50. doi: 10.1039/b500266d. [DOI] [PubMed] [Google Scholar]
- 71.Okamoto A, et al. Antibody-modified lipid nanoparticles for selective delivery of siRNA to tumors expressing membrane-anchored form of HB-EGF. Biochem Biophys Res Commun. 2014;449(4):460–5. doi: 10.1016/j.bbrc.2014.05.043. [DOI] [PubMed] [Google Scholar]
- 72.Guo Z, et al. Targeting efficiency of RGD-modified nanocarriers with different ligand intervals in response to integrin alphavbeta3 clustering. Biomaterials. 2014;35(23):6106–17. doi: 10.1016/j.biomaterials.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 73.Hrkach J, et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4(128):128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 74.McGurk SL, et al. Screening the Biointeractions of Submicron Sized Particles Intended for Site-Specific Delivery Using Surface Plasmon Resonance. J Colloid Interface Sci. 1999;218(2):456–461. doi: 10.1006/jcis.1999.6435. [DOI] [PubMed] [Google Scholar]
- 75.Hong S, et al. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14(1):107–15. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 76.Choi SK, et al. Dendrimer-based multivalent vancomycin nanoplatform for targeting the drug-resistant bacterial surface. ACS Nano. 2013;7(1):214–28. doi: 10.1021/nn3038995. [DOI] [PubMed] [Google Scholar]
- 77.Moghimi SM, Pavey KD, Hunter AC. Real-time evidence of surface modification at polystyrene lattices by poloxamine 908 in the presence of serum: in vivo conversion of macrophage-prone nanoparticles to stealth entities by poloxamine 908. FEBS Lett. 2003;547(1–3):177–82. doi: 10.1016/s0014-5793(03)00707-5. [DOI] [PubMed] [Google Scholar]
- 78.Munoz EM, et al. Probing the relevance of lectin clustering for the reliable evaluation of multivalent carbohydrate recognition. J Am Chem Soc. 2009;131(49):17765–7. doi: 10.1021/ja9074826. [DOI] [PubMed] [Google Scholar]
- 79.Reynolds M, et al. Influence of ligand presentation density on the molecular recognition of mannose-functionalised glyconanoparticles by bacterial lectin BC2L-A. Glycoconj J. 2013;30(8):747–57. doi: 10.1007/s10719-013-9478-6. [DOI] [PubMed] [Google Scholar]
- 80.Munoz EM, et al. Real-time evaluation of binding mechanisms in multivalent interactions: a surface plasmon resonance kinetic approach. J Am Chem Soc. 2013;135(16):5966–9. doi: 10.1021/ja400951g. [DOI] [PubMed] [Google Scholar]
- 81.Song G, et al. Nanoparticles and the mononuclear phagocyte system: pharmacokinetics and applications for inflammatory diseases. Curr Rheumatol Rev. 2014;10(1):22–34. doi: 10.2174/1573403x10666140914160554. [DOI] [PubMed] [Google Scholar]
- 82.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61(2):158–71. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim AJ, et al. Use of single-site-functionalized PEG dendrons to prepare gene vectors that penetrate human mucus barriers. Angew Chem Int Ed Engl. 2013;52(14):3985–8. doi: 10.1002/anie.201208556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mastorakos P, et al. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc Natl Acad Sci U S A. 2015;112(28):8720–5. doi: 10.1073/pnas.1502281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nance EA, et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med. 2012;4(149):149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woodworth GF, et al. Emerging insights into barriers to effective brain tumor therapeutics. Front Oncol. 2014;4:126. doi: 10.3389/fonc.2014.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–64. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Monopoli MP, et al. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7(12):779–86. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 89.Nel AE, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–57. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 90.Casals E, V, Puntes F. Inorganic nanoparticle biomolecular corona: formation, evolution and biological impact. Nanomedicine (Lond) 2012;7(12):1917–30. doi: 10.2217/nnm.12.169. [DOI] [PubMed] [Google Scholar]
- 91.Pearson RM, Juettner VV, Hong S. Biomolecular corona on nanoparticles: a survey of recent literature and its implications in targeted drug delivery. Front Chem. 2014;2:108. doi: 10.3389/fchem.2014.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]