

Summary
Alterations in mitophagy have been increasingly linked to aging and age-related diseases. There are, however, no convenient methods to analyze mitophagy in vivo. Here, we describe a transgenic mouse model in which we expressed a mitochondrial-targeted form of the fluorescent reporter Keima (mt-Keima). Keima is a coral-derived protein that exhibits both pH-dependent excitation and resistance to lysosomal proteases. Comparison of a wide range of primary cells and tissues generated from the mt-Keima mouse revealed significant variations in basal mitophagy. In addition, we have employed the mt-Keima mice to analyze how mitophagy is altered by conditions including diet, oxygen availability, Huntingtin transgene expression, the absence of macroautophagy (ATG5 or ATG7 expression), an increase in mitochondrial mutational load, the presence of metastatic tumors and normal aging. The ability to assess mitophagy under a host of varying environmental and genetic perturbations suggests that the mt-Keima mouse should be a valuable resource.
Graphical Abstract
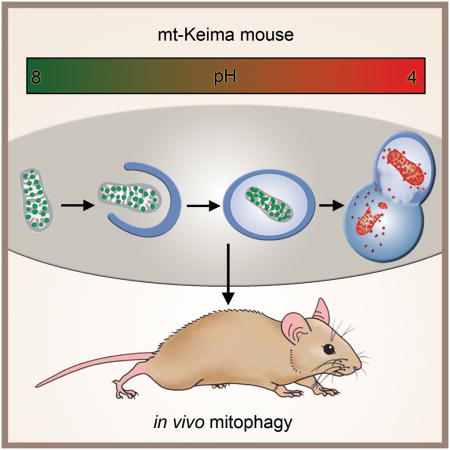
Introduction
Mitochondria play an essential role in cellular physiology as these organelles are essential for maintaining cellular bioenergetics, shaping intracellular calcium dynamics, as well as modulating the threshold for cell death. Moreover, although not necessarily causative, various age-related pathologies are accompanied by a decline in mitochondrial function, or the development of frank mitochondrial dysfunction (Bratic and Larsson, 2013). As such, the mechanisms that regulate mitochondrial quality control are receiving increased attention. These mechanisms include resident mitochondrial chaperones and proteases that help re-fold or degrade misfolded mitochondrial proteins, as well as the process of fusion and fission, that seems essential to maintain mitochondrial homeostasis (Baker et al., 2014; Burte et al., 2015).
Mitophagy is another critical mechanism necessary to maintain mitochondrial quality. While many questions remain, mitophagy is believed to be a selective pathway to remove damaged, old or dysfunctional mitochondria through a process, which in most cases, requires a functional macroautophagy machinery. Cell biological observations suggest however that several distinct forms of mitophagy may exist (Lemasters, 2014). These forms appear to differ in their requirement for mitochondrial membrane depolarization, as well as, whether all or only part of the mitochondria is engulfed by the lysosome. One recently described form of mitophagy, involves a process in which parts of mitochondria can bud off and transit to multivesicular bodies, that in turn ultimately fuse with the lysosome (Lemasters, 2014; Soubannier et al., 2012; Sugiura et al., 2014). This process appears to be independent of the normal autophagic machinery (e.g. ATG5 and LC3-independent) while still requiring PINK1 and Parkin function (McLelland et al., 2014). Similarly, the removal of mitochondria can apparently proceed in the absence of the conventional macroautophagy machinery as observed during the process of reticulocyte maturation (Honda et al., 2014; Zhang et al., 2009), or when cells are exposed to certain specific stresses (Nishida et al., 2009).
Interest in mitophagy has been spurred by observations linking this process to inherited forms of early-onset Parkinson’s disease. In particular, pharmacological depolarization of the mitochondrial membrane potential has been shown to result in the stabilization of the phosphatase and tensin homologue (PTEN)-induced kinase 1 (PINK1), that in turn recruits the E3 ubiquitin ligase Parkin to mitochondria which can triggger mitophagy (Geisler et al., 2010; Matsuda et al., 2010; Narendra et al., 2008). The realization that these two proteins can coordinate mitophagic flux, along with the observation that PINK1 and Parkin appear to function within the same genetic pathway for early-onset Parkinson’s disease (Park et al., 2006), strengthens the notion that mitophagy may play an important role in aging and age-related diseases. Nonetheless, the lack of robust methods to assess in vivo mitophagy has hampered progress in determining the precise role mitophagy plays in normal physiology and in various disease states.
Design
At present, the ability to monitor mitophagy is limited. Traditional methods such as electron microscopy can sometimes recognize mitochondria remnants within autophagosomes, however, these methods sample only a small fraction of any cell or tissue, making it difficult to quantify. Indirect measurements can be made by analyzing mitochondrial protein turnover rates (Kim et al., 2012). Nonetheless, these approaches provide no assessment of tissue architecture and cannot easily distinguish individual proteins degraded by intrinsic mitochondrial proteases, from whole mitochondria that are degraded following lysosomal delivery. Fluorescent-based strategies may provide the most robust approach. Most of these approaches have employed dual fluorescent labeling of both the mitochondria, along with some element of the autophagic machinery, often LC3 (Dolman et al., 2013). Assessment of mitophagy requires the co-localization of these dual fluorescent probes. Given the transient nature of the interaction between autophagosome and mitochondria and the fact that LC3-based fluorescent probes may have a high rate of false positive signals (Kuma et al., 2007), these methods can suffer from a lack of sensitivity and specificity. One significant refinement may come from proteins whose fluorescence exhibits pH dependence. Several examples of such fluorescent molecules have been described including Rosella and Keima (Katayama et al., 2011; Rosado et al., 2008). In this regard, Keima’s properties are particularly attractive as this coral-derived molecule is also resistant to lysosomal degradation. This property allows for the mitophagy signal to be integrated over time allowing some qualitative assessment of mitophagic flux. Moreover, the pH-dependent fluorescent properties of Keima allow rapid determination as to whether the protein is in the mitochondria (pH ~ 8.0) or the lysosome (pH ~ 4.5). These properties has led to a mitochondrial-targeted form of Keima (mt-Keima) being increasingly employed to measure mitophagy in vitro (Bingol et al., 2014; Kageyama et al., 2014; Katayama et al., 2011; Mizumura et al., 2014). Here, we have extended this approach and describe the generation of a transgenic mouse that expresses mt-Keima and demonstrate that this model can provide the first robust assessment of in vivo mitophagy under a wide-range of experimental conditions.
