

Abstract
Sepsis and septic shock are enormous public health problems with astronomical financial repercussions on health systems worldwide. The central nervous system (CNS) is closely intertwined in the septic process but the underlying mechanism is still obscure. AMP-activated protein kinase (AMPK) is a ubiquitous energy sensor enzyme and plays a key role in regulation of energy homeostasis and cell survival. In this study, we hypothesized that activation of AMPK in the brain would attenuate inflammatory responses in sepsis, particularly in the lungs. Adult C57BL/6 male mice were treated with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR, 20 ng), an AMPK activator, or vehicle (normal saline) by intracerebroventricular (ICV) injection, followed by cecal ligation and puncture (CLP) at 30 min post-ICV. The septic mice treated with AICAR exhibited elevated phosphorylation of AMPKα in the brain along with reduced serum levels of aspartate aminotransferase, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6), compared with the vehicle. Similarly, the expressions of TNF-α, IL-1β, keratinocyte-derived chemokine and macrophage inflammatory protein-2 as well as myeloperoxidase activity in the lungs of AICAR-treated mice were significantly reduced. Moreover, histological findings in the lungs showed improvement of morphologic features and reduction of apoptosis with AICAR treatment. We further found that the beneficial effects of AICAR on septic mice were diminished in AMPKα2 deficient mice, showing that AMPK mediates these effects. In conclusion, our findings reveal a new functional role of activating AMPK in the CNS to attenuate inflammatory responses and acute lung injury in sepsis.
INTRODUCTION
Sepsis and septic shock are life-threatening medical conditions, characterized by uncontrolled or overwhelming systemic inflammatory response to an infection and accounts for more than 750,000 cases annually in the United States (1). Despite significant advances and extensive therapeutic approaches, sepsis remains a major health concern and leading cause of high mortality worldwide (2–4). Human and animal studies have demonstrated that progressive multiple organ failure is the most common cause of death following sepsis, with the lungs usually representing the first organ to fail (5). Predisposition of the immune response has been shown to be critical in the development of an exaggerated inflammatory reaction and ensuing acute lung injury (ALI) (6). The clinical pathology of ALI includes increased vascular permeability, inflammation, oxidative stress, apoptosis, pulmonary edema and the accumulation of activated neutrophil in lung tissue and eventually cell death (7–11).
AMP-activated protein kinase (AMPK) is a serine/threonine kinase that functions as an energy sensing enzyme and is activated in response to an increase of the AMP/ATP ratio during hypoxia, glucose deprivation, heat shock or reduction in mitochondrial oxidative phosphorylation (12,13). AMPK is a heterotrimeric complex consisting of three subunits, α catalytic subunit and regulatory β and γ subunits (14). Two isoforms have been identified for both α subunit (α1 and α2) and β subunit (β1 and β2), and three isoforms have been reported for the γ subunit (γ1, γ2 and γ3) (14). Most recently, several reports indicate that the function of AMPK is not only restricted to the maintenance of energy metabolism, but also to coordination of several housekeeping mechanisms and involvement in modulating oxidative stress and inflammatory mediators (15–18). An adenosine monophosphate analog, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), has been used as a pharmacological activator to upregulate AMPK activity (19). Administering AICAR has been shown to inhibit inflammation and to alleviate organ injury in rodents (13,20–22).
There is growing evidence that the central nervous system (CNS) plays an important and functionally relevant role in regulating the inflammatory response. For example, the vagus nerve and cholinergic pathway have been demonstrated to be involved in modulating proinflammatory cytokine-induced injury locally and systemically in infectious diseases (23–26). However, the molecular mechanisms of the CNS in regulating systemic inflammation are still not well characterized. The hypothalamus, a key region in the CNS, controls several peripheral physiological activities through a broad network of hormonal and neuronal communication (27). AMPKα is expressed in hypothalamic neurons as a fuel sensor and counteracts energy deficits in the brain (28). More specifically, the AMPKα1 is primarily cytoplasmic, whereas AMPKα2 is predominantly nuclear and plays a role in transcriptional regulation (29). In addition, the α2 catalytic subunit is highly expressed in neurons in comparison with α1 (30). Thus, we hypothesized that activation of AMPK in the CNS could attenuate inflammatory responses and reduce ALI in sepsis.
In this study, we examined the mechanistic and beneficial effect of activating centrally located AMPK in septic mice induced by cecal ligation and puncture (CLP). AICAR administration to the brain through intracerebroventricular (ICV) injection attenuated the peripheral inflammation, as evidenced by decreased production of cytokines and chemokines, and reduced lung injury in septic mice. We also provided the evidence that the protective effect of AICAR is mediated though the presence of AMPKα2 in the brain by using AMPKα2 deficient mice.
