

Abstract
AIM: To develop a rapid detection method of enteroviruses and Hepatitis A virus (HAV).
METHODS: A one-step, single-tube consensus primers multiplex RT-PCR was developed to simultaneously detect Poliovirus, Coxsackie virus, Echovirus and HAV. A general upstream primer and a HAV primer and four different sets of primers (5 primers) specific for Poliovirus, Coxsacki evirus, Echovirus and HAV cDNA were mixed in the PCR mixture to reverse transcript and amplify the target DNA. Four distinct amplified DNA segments representing Poliovirus, Coxsackie virus, Echovirus and HAV were identified by gel electrophoresis as 589-, 671-, 1084-, and 1128 bp sequences, respectively. Semi-nested PCR was used to confirm the amplified products for each enterovirus and HAV.
RESULTS: All four kinds of viral genome RNA were detected, and producing four bands which could be differentiated by the band size on the gel. To confirm the specificity of the multiplex PCR products, semi-nested PCR was performed. For all the four strains tested gave positive results. The detection sensitivity of multiplex PCR was similar to that of monoplex RT-PCR which was 24 PFU for Poliovrus, 21 PFU for Coxsackie virus, 60 PFU for Echovirus and 105 TCID50 for HAV. The minimum amount of enteric viral RNA detected by semi-nested PCR was equivalent to 2.4 PFU for Poliovrus, 2.1 PFU for Coxsackie virus, 6.0 PFU for Echovirus and 10.5 TCID50 for HAV.
CONCLUSION: The consensus primers multiplex RT-PCR has more advantages over monoplex RT-PCR for enteric viruses detection, namely, the rapid turnaround time and cost effectiveness.
INTRODUCTION
Enteroviruses include Poliovirus, Coxsackie virus and Echo virus. Enterovuses and HAV can bring about many diseases[1]. The enteroviruses and HAV transmit through water or foods and constitute a risk to public health, even when their concentrations are very low. Therefore, to establish a set of fast and dependable technique for detecting enteroviruses and HAV in water is of great importance for preventing outbreak of virus diseases through water and, at the same time laying a foundation for working out virology sanitation standards for drinking water.
The traditional technique for detecting viruses at present is cell culture[2] and it is characterized by its large water volume treated and high sensitivity. Normally viruses can be cultured and reproduced even if there is only one infectious virus existing in water. But the technique also has some shortcomings: (1) it needs great efforts and is time-consuming. There are high technical demands on cell culture and its result is unstable. It takes more than 3 d from inoculating virus to the time when visible pathological cell change effect (PCE) appears. For HAV, there is no pathological cell change effect and it takes 6-8 wk to carry out the test. (2) it is poor in specificity. It is impossible to draw a clear identification among enteroviruses on the basis of pathological cell changes.
In recent years, PCR has been adopted extensively in detecting enterovirus or HAV in environment owing to its high specificity and simpler operation[1,3-5]. But it also has its shortcomings. Firstly, the sensitivity is low. Although PCR itself possesses high sensitivity and it can pick out even there is only one virus, its sensitivity still does not come up to the standards for testing virus in water because the size of added samples is extremely small (about 10 mL). Even the viruses in the water are concentrated, the volume is still too large for PCR. Secondly, the testing capability is inconsistent with infectivity of the virus. It is unable to decide whether the virus is infectious even if the result is positive. Besides, conventional PCR can only detect one type of virus at one time. There are many types of viruses in water so it can't meet the needs of practical application. Tsai et al[6] used multiplex-PCR to detect poliovirus, hepatitis A virus and rotavirus at one time. Their technique requires more primers and these primers interfere with each other, making it more difficult to test the viruses over three types. Zoll et al[7] employed general primer PCR to detect enterovirus but their method can only show whether there is existence of enterovirus or not. It can not distinguish the types of viruses.
The combination of the cell culture with PCR technique is implemented organically in our study for the purpose of setting up a set of fast and dependable methods for testing viruses in water. This method can either preserve the advantages of the cell culture and PCR technique or overcome the shortcomings of each technique. Our technique is basically divided into three steps: first, to make collection and recovery of viruses in water; second, to make short-term cultivation of viruses; and third, to detect viral nucleic acid using multiplex-PCR and identified by semi-nested PCR.
MATERIALS AND METHODS
Viruses and viruses assays
Plaque-purified Poliovirus type 1, strain LSc, Cox B1, 3 and 4 or Echo 7, 9, 11, 12 virus were used as the model for the enteroviruses that may be present in the water. These viruses were grown and assayed by using African green monkey kidney (Vero) cells as previously described[1]. The viruses were titrated by the plaque method and expressed as plaque-forming units (PFU). The NJ-3 strain of HAV (Institute of Military Medical Research of Nanjing) was adopted to the human hepatoma cell line PLC/PRF/5 by serial passages. The HAV antigen was detected by ELISA method.