Results
mt-Keima is a robust reporter for mitophagy
Using a mitochondrial targeting sequence from COX VIII, Keima can be directed to the mitochondrial matrix (Katayama et al., 2011). The alkaline nature of this organelle contrasts with the highly acidic environment of the lysosome. This intrinsic pH difference, along with the lysosomal protease-resistance of Keima, allows the ratio of the 561 nm/458 nm excited Keima fluorescence to reflect the underlying level of mitophagy. In our experiments, we chose to depict the mt-Keima fluorescence signal from 561 nm laser excitation (acidic) in red and the signal from 458 nm laser excitation (neutral pH) in green and defined the level of mitophagy as the total number of red pixels divided by the total number of all pixels (Figure S1A–D). Alternatively, the data can be represented as a ratiometric heat map (Figure S1E). As expected, in permeabilized cells, mt-Keima exhibited a clear pH-dependent fluorescence (Figure 1A). We noted an approximate 7–8 fold increase in the ratio of 561nm/458nm (red/green) fluorescence as the cellular pH was engineered to go from pH 7 to pH 4 (Figure 1B). Prolonged hypoxia is known to stimulate mitophagy (Liu et al., 2012). When mt-Keima was stably expressed in HeLa cells, we noted that hypoxia induced a marked rise in the red-to-green fluorescence ratio (Figure 1C). When assessed by Western blot analysis, the hypoxic increase in mitophagy, and hence increased mitochondrial turnover, was accompanied by the expected decline in the level of mitochondrial proteins relative to the cytosolic protein β-actin (Figure S1F). Under these conditions, mitophagy could also be readily assessed in mt-Keima expressing cells using a FACS-based approach in which an arbitrary threshold of red fluorescence was used to indicate a mitophagic positive cell (Figure 1D). Similarly, time lapse imaging of HeLa cells expressing both YFP-Parkin and mt-Keima and subsequently treated with FCCP and oligomycin revealed an increase in mitophagy beginning approximately 15 minutes after Parkin first localized to the mitochondria (Figure 1E). This treatment strategy eventually culminated in the near total loss of green mt-Keima fluorescence (as well as a loss of YFP-Parkin) and a marked increase in overall red mt-Keima fluorescence (Supplemental Video 1). In contrast, untreated HeLa cells expressing mt-Keima and YFP-Parkin maintained Parkin in the cytosol and exhibited a stable red to green fluorescence ratio over 18 hours of imaging (Figure S1G and Supplemental Video 1).
Figure 1.

Detection of in vitro mitophagy using mt-Keima. A) HeLa cells expressing mt-Keima were permeabilized and equilibrated with buffers at pH 4 and pH 7. Levels of green and red signals are shown as a function of cellular pH. Scale bar=10 μm. B) mt-Keima demonstrates a greater than 7-fold change in ratiometric fluorescence over three pH units. Values are normalized to the red/green signal at pH 7 (n=3 per condition, **p<0.01). C) Representative confocal images of HeLa cells expressing mt-Keima in a normoxic environment (21% oxygen) or after 24 hours of hypoxia (1% oxygen). Scale bar=10 μm. D) Representative FACS analysis of hypoxia-induced mitophagy using red-to-green fluorescence. E) Time lapse analysis of a HeLa cell line expressing YFP-Parkin and mt-Keima treated with FCCP/oligomycin (5 μM each). Parkin appears to be recruited to the mitochondria rapidly after FCCP/oligomycin addition and an increase in mitophagy (red fluorescence) begins roughly 15 minutes after Parkin recruitment. After 18 hours, nearly all the green mt-Keima and YFP-Parkin fluorescence has vanished, with cells now exhibiting predominantly red mt-Keima fluorescence.
In vivo assessment of mitophagy with mt-Keima mice
These results, as well as the recent observations of others (Bingol et al., 2014; Kageyama et al., 2014; Mizumura et al., 2014), demonstrate that mt-Keima can faithfully measure mitophagy. We therefore constructed a mt-Keima reporter mouse by inserting a single copy of the transgene into the Hipp11 locus on chromosome 11. This site-specific integration has been previously shown to result in consistent transgene expression without any deleterious effects to mice (Tasic et al., 2011). When mouse embryonic fibroblasts (MEFs) derived from the mt-Keima mice were imaged via 458 nm excitation, they demonstrated characteristic tubular mitochondria (Figure 2A, shown in green). When these cells were imaged via 561 nm excitation, the red fluorescence was largely confined to multiple, small, round punctate structures (Figure 2A and 2B). The localization of these punctate structures overlapped with lysosomes, as evidenced when cells were simultaneously assessed with a lysosomal probe (LysoSensor, shown in cyan, Figure 2C&D).