MATERIALS AND METHODS
Animals
Male age-matched wild-type C57 BL/6 mice (25–30 g) (Taconic) and AMPKα2 knockout (Prkaa2–/–) mice were obtained from Benoit Viollet (INSERM) and were maintained at the Feinstein Institute for Medical Research (14). They were housed in a temperature-controlled room on a 12-h light–dark cycle and fed a standard mouse diet. All experiments were performed in accordance with the National Institutes of Health guidelines for use of experimental animals (Guide for the Care and Use of Laboratory Animals, 8th edition, 2011), and this study was approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research.
Intracerebroventricular (ICV) Injection
Stereotactic and surgical materials were obtained from Stoelting and Hamilton. Animals were lightly anesthetized with inhalational isoflurane and the head of the animal was secured on the stereotactic apparatus. A thermal pad was used to maintain core body temperature during the procedure. The head of the animal was shaved and cleansed with 10% povidone-iodine wash. A midline incision was made, extending from the eyes to the back of the cranium to expose the bregma. Either 20 ng AICAR (Sigma-Aldrich) or 2 μL vehicle (normal saline) was injected into the lateral ventricle area by using predetermined coordinates (AP, 0.34 mm from bregma; lateral, 1.0 m; and vertical, 2.2 mm).
Animal Model of Sepsis
Thirty minutes after ICV injection, the animal was removed from the stereotactic apparatus and placed in supine position. The ventral abdomen was shaved and cleansed with 10% povidone–iodine wash and cecal ligation and puncture (CLP) was performed as previously described (31). One- to 2-cm midline incision was performed to allow exposure of the cecum and tightly ligated ~1.0 cm from the tip with a 3-0 silk suture. A through double puncture of the cecum was performed using a 22-gauge needle. A small amount of feces was expressed from the perforated sites and returned to the peritoneal cavity. The laparotomy site was then closed with 6-0 silk suture. Sham-operated animals underwent the same procedure with the exception that the cecum was neither ligated nor punctured. The CLP animals subcutaneously received 1 mL isotonic normal saline immediately after the surgery.
Western Blotting
Hypothalamus and surrounding tissue were lysed and homogenized in 300 μL lysis buffer (10 mmol/L Tris-HCL pH 7.5, 120 mmol/L NaCl, 1% NP-40, 1% sodium deoxycholate and 1% Triton X-100) containing protease and phosphatase inhibitor cocktails (Roche) using a sonic dismembrator on ice. Samples were centrifuged at 14,000g for 20 min at 4°C, and the supernatant were collected. Following measurement of sample protein concentration by Pierce BCA protein assay kit (Pierce Biotechnology), 60 μg samples were separated on 4% to 12% Bis-Tris gradient gels and transferred to nitrocellulose membranes. Membranes were blocked by incubation with 0.1% casein followed by incubation with primary antibody against p-AMPKα, t-AMPKα (Cell Signaling), or β-actin (Sigma-Aldrich). After washing, membrane was incubated with appropriate fluorescent secondary antibodies. Bands were detected using the Odyssey FC Dual-Mode Imaging system 2800 (LI-COR).
Measurements of Serum Liver Enzymes and Cytokines
Whole-blood samples were centrifuged at 4,000g for 12 min to collect serum, which was then stored at –80°C before use. The activity of aspartate aminotransferase (AST) was determined by a commercial assay kit from Pointe Scientific. Serum tumor necrosis factor-α (TNF-α), interleukin 1β (IL-1β) and interleukin-6 (IL-6) levels were determined by an enzyme-linked immunosorbent assay kit specific for mouse (BD Biosciences). The assays were carried out according to the instructions provided by the manufacturer.
Real-Time Reverse Transcriptase–Polymerase Chain Reaction (RT-PCR) Analysis
Total RNA was extracted from lung tissue by TRIzol reagent (Invitrogen) and reverse-transcribed in to cDNA by using murine leukemia virus reverse transcriptase (RT) (Applied Biosystems). A PCR reaction was carried out in 25 μL final volume containing 0.08 μmol of each forward and reverse primer, 5 μL cDNA, 6.5 μL H2O and 12.5 μL SYBR Green PCR Master Mix (Applied Biosystems). Amplification was conducted in an Applied Biosystems 7300 real-time PCR machine under the thermal profile of 50°C for 2 min and 95°C for 10 min, followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. The level of mouse β-actin mRNA was used for normalization and each specific mRNA was conducted in duplicate. Relative expression of mRNA was calculated by the 2–ΔΔCt method, and results were expressed as fold change in comparison with controls. The sequences of primers for this study are listed in Table 1.
Table 1.