Virus seeding
A virus mixture containing 1.0 × 105 PFU of Poliovirus type 1, strain LSc, Cox B3 or Echo 9 virus, and 1.0 × 105 TCID50 of HAV was seeded into 10 liters of water samples. The seeded and unseeded samples were concentrated by electropositive filter media particle as described previously by Jun-Wen Li, and the recovery of seeded viruses was 88.7%[8]. The final concentrates were 1.0 mL, If needed, which was cultured for three days for HAV or one day for other enterovirues. The sensitivities of RT-PCR were evaluated with the ten-fold dilutions of HAV or other enteroviruses which were made with HPLC grade water.
RNA extraction and purification
Viral genome RNA was extracted and purified by the TRIzol Reagent kit (Life Technologies) according to the manufacturer's recommendation, and the RNA was stored at -20 °C for future analysis.
Primers
The consensus primer of enteroviruses was from the 5’ noncoding region because of their presence in many enteroviruses serotypes[7,9]; the specific primers for enteric viruses, including HAV, were selected from the 5’ coding region; and the semi-nested PCR primers were designed within the fragment of the first PCR products. The information of primers was summarized in Table 1.
Table 1.
Nucleotide sequences and positions of primers
| Primers | Sequence (5’-3’) | Position | band sizes (bp) |
| CE (+) | ATTGGATTGGCCATCCGGTG | 620-639 | |
| P (-)1 | TCCACCACCACCCTCTCGACTC | 1169-1190 | 571 |
| P (-)2 | GCATTACACTGTACGTGCAC | 1271-1290 | 671 |
| C (-)1 | CACCAACACCCATACCTGCA | 1470-1489 | 870 |
| C (-)2 | GCGCACATTGGCGCAATTGTG | 1683-1703 | 1084 |
| E (-)1 | CGCACCCCATCTCAGCTTCA | 1330-1349 | 730 |
| E (-)2 | TTCATTGTCGGCAAACCTTGA | 1727-1747 | 1128 |
| H (-) | CCAATCTTCCTGATCCAAAGC | 1557-1577 | |
| H (+)1 | AACCCTACACCTTTCCAACA | 1146-1165 | 432 |
| H (+)2 | GATTCATTCTGCAGATTGGC | 989-1008 | 589 |
+; upstream primer, -; downsream primer, 1: semi-nested primer; 2: the first PCR primer, CE; general primer, P: primers of Polioviruses; C: primers of Coxsackie viruses; E: primers of Echoviruses; H: primers of HAV
One-step, single-tube multiplex PCR
2-10 μL of prified viral RNAs were added in a final 100 μL containing 1 × PCR buffer with 1.5 mM MgCl2, 0.2 mM each of dNTP, 0.2 μM each of the primers, 10 U Rnase inhibitor, 5 mM of DTT, 1 μL of mixture of reverse trancriptase and DNA polymerase. RT was carried out at 50 °C for 30 min. This was followed by an initial denaturation at 94 °C for 3 min, and 30 PCR cycles of denaturation at 94 °C for 0.5 min, annealing at 60 °C for 0.5 min and extension at 68 °C for 1 min with a fixed ramp time of 3 s for each cycle[10].
Semi-nested PCR
The semi-nested PCR was carried out on a final 50 μL containing 1 × PCR buffer with 2.5 mM MgCl2, 0.2 mM each of dNTP, 0.2 μM each of primers, 1 μL of the first PCR product and 2.5 U of Taq polymerase. This was followed by an initial denaturation at 94 °C for 3 min, and 30 PCR cycles of denaturation at 94 °C for 0.5 min, annealing at 60 °C for 0.5 min and extension at 68 °C for 1 min[12,13].
Analysis of PCR products
Ten μL of each amplified product was electrophoresed on a 1.5% agarose gel in 1 × TBE buffer containing 0.5 μg/mL ethidium bromide. The amplified bands were directly visualized under UV light[7,14].
RESULTS
Specificities of the primers
First, only one kind of viral RNA extracted from cell culture was tested in the multiplex PCR with all the primers. It is shown that only one amplicon was yielded, which was in agreement with the information on the designed primers (results not shown).
Secondly, two kinds of viral RNA were added to the multiplex PCR, it is expected that there were two PCR products on the gel which were conformed to the theoretical results (Figure 1).
Figure 1.