Figure 2.

The mt-Keima mouse allows for detection of in vivo mitophagy. A) Confocal images of overlayed red and green fluorescence in MEFs expressing mt-Keima. Green structures are consistent with tubular mitochondria and differ morphologically from the red punctate structures. B) Red fluorescence only from this image. C) Lysosomal staining using LysoSensor (depicted in cyan). D) Magnified image demonstrating red mt-Keima signal is wholly contained within lysosomes. Scale bar=10 μm. E) Representative confocal images showing superimposed red/green signals in the liver of mt-Keima mouse. Scale bar=10 μm. F) Red signal only from this image representing mitochondria within the acidic compartment of hepatocytes and G) dextran cascade blue fluorescence that labels the late endosomal/lysosomal compartment within hepatocytes. H) Overlay of red mt-keima and dextran blue fluorescence demonstrating that in vivo the red mt-Keima signal co-localizes with the hepatic lysosomal compartment. I) Levels of mitophagy in embryonic cortex (E15.5) of wild type (Atg7+/+) mice. J) mt-Keima signal in the cortex of an Atg7−/− littermate. Scale bar=10 μm. K) Relative mt-Keima signal in WT or Atg7−/− (E14.5-E18.5) cortex. Values are normalized to WT levels of mitophagy (n=5 per genotype, **p<0.005).
Tissue analysis of mt-Keima mice revealed that basal mitophagic levels could be visualized, as is apparent by the green and red punctate seen in images obtained from hepatic sections (Figure 2E). When mice were given an injection of dextran cascade blue, a fluorescent compound that accumulates in the late endosomes/lysosomes of hepatocytes (Chen et al., 1998; Koval and Pagano, 1990), there was co-localization of the red (561 nm excitation) mt-Keima signal with the hepatic lysosomal signal (Figure 2F–H). A similar overlap between the 561 nm excitation signal and lysosomes was observed in a range of other tissues (Figure S2A–I). In contrast, treatment of mt-Keima mice with the lysosomal acidification inhibitor chloroquine resulted in a marked diminution of the red signal in tissues (Figure S2K&L). Thus, in vivo, as we observed in cells, the red mt-Keima signal is largely, if not exclusively, confined to the acidic, lysosomal compartment. To further verify the fidelity of this model, we next crossed the mt-Keima mice with mice lacking the essential autophagy gene ATG7. Mice deficient in macroautophagy usually die shortly after birth (Kuma et al., 2004). To circumvent this post-natal lethality, we analyzed the embryonic brain of mt-Keima mice with and without ATG7 expression. Consistent with our observations in liver, the brain of wild-type mt-Keima mice had readily detectable levels of basal mitophagy (Figure 2I). This signal was, however, dramatically reduced, but not completely eliminated, in mice lacking ATG7 (Figure 2J and 2K).
Analyzing an array of different cell types, including cardiomyocytes, neural stem cells, hepatocytes and spermatozoa, we noted a wide range of basal mitophagy in primary cells derived from the mt-Keima mice, suggesting cell autonomous aspects of this process (Figure 3A–E). Given these cell-type-dependent differences, we reasoned that primary cells derived from the mt-Keima mouse might, in themselves, be useful reagents. Since there is a growing interest in stimulating autophagy and mitophagy as a means to treat neurodegenerative conditions (Martin et al., 2015; Schapira et al., 2014), we decided to conduct a small, pilot chemical screen using embryonic neural stem cell (NSCs) cultures derived from mt-Keima mice. Previous observations demonstrated that the antibiotic chloramphenicol can stimulate autophagy, at least in part, by inhibiting mitochondrial protein synthesis (Sala-Mercado et al., 2010; Xie et al., 2012). We hypothesized that other antibiotics might also stimulate mitophagy. Using NSCs derived from mt-Keima mice as a potentially relevant cell type, we screened 16 different antibiotics for their ability to stimulate mitophagy. We found that one of the antibiotics tested, actinonin, was in fact a potent activator (Figure 3F). Actinonin has recently been demonstrated to induce a specific mitochondrial ribosomal and RNA decay pathway in cells (Richter et al., 2013). Our results suggest that triggering this pathway may also be linked to the induction of mitophagy.
Figure 3.

Assessment of mitophagy in primary cells derived from the mt-Keima mouse. A) Mitophagy levels in an adult cardiomyocyte (scale bar=10 μm), B) isolated embryonic neural stem cell (Scale bar=10 μm), C) hepatocytes (scale bar=50 μm), D) a neurosphere derived from embryonic neural stem cells (Scale bar=20 μm) and E) an isolated spermatozoa (scale bar=10 μm). F) Small targeted chemical screen treating embryonic neural stem cells expressing mt-Keima with various antibiotics (each at 100 μM concentration). Twenty-four hours after antibiotic exposure, cells expressing mt-Keima were analyzed by FACS. The antibiotic actinonin (structure shown in inset) functions as an inducer of mitophagy. The chemical uncoupler FCCP (2 μM) was used as a positive control. **p<0.01 compared to control.