A list of primer sequences used in this study.
| Name | GenBank | Forward | Reverse |
|---|---|---|---|
| TNF-α | X_02611 | AGACCCTCACACTCAGATCATCTTC | TTGCTACGACGTGGGCTACA |
| IL-1β | NM_008361 | CAGGATGAGGACATGAGCACC | CTCTGCAGACTCAAACTCCAC |
| KC | NM_008176 | GCTGGGATTCACCTCAAGAA | ACAGGTGCCATCAGAGCAGT |
| MIP-2 | NM_009140 | CCCTGGTTCAGAAAATCATCCA | GCTCCTCCTTTCCAGGTCAGT |
| β-Actin | NM_007393 | CGTGAAAAGATGACCCAGATCA | TGGTACGACCAGAGGCATACAG |
Myeloperoxidase (MPO) Activity Assay
Lung tissues were sonicated in 50 mmol/L potassium phosphate buffer containing 0.5% hexadecyl trimethyl ammonium bromide. After the centrifugation, the supernatant was diluted in reaction solution containing o-dianisidine hydrochloride and H2O2. The rate of change in optical density (OD) for 1 min was measured at 460 nm to calculate MPO activity as described previously (32).
Histological Evaluation and Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Staining of the Lungs
Lung tissues were taken from the upper and lower lobes 20 h following CLP and stored in 10% formalin before being fixed in paraffin. Tissues were then sectioned to 4-μm cuts and stained with hematoxylin-eosin. For TUNEL staining, fluorescence staining was performed using a commercially available in situ Cell Death Detection Kit (Roche). The assay was conducted according to the manufacturer’s instructions. The nucleus was stained with propidium iodide. Results were expressed as the average number of TUNEL-positive staining cells per 10 microscopic fields.
Statistical Analysis
All data are expressed as a mean ± SE (n = 6–8/group) and compared by one-way analysis of variance (ANOVA) and the Student-Newman-Keuls (SNK) test. Differences in values were considered significant if P < 0.05.
RESULTS
AICAR Activates AMPKα in the Brain of Septic Mice
We first verified the effect of centrally administered AICAR on the activation of AMPKα. At 20 h after CLP, the hypothalamus sections of the mice were harvested and subjected to Western blot analysis. The phosphorylated level of AMPKα in the brain remained unchanged after CLP, while its level increased by 2.18-fold in the AICAR treatment group in comparison with the vehicle treated septic mice (Figure 1). This result indicated that ICV-injection of IACAR effectively stimulated AMPK activity in the brain of septic mice.
Figure 1.
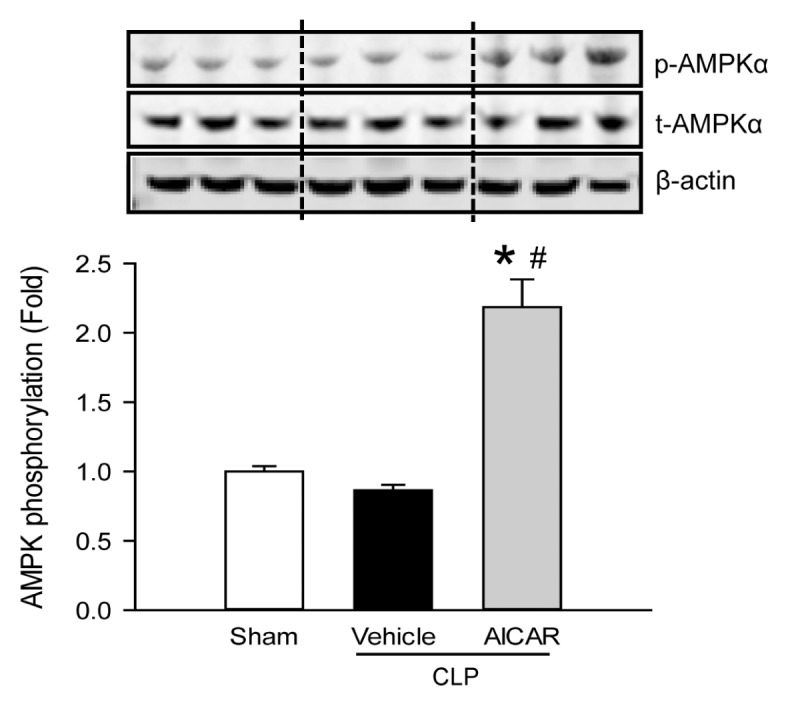
Phosphorylation of AMPK in the brain. The hypothalamus and surrounding tissue of sham, vehicle- and AICAR-treated mice were harvested at 20 h after CLP. The tissue was lysed and subjected to Western blotting against phosphorylated (p-AMPKα) and total AMPK (t-AMPKα). Histogram shows mean densitometric analysis of bands after normalizing with t-AMPK and β-actin. Expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SE (n = 6–8 per group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Attenuates Organ Injury and Systemic Inflammation in Septic Mice
We then examined the effect of AICAR treatment on the tissue damage and inflammatory response in sepsis. At 20 h after CLP, the serum level of AST, an organ injury marker, increased by 7.4-fold in the vehicle, compared with the sham (Figure 2A). By contrast, AICAR treatment significantly decreased the AST level by 36.7% as compared with the vehicle group (Figure 2A). Similarly, we also found significantly increased serum levels of proinflammatory cytokines, TNF-α (25.3-fold), IL-1β (24.3-fold) and IL-6 (6.8-fold) in the vehicle group as compared with the respective sham groups. Importantly, AICAR treatment significantly decreased the level of TNF-α by 70.5%, IL-1β by 90.3%, and IL-6 by 42.2% as compared with the vehicle (Figures 2B–D).