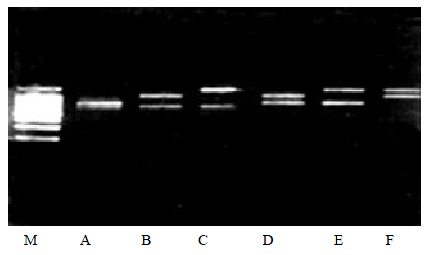
PCR products of two types of viruses. M:DNA ladder, A: HAV and PV, B: HAV and CV, C: HAV and EV; D: PV and CV, E: PV and EV, F: CV and EV.
Thirdly, three kinds of viral RNA were tested in the multiplex PCR, and given similar results (Figure 2).
Figure 2.
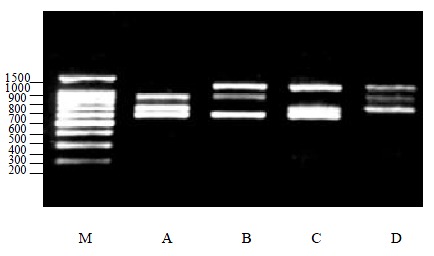
PCR products of three types of viruses. M:DNA ladder, A: HAV, PV and CV; B: HAV, CV and EV; C: HAV, PV and EV; D: PV, CV and EV.
Fourthly, all four kinds of viral genome RNA were detected, and produced four bands which could be differentiated by the band size on the gel (Figure 3).
Figure 3.
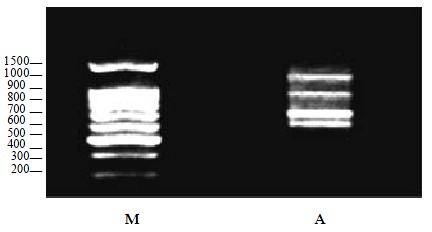
PCR products of four types of viruses. M:DNA ladder, A: poliovirus 1 (671 bp), Coxsackie virus B3 (1084 bp), Echo virus 9 (1128 bp), and HAV (589 bp).
To confirm the specificity of the multiplex PCR products, semi-nested PCR was performed. For all the four strains tested gave positive results (Figure 4).
Figure 4.
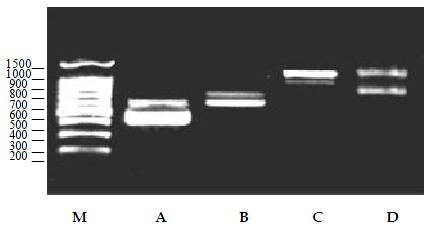
Semi-nested PCR products of four types of viruses. M:DNA ladder, A: HAV (589, 432 bp), B: Poliovirus 1 (671, 571 bp), C: Coxsackie virus B3 (1084, 870 bp), D: Echo virus 9 (1128, 730 bp)
Sensitivity of PCR
The detection sensitivity of multiplex PCR was similar to that of monoplex RT-PCR which was 24 PFU for Poliovrus, 21 PFU for Coxsackie virus, 60 PFU for Echovirus and 105 TCID50 for HAV. The minimum amount of enteric viral RNA detected by semi-nested PCR was equivalent to 2.4 PFU for Poliovrus, 2.1 PFU for Coxsackie virus, 6.0 PFU for Echovirus and 10.5 TCID50 for HAV (Table 2). The method achieved a 10 folds higher sensitivity than that of multiplex PCR.
Table 2.
Sensitivity of semi-nested-PCR for different viruses
| Viruses |
Virus concentration (PFU) |
||||
| 103 | 102 | 101 | 100 | 10-1 | |
| Poliovirus | + | + | + | + (2.4) | - |
| Coxsackie virus B3 | + | + | + | + (2.1) | - |
| Echovirus 9 | + | + | + | + (6.0) | - |
| Three viruses | + | + | + (10.5) | - | - |
Detection of viral genome after concentration
A tap water with and without seeding enteroviruses were collected for testing. Viruses added in the 10 liters of water which were not concentrated in primary could not be detected by the multiplex PCR or semi-nested PCR, even when 200-1000 PFU or TCID50 of viruses were seeded. However, viruses seeded in the waters which were concentrated with electropositive filter media could be detected by multiplex PCR and semi-nested PCR and obtained the similar sensitivity with that from cell cultures.
Fifty liters of tap water, or river water, or 5 liters of sewage, or 10 liters of ocean water were concentrated with electropositive filter media, and the virus RNAs were extracted from the concentrates. The multiplex PCR and semi-nested PCR were used to amplify the enteric viruses RNA. It is found that only Poliovirus RNA could be detected from the concentrate from 50 liters river water, other enteroviruses were not detected.