We next sought to assess basal levels of mitophagy in various mouse tissues. Similar to what we observed in isolated cells, we observed tissue-specific differences in mitophagy with a low signal observed in tissues such as the thymus, and high rates seen in organs such as the heart (Figure 4A and 4B). Interestingly, within a given tissue there was often considerable heterogeneity. In the brain, the cortex, striatum and substantia nigra had seemingly modest levels of basal mitophagy (Figure 4C and Figure S3A–C). In contrast, mitophagy was enhanced in the dentate gyrus and the lateral ventricle, areas known to be enriched for neural stem cells (Figure 4D&E, Figure S3D&E). The Purkinje cell layer within the cerebellum had a similar high level of mitophagy (Figure 4F and Figure S3F). When we conditionally deleted ATG5 in the brain using Nestin-Cre, we again noted a marked diminution but not complete absence of the mt-Keima red signal in regions with increased rates of mitophagy such as the dentate gyrus and Purkinje cell area of the cerebellum (Figure 4G and 4H and Figure S4A). We estimate that deletion of ATG5 resulted in an approximate 80% reduction in mitophagy (Figure S4B). This diminution seen with conditional deletion of ATG5, or previously observed with complete ATG7 deletion, was not a result of any apparent disruption of brain architecture or neuronal cell loss (Figure S4C–F) and presumably represents the degree of ATG5/LC-3-independent mitophagy that occurs within this tissue.
Figure 4.

Mitophagy varies between and within tissues. A) Assessment of basal mitophagy in the thymus (scale bar=50 μm) and B) heart (scale bar=10 μm). C) Level of mitophagy in the mouse cortex (scale bar=50 μm), D) the dentate gyrus region (scale bar=100 μm) and E) the lateral ventricle (LV; scale bar=50 μm). F) High rates of mitophagy is observed in the Purkinje (labeled P) cell layer of the cerebellum with less mitophagy evident in the granular (labeled G) layer (scale bar=50 μm). G) Representative confocal images showing levels of mitophagy in the dentate gyrus region of a four week old control mouse (Nestin-Cre+/Atg5WT/WT) versus H) a littermate lacking neuronal ATG5 expression (Nestin-Cre+/Atg5fl/fl; scale bar=50 μm).
Mitophagy assessed using STED microscopy
We next took advantage of recent improvements in light microscopy techniques to see if mt-Keima could be assessed using Stimulated Emission Depletion (STED) microscopy. We studied MEFs derived from mt-Keima embryos with STED microscopy to obtain high resolution sub-cellular images of the mt-Keima compartments (Figure 5A). When combined with a stain for lysosomes, the red mt-Keima signal again consistently appeared to be located exclusively within the acidic compartment (Figure 5B and 5C). With higher resolution of these images, mt-Keima expression along with STED microscopy allowed for the detection of red mt-Keima signal both within lysosomes, as well as within larger acidic structures, that were consistent with autolysosomes (Figure 5D). Similar STED-based approaches were employed to assess various tissues of the mt-Keima mouse. Our laser sources for this microscope were 480 nm and 561 nm rather than the 458 nm and 561 nm used for confocal, resulting in some overlap of the ‘red’ signal into the ‘green’ images. Nonetheless, these images revealed significantly enhanced tissue morphology in both cardiac and skeletal muscle (Figure S5). Indeed, the increased resolution of STED microscopy allowed for delineation of mitochondrial morphology within these tissues and the appearance of cristae-like structures within the organelle, a level of detail that is usually confined to electron microscopic approaches (Figure S5A–F).
Figure 5.

Assessment of mitophagy using mt-Keima and Stimulated Emission Depletion (STED) microscopy. A) Representative STED image of mt-Keima MEFs showing merged red and green fluorescence. B) The red only mt-Keima signal. C) Merged red mt-Keima signal with LysoSensor fluorescence (depicted in Cyan; scale bar=2 μm). D) Higher magnification of the mt-Keima red signal within lysosomes. Arrows indicate what appears to be a recently ingested autophagosome decorated with mt-Keima and contained within a presumed newly formed autolysosome (scale bar=1 μm).
Aging and HTT expression reduce mitophagy
There is growing interest in the potential role of mitophagy in aging and age-related conditions (Burte et al., 2015; Madeo et al., 2015). We therefore sought to characterize whether normal, physiological aging impacted mitophagy. We chose to concentrate on the dentate gyrus (DG) region of the brain because of the importance of this area for memory and learning, and because we had demonstrated that this region had a pronounced mitophagy signal (Figure 4D). As expected, in 3 month old mice, the DG area demonstrated high level of mitophagy (Figure 6A). A similar analysis in older mice (age 21 months) revealed a markedly diminished signal (Figure 6B). Quantification from multiple young and old mt-Keima mice revealed that mitophagy was reduced by approximately 70% in old mice (Figure 6E). We cannot exclude that some of this reduction comes from alterations in cellular composition or a small decrease in neural stem cell numbers that can happen as animals age (Rolando and Taylor, 2014). Nonetheless, at least by DAPI staining, cellular abundance in the dentate gyrus region appears similar in young and old animals (Figure 6C and 6D).
Figure 6.

Assessment of mitophagy in the dentate gyrus region and the effects of age and HTT expression. A) Levels of mitophagy in the dentate gyrus region of a three month old mt-Keima mouse (scale bar=50 μm). B) Similar analysis of a 21 month animal. C) Corresponding DAPI fluorescence in the same young and D) old mouse. E) Quantification of mt-Keima fluorescence in young (2–3 months) and old (21–23 months) mice. (n=5 per age group, **p<0.005). F) Representative confocal image of mitophagy in the dentate gyrus region of a 24 week old WT control mt-Keima mouse or G) in a similar aged mt-Keima mouse expressing the HTT transgene. H) Corresponding DAPI staining in the control animal or I) the HTT expressing animal. J) Quantification of the mt-Keima in control or HTT expressing animals. (n=5 per genotype, **p<0.005).