Figure 2.

Alterations in serum levels of organ injury marker and proinflammatory cytokines. Blood of sham, vehicle- and AICAR-treated mice were harvested at 20 h after CLP for measuring (A) AST, (B) TNF-α, (C) IL-1β, and (D) IL-6. Data are expressed as mean ± SE (n = 6–8 per group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Ameliorates Lung Damage and Apoptosis in Septic Mice
The lungs are very vulnerable in sepsis (33). We examined the lung tissue at 20 h after CLP with hematoxylin and eosin staining. As shown in Figure 3, CLP induced severe lung injury shown with septal thickening, proteinaceous exudate, pulmonary edema and enhanced inflammatory infiltrates as compared with the sham. By contrast, the severity of lung damage was ameliorated by AICAR treatment, shown with a better integrity of microscopic structure as compared with the vehicle-treated animals (Figure 3).
Figure 3.

Morphological changes in the lungs. The lung tissues of sham, vehicle- and AICAR-treated mice were harvested at 20 h after CLP and subjected to histological analysis with hematoxylin–eosin staining. Representative images of lung histology from n = 6–8 per group. Original magnification 100× (top panels); 200× (bottom panels).
Apoptosis plays a major role in the pathogenesis of sepsis-induced organ injury (34). We applied TUNEL assay to examine the apoptosis in the lungs by immunofluorescence. In the vehicle group, the TUNEL-positive cells (green fluorescence) were well detected, while they were barely observed in the sham (Figure 4A). On the other hand, the number of TUNEL-positive cells in the lung tissues of the AICAR-treated mice was reduced significantly (63.3%) as compared with the vehicle group (Figure 4B).
Figure 4.

Apoptosis in the lungs after CLP. The lung tissues of sham, vehicle- and AICAR-treated mice were harvested at 20 h after CLP and subjected to TUNEL assay. (A) Representative images of TUNEL staining (green fluorescent) and nuclear counterstaining (red fluorescent) of lung sections. Original magnification 100×. Scale bar, 50 μm. (B) The number of apoptotic cells in the lung quantified from TUNEL staining. Data are expressed as mean ± SE (n = 6–8 per group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Suppresses Lung Inflammation and Neutrophil Infiltration in Septic Mice
Excessive production of proinflammatory cytokines is one of the important contributing factors for the lung injury (35). Lung tissue was harvested at 20 h after CLP and subjected to RT-PCR and ELISA analysis. The mRNA level of TNF-α was increased by 53.1-fold in vehicle-treated animal as compared with the sham. Parallel to mRNA expression, we found increased protein levels of TNF-α in vehicle-treated animal as compared with the sham (Figures 5A, B). AICAR treatments significantly suppressed mRNA and protein levels of TNF-α by 72.5% and 44.2%, respectively, compared with the vehicle (Figures 5A, B). Similarly, the mRNA and protein levels of IL-1β were elevated significantly in the vehicle group as compared the sham, while their levels were reduced by 76.9% and 47.4%, respectively, compared with the vehicle (Figures 5C, D).
Figure 5.

Alterations in expression of proinflammatory cytokines in the lungs. The lung tissues of sham, vehicle- and AICAR-treated mice were harvested at 20 h after CLP for measuring the mRNA levels of (A) TNF-α and (C) IL-1β by real-time RT-PCR as well as protein levels of (B) TNF-α and (D) IL-1β by ELISA. The mRNA expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SE (n = 6–8 per group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
Chemokines keratinocyte derived-chemokine (KC) and macrophage inflammatory protein-2 (MIP-2) both have been shown to participate in the pathogenesis of lung injury (36,37). The mRNA levels of KC and MIP-2 increased by 748- and 1415-fold, respectively, in the vehicle as compared with the sham (Figures 6A, B). By contrast, a significant reduction of KC mRNA by 76.2% and MIP-2 mRNA by 64.2% was observed in lung tissues of AICAR-treated mice (Figures 6A, B). We further measured the myeloperoxidase activity (MPO) in the lungs to evaluate the neutrophil infiltration. The MPO activity increased by 3.1-fold in vehicle-treated group as compared with the sham, while AICAR treatment decreased the MPO activity by 42.4% as compared with the vehicle (Figure 6C).
Figure 6.

Alterations in expression of chemokines and myeloperoxidase activity in the lungs. The lung tissues of sham, vehicle- and AICAR-treated mice were harvested at 20 h after CLP for measuring the mRNA levels of (A) KC and (B) MIP-2 by real-time RT-PCR. (C) Lung myeloperoxidase (MPO) activity was determined spectrophotometrically. The mRNA expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SE (n = 6–8 per group). *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Activity Is Mediated by AMPKα2 in the Brain of Septic Mice
To investigate the molecular mechanism by which AICAR treatment to the CNS attenuated organ injury and inhibited inflammation, we undertook a genetic approach. AMPKα2 is highly expressed in the neurons of the brain. Thus, we performed the CLP in AMPKα2 knockout (KO) mice after ICV injection of either vehicle or AICAR as described in the wild-type (WT) mice. We first measured the serum AST level and observed its elevation in the vehicle group of AMPKα2 KO mice, which was similar to that in the WT mice (Figure 7A). AICAR treatment only reduced AST levels by 13.8% without statistical significance as compared with the vehicle (Figure 7A). Similarly, there was an increase in serum levels of TNF-α and IL-1β in septic AMPKα2 KO mice, however, AICAR treatment did not significantly change their levels as compared with the vehicle (Figures 7B, C).
Figure 7.

Alterations in serum levels of organ injury marker and proinflammatory cytokines in AMPKα2 KO mice. Blood of sham, vehicle- and AICAR-treated AMPKα2 KO mice were harvested at 20 h after CLP for measuring (A) AST, (B) TNF-α and (C) IL-1β. Data are expressed as mean ± SE (n = 6–8 per group). P = nonsignificant (NS).
We also examined the mRNA levels of proinflammatory cytokines and chemokines in the lungs by RT-PCR. There was an increase in mRNA levels of TNF-α, IL-1β, KC and MIP-2 in the lungs of vehicle-treated AMPKα2 KO mice as compared with their respective sham (Figures 8A–D). However, AICAR-treated AMPKα2 KO septic mice did not show any significant changes in the expression levels of these cytokines and chemokines as compared with the vehicle-treated AMPKα2 KO septic mice (Figures 8A–D). Taken together, these results suggest that the protective effects of administered ICV with AICAR in the WT mice may mainly mediate through AMPKα2-dependent mechanisms in the brain.
Figure 8.

Alterations in expression of proinflammatory cytokines and chemokines in the lungs of AMPKα2 KO mice. The lung tissues of sham, vehicle- and AICAR-treated AMPKα2 KO mice were harvested at 20 h after CLP for measuring (A) TNF-α, (B) IL-1β, (C) KC and (D) MIP-2 by real time RT-PCR. The expression in the sham group is designated as 1 for comparison. Data are expressed as mean ± SE (n = 6–8 per group). P = nonsignificant (NS).
DISCUSSION
Sepsis is the leading cause of death among critically ill patients and is the most common risk factor for ALI. Ungoverned inflammation, characterized by cytokine storm and subsequent neutrophil sequestration is an underlying component of sepsis associated organ failure (38). Thus, elucidation of novel molecular and cellular pathways that influence the pathogenesis of sepsis and its associated complication could provide greater insight into the mechanisms of disease pathology.
The significance of the brain AMPK stimulation in regulation of inflammation in sepsis is not known. We have used a combination of pharmacological and genetic approaches for the study. First, we demonstrate that ICV injection of AICAR markedly increases the phosphorylation of AMPK in the brain of CLP-induced septic mice. Such AICAR administration reduced organ injury and inhibited systemic inflammation. In more specific organs, both the severity of damage and the induction of apoptosis in the lungs of septic mice are attenuated by AICAR treatment, which is associated with a reduction of cytokine and chemokine production as well as neutrophil infiltration. On the other hand, AICAR-mediated protective effect on lowering organ injury and inflammation in septic WT mice is diminished in septic AMPKα2 KO mice, suggesting that brain AMPKα2 may be a key player to mediate AICAR activity.