We next asked whether a similar decline could be observed in genetic models of neurodegeneration. Evidence suggests that in Huntington’s disease, mutant Huntingtin protein has a direct, deleterious effect on mitochondrial function, perhaps by impairing mitochondrial calcium homeostasis (Panov et al., 2002; Zuccato et al., 2010). Moreover, evidence suggest that in mouse models of Huntington’s disease there is impaired targeting of cytosolic and mitochondrial cargo to the autophagosome, leading to impaired removal of damaged proteins and organelles (Martinez-Vicente et al., 2010). For these reasons, there is increasing interest in stimulating mitophagy as a treatment for Huntington’s disease, as well as for other neurodegenerative conditions (Schapira et al., 2014). We therefore analyzed control mt-Keima mice, or mice expressing mt-Keima along with the human Huntingtin’s transgene (HTT). Analysis of the DG region demonstrated that HTT expressing mice had markedly reduced levels of mitophagy (Figure 6F–J).
Environmental and genetic perturbations of mitophagy
We next sought to analyze how environmental and genetic perturbations affect mitophagy in other tissues. We focused on the liver because of this organ’s central role in metabolism. We first analyzed hepatic mitophagy in mice fed a control diet compared to mice fed a high fat diet for 18–20 weeks. Previous observations have demonstrated that overall hepatic autophagic flux is reduced following chronic high fat feeding (Papackova et al., 2012; Yang et al., 2010). Consistent with such observations, we found that levels of mitophagy were also markedly reduced under these conditions (Figure 7A and 7B). In contrast, placing mice in a low oxygen (10% oxygen) environment for 10 days resulted in a significant increase in hepatic mitophagy (Figure 7C and 7D). This increased mitophagy is consistent with what is observed with hypoxic cells in culture (Figure 1D). We also analyzed the effects of mitochondrial damage on mitophagy rates. We crossed the mt-Keima mice with mice containing a mutated, proofreading-deficient form of the mitochondrial DNA polymerase POLGγ. Mitochondria contain a single DNA polymerase and mice expressing the knockin form of the proofreading-deficient POLGγ spontaneously accumulate high rates of mitochondrial mutations and subsequent mitochondrial damage (Kujoth et al., 2005; Trifunovic et al., 2004; Vermulst et al., 2008). When we analyzed the livers of these animals, we noted that these mice also exhibited increased hepatic mitophagy (Figure 7E and 7F). Finally, there is a growing interest concerning mitochondria and tumorgenesis (Gaude and Frezza, 2014; Weinberg and Chandel, 2015). In the course of our experiments, we imaged a mouse that for unknown reasons was losing weight and exhibiting a failure-to-thrive phenotype. Images obtained from this animal revealed markedly increased hepatic mitophagy. Subsequent necropsy revealed a non-metastatic, malignant brain tumor. This raised the possibility that animals with tumors might have augmented mitophagy, perhaps coincident with the observed systemic cachexia and presumably due to the secretion of cytokines or other circulating factors. To more formally pursue this possibility, we analyzed mice given a tail vein injection of B16 murine melanoma cells or control mice not given any tumor cells. Following tail vein injection, B16 melanoma cells go predominantly to the lung. Fifteen days after injection, when the tumor injected mice had begun to lose weight, we harvested the mice and analyzed their levels of hepatic mitophagy. Interestingly, while the liver contained no macroscopically visible tumor, we observed a marked increase in the hepatic mitophagy signal in tumor bearing animals (Figure 7G and 7H).
Figure 7.

Hepatic mitophagy is sensitive to environmental and genetic perturbations. A) mt-Keima signal in the liver of mice fed a normal chow (NC) diet or fed a high fat diet (HFD; scale bar=20 μm) for 18 weeks with corresponding lipid staining (Oil Red O) in the lower panels (scale bar=100 μm). B) Quantification of the decline in mitophagy induced by a high fat diet (n=5 per diet, **p<0.005). C) Levels of mitophagy in liver of mice maintained under normoxic conditions (21% oxygen) versus mice maintained in 10% oxygen for 10 days (Scale bar=50 μm). D) Quantification of the increase in mitophagy in livers after hypoxic stress (n=4 per condition, **p<0.01). E) Levels of mitophagy in control mice or mice containing a knockin mutation of mitochondrial polymerase γ that lacks proof reading function (POLG; scale bar=50 μm). F) Quantification of mitophagy in the control and POLG mice (n=6 per genotype, **p<0.005). G) Levels of hepatic mitophagy in control mice or mice who received a tail vein injection of B16 murine melanoma cells (scale bar=50 μm). Gross examination of the liver revealed no evidence of tumors that were however readily apparent in the lungs of these animals. H) Quantification of hepatic mitophagy in control or B16 melanoma-injected mice (n=6 per condition, **p<0.005).
Discussion
Limitations
We describe a new transgenic mouse model that allows for measurement of in vivo mitophagy. Our data demonstrates that differences in mitophagy rates exist between and within tissues. Moreover, we demonstrate that in vivo mitophagy is sensitive to environmental conditions (e.g. high fat diet, hypoxia, age), as well as to genetic perturbations (e.g. HTT expression, POLGγ mutation, ATG7 or ATG5 deletion). The ability to visualize and quantify mitochondrial turnover, in a setting where tissue architecture is maintained, suggests that the mt-Keima mouse will be a useful model to better understand how mitophagy impacts a wide range of normal physiologic responses, and how it contributes to a number of disease states.