Activation of AMPK by systemic administration of AICAR has been shown to suppress inflammatory responses in other studies. Zhao and coworkers demonstrate that AICAR reduces the proinflammatory activity of neutrophils and decreases the severity of ALI through AMPK activation, linking to cellular responses to metabolic stress (13). Escobar et al. demonstrate that activation of AMPK by AICAR ameliorates liver and renal injury in septic mice, which is associated with a decrease in circulating cytokines and tissue inflammation (39). In this study, we provide new evidence of activating the CNS in regulating the systemic responses. By directly injecting AICAR into the brain, we have observed a significant reduction of serum levels of proinflammatory cytokines TNF-α, IL-1β and IL-6 in septic mice.
Another important aspect of our study is to observe an improvement of lung morphology and inhibition of lung apoptosis in septic mice with central AICAR injection. As known, lung tissue damage is observed in 90% of patients dying from sepsis and its associated complication (40). The histopathological features of lung injury in septic mice demonstrated here are resembled in the clinical condition of patients with sepsis (41). Other studies have indicated that apoptosis plays an important role on progression of sepsis (42,43). Oberholzer and colleagues suggested that targeting signaling pathways that lead to apoptosis would represent a new therapeutic target for the patient with sepsis or other related clinical conditions (34). By systemic administration, AICAR has been shown to decrease the apoptosis in different cells associated with various disease conditions (44–46). Rossi and Lord describe that AICAR treatment decreases the neutrophil apoptosis through the AMPK activation, suggesting an important role of AMPK in normal function of neutrophils (45). Similarly, Kim et al. demonstrate that AICAR reduces the palmitate-induced apoptosis in neuron cells by activation of AMPK (46).
In addition to inhibiting systemic inflammation, central AICAR administration also decreases local inflammation in the lungs. We have demonstrated a significant decrease in expression of cytokines TNF-α.and IL-1β in the lungs with AICAR treatment. Moreover, central AICAR administration also effectively inhibits the expression of KC and MIP-2 in the lungs. It has been reported that elevated expression of KC and MIP-2 potently stimulate neutrophil influx in the lungs, while suppression of these chemokines markedly inhibits neutrophil sequestration in the lungs (47). Consistent with this mechanism, we have detected decreased MPO activity, a marker for neutrophil infiltration (48). Considered together, our findings suggest that activation of the brain AMPK by AICAR for a protective milieu to the lung in sepsis may confer inhibition of the production of inflammatory mediators.
We then further identify the specific target in the CNS for AICAR’s activity in regulating systemic inflammation. When comparing the serum levels of AST, TNF-α and IL-1β in septic AMPKα2 KO mice between vehicle and AICAR ICV injections, the beneficial effects of AICAR observed in the septic WT mice are diminished in the AMPKα2 KO mice.
Moreover, the AICAR’s effect on inhibiting the expression of cytokines and chemokines in the lungs of the septic WT mice disappeared in the septic AMPKα2 KO mice. These results clearly indicate that AMPKα2 in the brain is majorly responsible for central AICAR stimulation. As it has been indicated that the CNS can modulate the systemic inflammation and cytokines production by: 1) activation of the sympathetic nervous system and release of norepinephrine which target the immune cells expressing adrenergic receptors (49,50); 2) stimulation of the vagus nerve-mediated cholinergic antiinflammatory pathway (23,51,52); and 3) activation of the hypothalamic-pituitary-adrenal (HPA) axis for glucocorticoids secretion in response to adrenocorticotropic hormone and immune suppression (53,54). However, which pathway(s) serves as downstream-of-brain AMPK activation for modulating the systemic and peripheral tissue inflammation following sepsis needs further investigation.
By the same token, Giri et al. present the evidence that activation of AMPKα2 by AICAR attenuates the LPS-mediated induction of proinflammatory mediators in rat primary astrocytes and microglia cells through inhibiting NF-κB and C/EBP transcription factors (22). A study by Bernik et al. demonstrates that intracerebral administration of a tetravalent guanylhydrazone molecule that inhibited TNF-α production increases efferent vagus nerve activity and inhibits inflammation outside the CNS (55). Moreover, it has been shown that activation of hypothalamic AMPK can prevent LPS-induced hypoglycemia in mice liver, suggesting centrally located AMPK in the regulation of peripheral physiological activities (56).
CONCLUSION
Our results suggest that centrally administered AICAR protects against sepsis-induced pulmonary injury and apoptosis, and that this protection is associated with a decrease in circulating proinflammatory cytokines. Furthermore, hypothalamic AMPKα2 mediates AICAR activity to regulate systemic inflammatory response in sepsis. Thus, further understanding the interaction between the CNS and systemic activity will provide another direction for sepsis treatment.
ACKNOWLEDGMENTS
Supported in part by National Institutes of Health (NIH) grants GM057468 and GM053008 (to P Wang). The authors thank Benoit Viollet (INSERM, Institut Cochin, Paris, France) for generously providing the AMPKα2 knockout mice.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Mulchandani N, et al. (2015) Stimulation of brain AMP-activated protein kinase attenuates inflammation and acute lung injury in sepsis. Mol. Med. 21:637–44.
REFERENCES
- 1.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41:1167–74. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 2.Khan MM, Yang WL, Wang P. Endoplasmic reticulum stress in sepsis. Shock. 2015;44:294–304. doi: 10.1097/SHK.0000000000000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Z, et al. Ursolic acid improves survival and attenuates lung injury in septic rats induced by cecal ligation and puncture. J Surg Res. 2015;194:528–36. doi: 10.1016/j.jss.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 4.Sharma A, Matsuo S, Yang WL, Wang Z, Wang P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit Care. 2014;18:R142. doi: 10.1186/cc13970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann B, et al. Mechanisms of acute inflammatory lung injury induced by abdominal sepsis. Int Immunol. 1999;11:217–27. doi: 10.1093/intimm/11.2.217. [DOI] [PubMed] [Google Scholar]
- 6.Ayala A, et al. Shock-induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am J Pathol. 2002;161:2283–94. doi: 10.1016/S0002-9440(10)64504-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lomas-Neira JL, Chung CS, Wesche DE, Perl M, Ayala A. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J Leukoc Biol. 2005;77:846–53. doi: 10.1189/jlb.1004617. [DOI] [PubMed] [Google Scholar]
- 8.Filgueiras LR, Capelozzi VL, Martins JO, Jancar S. Sepsis-induced lung inflammation is modulated by insulin. BMC Pulm Med. 2014;14:177. doi: 10.1186/1471-2466-14-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannam P, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306:L604–19. doi: 10.1152/ajplung.00272.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 2013;93:329–42. doi: 10.1189/jlb.0912437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Botha AJ, et al. Early neutrophil sequestration after injury: a pathogenic mechanism for multiple organ failure. J Trauma. 1995;39:411–7. doi: 10.1097/00005373-199509000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–83. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viollet B, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–8. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–23. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li XN, et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes. 2009;58:2246–57. doi: 10.2337/db08-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolinsky VW, Dyck JR. Role of AMP-activated protein kinase in healthy and diseased hearts. Am J Physiol Heart Circ Physiol. 2006;291:H2557–69. doi: 10.1152/ajpheart.00329.2006. [DOI] [PubMed] [Google Scholar]
- 18.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011;89:667–76. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan JE, et al. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–6. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- 20.Bai A, et al. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol. 2010;80:1708–17. doi: 10.1016/j.bcp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Hoogendijk AJ, Pinhancos SS, van der Poll T, Wieland CW. AMP-activated protein kinase activation by 5-aminoimidazole-4-carbox-amide-1-beta-D-ribofuranoside (AICAR) reduces lipoteichoic acid-induced lung inflammation. J Biol Chem. 2013;288:7047–52. doi: 10.1074/jbc.M112.413138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giri S, et al. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–87. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–96. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huston JM, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med. 2007;35:2762–8. doi: 10.1097/01.CCM.0000288102.15975.BA. [DOI] [PubMed] [Google Scholar]
- 25.Boeckxstaens G. The clinical importance of the anti-inflammatory vagovagal reflex. Handb Clin Neurol. 2013;117:119–34. doi: 10.1016/B978-0-444-53491-0.00011-0. [DOI] [PubMed] [Google Scholar]
- 26.Park J, Kang JW, Lee SM. Activation of the cholinergic anti-inflammatory pathway by nicotine attenuates hepatic ischemia/reperfusion injury via heme oxygenase-1 induction. Eur J Pharmacol. 2013;707:61–70. doi: 10.1016/j.ejphar.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 27.Cai D, Liu T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Ann N Y Acad Sci. 2011;1243:E1–39. doi: 10.1111/j.1749-6632.2011.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronnett GV, Aja S. AMP-activated protein kinase in the brain. Int J Obes (Lond) 2008;32(Suppl 4):S42–8. doi: 10.1038/ijo.2008.122. [DOI] [PubMed] [Google Scholar]