While the properties of Keima protein make it a useful reagent to measure mitophagy, it still has limitations as a probe. Unlike most ratiometric fluorescent molecules, Keima has a single emission peak at 620 nm with a bimodal excitation spectrum centered at 440 and 586 nm (Katayama et al., 2011). In the alkaline environment of the mitochondria, the shorter wavelength (green) excitation predominates, while the longer (red) excitation predominates within the acidic lysosome. As noted in Figure 1, over 3 pH units, the red-to-green fluorescence ratio changes 7–8 fold. Nonetheless, as seen in this figure, and as noted in the initial description (Katayama et al., 2011), the Keima excitation spectrums partially overlap. Thus, within the acidic environment of the lysosome, mt-Keima is predominantly excited by the red wavelength, although it still retains some modest capacity for green excitation. This property results in the mt-Keima localized to the lysosome sometimes looking orange rather than completely red. Future genetic alterations in the structure of Keima, that more completely separate the excitation spectra would therefore enhance the probe’s utility. The recent increase in structural information about Keima suggests that by using a rational design strategy, such modification should indeed be feasible (Tantama et al., 2011; Violot et al., 2009). While the sensitivity of detection of mitophagy might depend on the level of mt-Keima expression, we found that in general, levels of Keima appeared to reflect mitochondrial abundance in tissues (Figure S6A). We noted, however, little change in overall Keima expression in conditions in which we observed large differences in mitophagy (Figure S6B–D). Furthermore, the expression of mt-Keima did not appear to have any direct effect on mitochondrial function (Figure S6E). Finally, since the measurement of mitophagy represents a ratio of red pixels to total pixels, we believe that our assessment of mitophagy should be relatively independent of mitochondrial number or mt-Keima expression.
In this study, we imaged mt-Keima mouse tissues using confocal imaging, as well as STED microscopy. The latter method provides remarkable resolution of cellular structure and tissue architecture. Nonetheless, all these images were obtained on freshly excised organs imaged ex vivo. This leads to two potential limitations. First, in heterogeneous tissues such as the brain, unless there are clear morphological landmarks or the region of interest has been previously identified with a second fluorescent reporter, it is often difficult to know one’s precise location within the tissue. Second, since Keima is pH dependent, there are concerns that ex vivo imaging could lead to artifacts. It should be noted, however, that we noted little difference when we analyzed anesthetized mice with a surgical window and compared the true in vivo red to green fluorescence in liver to the ex vivo signal from this tissue (Figure S7A). Similarly, at least over an hour of observation, the ex vivo red to green fluorescence ratio remained stable (Figure S7B). We have also performed some preliminary experiments using intravital two-photon imaging. These experiments have demonstrated that such approaches are indeed viable with mt-Keima. As such, it should be possible using such approaches to assess mitophagic flux in multiple different organs in real-time and within a living animal.
The resistance of Keima to lysosomal proteases allows integration of mitophagic events and thus provides a qualitative snapshot of mitophagic flux. One limitation of the current technique is that the ultimate fate of lysosomal Keima is not entirely defined. For instance, differences in mt-Keima signal could reflect tissue-specific differences in lysosomal degradation rates. Over a 24 hour period however, we did not see a significant increase in red fluorescence when cells were treated with a cocktail of lysosomal protease inhibitors (Figure S7C&D). This would argue that the half-life of lysosomal Keima is relatively insensitive to normal lysosomal proteolytic degradation. Turnover of lysosomal Keima may require slower processes such as lysosomal exocytosis (Xu and Ren, 2015). This lysosomal turnover rate is presumably matched with the generation of new ‘green’ mitochondria via mitochondrial biogenesis. The end result is that in the absence of genetic or environmental perturbation, the tissue red to green fluorescent ratio appears stable.
Assessing mitophagy with the mt-Keima mouse
We observed that in the absence of whole body ATG7 expression, or following tissue-specific ATG5 deletion, the level of mitophagy was markedly diminished but was not completely absent. This was also seen in previous studies employing mt-Keima in cell culture systems (Katayama et al., 2011). Such observations are consistent with other studies that have described different forms of mitophagy, some of which appear to be independent of the macroautophagy machinery and hence not dependent on ATG5 or ATG7 expression (Lemasters, 2014). It is possible that comparing the level of mt-Keima signal in the presence or absence of ATG5 or ATG7 expression represents a potential way to assess the balance between these various forms of mitophagy.
There is a growing appreciation that alterations in mitophagic flux might be important in a wide range of physiological and pathophysiological conditions. Nonetheless, the inability to measure mitophagy in the in vivo context has limited progress. There is particular interest in the role of autophagy and more specifically mitophagy as a contributor to neurodegenerative disease, and to aging itself (Madeo et al., 2015; Rubinsztein et al., 2011; Wong and Cuervo, 2010). In this regard, our observations that mitophagy appears to decline with age is consistent with other observations that overall autophagic flux, for unclear reasons, appears to decline with age (Del Roso et al., 2003). Aging is also accompanied by an increase in damaged and dysfunctional mitochondria (Balaban et al., 2005; Lopez-Otin et al., 2013), a situation that might, by itself, be expected to result in increased mitophagy. The fact that mitochondrial mutations give rise to increased rates of mitophagy is supported by our observations with POLGγ knockin mice, that accumulate high rates of mitochondrial mutations and in which we see higher rates of mitophagy. Taken together, these observations would suggest that the age-dependent decline in autophagy/mitophagy is physiologically more important than the age-dependent increase in mitochondrial dysfunction. It leaves open the possibility that the age-dependent decline in mitophagy is actually the primary cause or a major contributor to the observed age-dependent increase in mitochondrial dysfunction. This hypothesis would posit that the observed age-dependent increase in mitochondrial ROS, dysfunction and mutational load is not primarily a result of increased mitochondrial damage as we age, but rather a consequence of an inability to rid tissues of these damaged organelles due to the age-dependent decline in mitophagy.