- 29.Turnley AM, et al. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–16. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 30.Amato S, Man HY. Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011;10:3452–60. doi: 10.4161/cc.10.20.17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giangola MD, et al. Growth arrest-specific protein 6 attenuates neutrophil migration and acute lung injury in sepsis. Shock. 2013;40:485–91. doi: 10.1097/SHK.0b013e3182a588c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirano Y, et al. Neutralization of osteopontin attenuates neutrophil migration in sepsis-induced acute lung injury. Crit Care. 2015;19:782. doi: 10.1186/s13054-015-0782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudson LD. New therapies for ARDS. Chest. 1995;108:79S–91S. doi: 10.1378/chest.108.2_supplement.79s. [DOI] [PubMed] [Google Scholar]
- 34.Oberholzer C, Oberholzer A, Clare-Salzler M, Moldawer LL. Apoptosis in sepsis: a new target for therapeutic exploration. FASEB J. 2001;15:879–92. doi: 10.1096/fj.00-058rev. [DOI] [PubMed] [Google Scholar]
- 35.Liu D, Zienkiewicz J, DiGiandomenico A, Hawiger J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Mol Ther. 2009;17:796–802. doi: 10.1038/mt.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmal H, Shanley TP, Jones ML, Friedl HP, Ward PA. Role for macrophage inflammatory protein-2 in lipopolysaccharide-induced lung injury in rats. J Immunol. 1996;156:1963–72. [PubMed] [Google Scholar]
- 37.Lomas JL, et al. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock. 2003;19:358–65. doi: 10.1097/00024382-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Chong DL, Sriskandan S. Pro-inflammatory mechanisms in sepsis. Contrib Microbiol. 2011;17:86–107. doi: 10.1159/000324022. [DOI] [PubMed] [Google Scholar]
- 39.Escobar DA, et al. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J Surg Res. 2015;194:262–72. doi: 10.1016/j.jss.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torgersen C, et al. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth Analg. 2009;108:1841–7. doi: 10.1213/ane.0b013e318195e11d. [DOI] [PubMed] [Google Scholar]
- 41.Weiss YG, et al. Adenoviral vector transfection into the pulmonary epithelium after cecal ligation and puncture in rats. Anesthesiology. 2001;95:974–82. doi: 10.1097/00000542-200110000-00029. [DOI] [PubMed] [Google Scholar]
- 42.Aschkenasy G, Bromberg Z, Raj N, Deutschman CS, Weiss YG. Enhanced Hsp70 expression protects against acute lung injury by modulating apoptotic pathways. PLoS One. 2011;6:e26956. doi: 10.1371/journal.pone.0026956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 2009;234:361–71. doi: 10.3181/0811-MR-318. [DOI] [PubMed] [Google Scholar]
- 44.Kim JE, et al. AMPK activator, AICAR, inhibits palmitate-induced apoptosis in osteoblast. Bone. 2008;43:394–404. doi: 10.1016/j.bone.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 45.Rossi A, Lord JM. Adiponectin inhibits neutrophil apoptosis via activation of AMP kinase, PKB and ERK 1/2 MAP kinase. Apoptosis. 2013;18:1469–80. doi: 10.1007/s10495-013-0893-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Park YJ, Jang Y, Kwon YH. AMPK activation inhibits apoptosis and tau hyperphosphorylation mediated by palmitate in SH-SY5Y cells. Brain Res. 2011;1418:42–51. doi: 10.1016/j.brainres.2011.08.059. [DOI] [PubMed] [Google Scholar]
- 47.Shanley TP, et al. Requirement for C-X-C chemokines (macrophage inflammatory protein-2 and cytokine-induced neutrophil chemoattractant) in IgG immune complex-induced lung injury. J Immunol. 1997;158:3439–48. [PubMed] [Google Scholar]
- 48.Schmekel B, et al. Myeloperoxidase in human lung lavage. I. A marker of local neutrophil activity. Inflammation. 1990;14:447–54. doi: 10.1007/BF00914095. [DOI] [PubMed] [Google Scholar]
- 49.Bellinger DL, et al. Sympathetic modulation of immunity: relevance to disease. Cell Immunol. 2008;252:27–56. doi: 10.1016/j.cellimm.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cervi AL, Lukewich MK, Lomax AE. Neural regulation of gastrointestinal inflammation: role of the sympathetic nervous system. Auton Neurosci. 2014;182:83–8. doi: 10.1016/j.autneu.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 51.Huston JM. The vagus nerve and the inflammatory reflex: wandering on a new treatment paradigm for systemic inflammation and sepsis. Surg Infect (Larchmt) 2012;13:187–93. doi: 10.1089/sur.2012.126. [DOI] [PubMed] [Google Scholar]
- 52.Ji H, et al. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol. 2014;7:335–47. doi: 10.1038/mi.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. 2006;8:383–95. doi: 10.31887/DCNS.2006.8.4/ssmith. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernik TR, et al. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med. 2002;195:781–8. doi: 10.1084/jem.20011714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santos GA, et al. Hypothalamic AMPK activation blocks lipopolysaccharide inhibition of glucose production in mice liver. Mol Cell Endocrinol. 2013;381:88–96. doi: 10.1016/j.mce.2013.07.018. [DOI] [PubMed] [Google Scholar]