In summary, we provide a new resource that provides a convenient and robust strategy to measure mitophagy in vivo. This mouse model should have numerous potential applications in assessing both normal and diseased states where alterations in mitophagy are believed to play a significant role.
Experimental Procedures
Generation of mt-Keima transgenic mice
The mt-Keima transgenic mouse was generated using the TARGATTTM site-specific transgenic technology (Applied StemCell Inc.) according to the manufacturer’s protocol. Briefly, the mt-Keima cDNA was subcloned into a vector (pBT378) that contains the attB recombination site. DNA was purified and injected along with ϕC31 integrase mRNA into the pronuclei of fertilized eggs collected from FVB mice (Applied StemCell Inc.) which contain tandem attP recombination sites within the Hipp11 (H11) locus. After overnight culture, 2-cell embryos were implanted into the oviduct of pseudo-pregnant foster mother mice. After the pups were born, tail biopsies were screened for site-specific integration of mKeima gene by PCR. All animal studies were done in accordance and approval of the NHLBI Animal Care and Use Committee. Primers for mt-Keima genotyping included: forward primer, 5′-GAG CAG ACC GTG AAG CTG AC-3′ and reverse primer, 5′-GCC ATG TAG TCG TTG CCG AT-3′.
Mouse Tissue Collection
Fresh tissues were used for both confocal and STED microscopy studies. For the brain, tissues were rapidly dissected out, rinsed with cold PBS and then further processed into coronal sections (0.5–2 mm). These sections were placed into cold PBS and immediately analyzed or at indicated time points. For other tissues, following a rinse with cold PBS, tissues were cut into small pieces (approximately 100 mm3). The tissues were placed onto a 35 mm coverglass #1.5 bottom micro-well dish (MatTek) and analyzed immediately.
For hepatic intravital imaging, mice were anaesthetized with an intraperitoneal injection (4 μl/g) of 1: 1 (v/v) Xylazine (20 mg/ml): Ketamine (100 mg/ml). To expose the liver, a 2–3 cm incision was made along the ribcage starting from the xiphoid process and ending at the left lateral side of the abdomen. Mice were placed on a heated microscope stage and the left lateral lobe of the liver exposed. Carbomer gel was added from both sides of the liver to stabilize the organ. In these experiments, liver fluorescence was examined with a Leica SP5 microscope (confocal/multiphoton) and a 20x 0.75 N.A. objective lens that has a higher acquisition speed (resonant scanner 8000 Hz), in an effort to minimize in vivo motion artifacts. Sequential images were collected with 458 and 561 nm excitation at the indicated time points with a 570–700 nm emission bandwidth. After intravital imaging, the liver was rapidly dissected out and processed into coronal sections as above. Mean intensity of each channel was calculated using Zeiss ZEN software.
Confocal microscopy
Fluorescent samples were routinely examined with a Zeiss LSM 780 confocal microscope (Carl Zeiss MicroImaging) equipped with a Plan-Apochromat 20x/0.8 NA, 40x/1.4 NA and a 63x/1.40 NA oil immersion objective lens. Fluorescence of mt-Keima was imaged in two channels via two sequential excitations (458 nm, “green” and 561 nm, “red” respectively) and using a 570-695 nm emission range. Laser power was set at the lowest output that would allow clear visualization of the mt-Keima signal, and were individualized for each experimental condition. Imaging settings were maintained with the same parameters for comparison between different experimental conditions. In select experiments, DAPI fluorescence was imaged using a 405 nm excitation and a 415–470 nm emission filter, and LysoSensor Green was imaged using a 488 nm excitation and a 495–550 nm emission filter. Confocal time-lapse experiments were performed using a recently described HeLa cell line stably expressing both mt-Keima and YFP-parkin (Nezich et al., 2015) plated on 35 mm coverglass #1.5 chamber dishes (Mat-Tek), in DMEM containing 10% FBS supplemented with penicillin-streptomycin and incubated, where indicated, with FCCP (5 μM) and oligomycin (5 μM) to induce mitophagy. Cells were imaged every 15 minutes starting at ~ 10min after addition of FCCP/oligomycin. Images were obtained for up to 19 hours taking z-stacks at multiple positions using a 40x NA1.4 oil objective, in an environmental chamber maintained at 37°C and 5% CO2. YFP was imaged with a 514 nm excitation and 520–570 emission filters and mt-Keima was imaged using a “green” and “red” channel as described above. Time-lapse movies were created in Imaris software and assembled using Fiji (ImageJ) software. Representative confocal images were processed using Imaris software by contrast linear stretch only. When comparisons were made, the same linear processing parameters were applied.
Calculation of mitophagy based on mt-Keima signal was performed employing the original images using Zeiss ZEN software on a pixel by pixel basis. Every pixel in the image is plotted in the scatter diagram based on its intensity level from each channel. ‘Green’ intensity is shown on the x-axis and ‘red’ intensity is shown on the y-axis. As pixel intensity dictates its position on the scatter plot, pixels that have high red intensity are designated in the software by use of the crosshairs and quadrants. Pixels that have low intensity levels in both channels are referred to as background and are not taken into consideration for analysis. The fold change of mitophagy was calculated by comparing in matched images the numbers of pixels that have high red intensity over the number of all pixels (Figure S1). The average of four images from each tissue sample was taken and the values were normalized to the average value seen in the controls, assigned the value of one. In each experimental model, all imaging parameters remain the same for all data acquisition. We performed all image analysis using Zeiss Zen software, however, we believe other image analysis software would be equally effective. The ratiometric heatmap images (fluorescence intensity 561nm/458nm) were generated and analyzed using an in-house custom program written in the IDL (Exelis Boulder, CO) programing language. To assess the pH dependence of mt-Keima, we permeabilized HeLa cells with 0.01% Triton X-100 and equilibrated these permeablized cells with DMEM (without phenol red) with pHs ranging from 4 to 7. The ratio of the mean intensity of signals following excitation at 561 and 458 nm was calculated and normalized to the red-to-green values of pH 7. Results are presented as the mean ± standard deviation. The statistical significance of differences between groups was evaluated by Student’s t test and analysis of variance (ANOVA).
Supplementary Material
Acknowledgments
We are grateful for Junhui Sun, Michele Allen (NHLBI phenotyping Core), Zuxi Yu (NHLBI Pathology Core), Phil McCoy (NHLBI FACS Core), Pradeep Dagur, Natalie Porat Shilom and Shihui Liu for experimental assistance. We thank T. Prolla and M. Komatsu for mice, R. Youle for the YFP-Parkin cell line and A. Miyawaki for the original mt-Keima construct. This work was supported by a grant from the Leducq Foundation, NIH Intramural Funds and a National Research Foundation of Korea Grant (2009-0093197). The mt-Keima mouse is being made available through The Jackson Laboratory Repository with the JAX Stock No. 028072.
Footnotes
Author Contribution: N.S., J.Y., J.L., D.M., C.L., K.M.H., M.M.F., C.A.C. performed the experiments, N.S., J.Y, Y.H.Y and C.A.C analyzed the data and N.S. and T.F. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker MJ, Palmer CS, Stojanovski D. Mitochondrial protein quality control in health and disease. Br J Pharmacol. 2014;171:1870–1889. doi: 10.1111/bph.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510:370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- Chen CS, Bach G, Pagano RE. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc Natl Acad Sci U S A. 1998;95:6373–6378. doi: 10.1073/pnas.95.11.6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp Gerontol. 2003;38:519–527. doi: 10.1016/s0531-5565(03)00002-0. [DOI] [PubMed] [Google Scholar]
- Dolman NJ, Chambers KM, Mandavilli B, Batchelor RH, Janes MS. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy. 2013;9:1653–1662. doi: 10.4161/auto.24001. [DOI] [PubMed] [Google Scholar]
- Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014;2:10. doi: 10.1186/2049-3002-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun. 2014;5:4004. doi: 10.1038/ncomms5004. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Kim TY, Wang D, Kim AK, Lau E, Lin AJ, Liem DA, Zhang J, Zong NC, Lam MP, Ping P. Metabolic labeling reveals proteome dynamics of mouse mitochondria. Mol Cell Proteomics. 2012;11:1586–1594. doi: 10.1074/mcp.M112.021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M, Pagano RE. Sorting of an internalized plasma membrane lipid between recycling and degradative pathways in normal and Niemann-Pick, type A fibroblasts. J Cell Biol. 1990;111:429–442. doi: 10.1083/jcb.111.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3) Redox Biol. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38:26–35. doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. Embo J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015;210:435–450. doi: 10.1083/jcb.201501002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Papackova Z, Dankova H, Palenickova E, Kazdova L, Cahova M. Effect of short- and long-term high-fat feeding on autophagy flux and lysosomal activity in rat liver. Physiol Res. 2012;61(Suppl 2):S67–76. doi: 10.33549/physiolres.932394. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Richter U, Lahtinen T, Marttinen P, Myohanen M, Greco D, Cannino G, Jacobs HT, Lietzen N, Nyman TA, Battersby BJ. A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr Biol. 2013;23:535–541. doi: 10.1016/j.cub.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Rolando C, Taylor V. Neural stem cell of the hippocampus: development, physiology regulation, and dysfunction in disease. Curr Top Dev Biol. 2014;107:183–206. doi: 10.1016/B978-0-12-416022-4.00007-X. [DOI] [PubMed] [Google Scholar]
- Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4:205–213. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr, Gottlieb RA, Przyklenk K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–184. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW, Greenamyre JT, Bezard E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet. 2014;384:545–555. doi: 10.1016/S0140-6736(14)61010-2. [DOI] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. Embo J. 2014;33:2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantama M, Hung YP, Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J Am Chem Soc. 2011;133:10034–10037. doi: 10.1021/ja202902d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, Luo L. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc Natl Acad Sci U S A. 2011;108:7902–7907. doi: 10.1073/pnas.1019507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Violot S, Carpentier P, Blanchoin L, Bourgeois D. Reverse pH-dependence of chromophore protonation explains the large Stokes shift of the red fluorescent protein mKeima. J Am Chem Soc. 2009;131:10356–10357. doi: 10.1021/ja903695n. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Le L, Fan Y, Lv L, Zhang J. Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition. Autophagy. 2012;8:1071–1084. doi: 10.4161/auto.20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol. 2015;77:57–80. doi: 10.1146/annurev-physiol-021014-071649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, Ney PA. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood. 2009;114:157–164. doi: 10.1182/blood-2008-04-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.