DISCUSSION
In our studies, cell culture and PCR are combined together to improve the detection sensitivity and specificity. For the cell culture method, it has the advantages of large volume of testing water and high sensitivity. Viable viruses can be fostered and reproduced even if there is existence of only one type. But it is poor in specificity[15]. Types of virus can's be distinguished according to pathological changes cell and immunology method is needed to make further identifications. So it is heavy, complicated and time consuming (at least 3 d; it will take 6-8 wk to identify hepatitis A virus) to implement the test. As for PCR, it has the advantages of high specificity and simpler operation. But its sensitivity can's come up to the testing standards owing to its small testing size, and the same positive results are obtained no matter the virus is infectious or not so long as nucleic acid is complete. The improved cell culturing-PCR technique combines the high sensitivity of cell culture with the high specificity of PCR together, and avoids the shortcomings of low specificity and long testing period of cell culture, only two or four days are needed to detect Poliovirus, Coxsackie virus, Echovirus or HAV (cell culture for one or three days), thus two thirds testing time of cell culture is saved. Furthermore, cell culture dilutes the substances which play inhibiting effect on PCR in water, which further improves the sensitivity of PCR. In addition, this technique is only used to test the infectious viruses in water, hence inconsistency of PCR results with actual infectivity can be avoided[16].
The accuracy and reliability are ensured by using semi-nested PCR to identify PCR products. The techniques used at present to determine PCR products are Southern hybridization, PCR- depended DNA fingerprint pattern, nucleic acid sequencing and nested PCR, etc.[3,4,6]. Southern blot is poor in sensitivity and very complicated to handle[17-24]. It is generally taken as tool enzyme in molecule cloning and demands enzyme chip points existing in the expanded segment; DNA fingerprint pattern is poor in specificity with unstable results; nucleic acid sequencing is complicated, costly and low in efficiency; while nested PCR has many strong points. It is not only good in specificity and sensitivity but also easy to handle and is low in price[4,25]; semi-nested PCR has similar principles and functions as that of nested PCR. It further cuts down the cost by saving a strip of primer.
The general primer multiplex-PCR is established for detection of four kinds of enteroviruses in our studies. There were many shortcomings in the researches prior to ours, such as, only one virus can be detected at one time using conventional PCR so it can's meet the needs of practical application; Tsai et al[6] used multiplex-PCR to detect Poliovirus, Hepatitis A virus and Rotavirus at one time. Their technique required more primers and these primers interfere with each other, making it more difficult to test viruses for three types or more; Zoll et al[7] employed general primer PCR technique to detect Poliovirus, Coxsackie virus, Echo virus and new enterovirus. But their method can only show whether there is existence of enterovirus or not. It can not distinguish the types of virus and is impossible to detect hepatitis A virus. Namely, general primer multiplex-PCR combines the general primer with multiplex PCR, giving consideration to the strong points of the two techniques and overcoming the weak points of each. Six primers are used in one reacting tube and four types of enteroviruses can be detected in one reaction while in conventional PCR, eight strips of primer are required.
The incidence of Poliovirus in developing countries is 4‰ while in developed countries, the infection of Poliovirus is of less clinical significance but it is generally used as an indicating virus in water[25,11]. Coxsackie virus and Echo virus can lead to diseases such as aseptic meningitis, viral myocarditis, diarrhea etc. Because group A Coxsackie virus is hard to culture and the virus source is very difficult to ensure, it is not tested hereon.
To sum up, the technique can be used to detect Poliovirus, group B Coxsackie virus, Echo virus and Hepatitis A virus. Nearly all viruses can be detected among enteroviruses except group A Coxsackie virus.
Footnotes
Edited by Wu XN
Supported by Natural Science Foundation of China, No.39570609
References
- 1.Egger D, Pasamontes L, Ostermayer M, Bienz K. Reverse transcription multiplex PCR for differentiation between polio- and enteroviruses from clinical and environmental samples. J Clin Microbiol. 1995;33:1442–1447. doi: 10.1128/jcm.33.6.1442-1447.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reynolds KA, Gerba CP, Pepper IL. Detection of infectious enteroviruses by an integrated cell culture-PCR procedure. Appl Environ Microbiol. 1996;62:1424–1427. doi: 10.1128/aem.62.4.1424-1427.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chapman NM, Tracy S, Gauntt CJ, Fortmueller U. Molecular detection and identification of enteroviruses using enzymatic amplification and nucleic acid hybridization. J Clin Microbiol. 1990;28:843–850. doi: 10.1128/jcm.28.5.843-850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Guyader F, Dubois E, Menard D, Pommepuy M. Detection of hepatitis A virus, rotavirus, and enterovirus in naturally contaminated shellfish and sediment by reverse transcription-seminested PCR. Appl Environ Microbiol. 1994;60:3665–3671. doi: 10.1128/aem.60.10.3665-3671.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gantzer C, Senouci S, Maul A, Levi Y, Schwartzbrod L. Enterovirus genomes in wastewater: concentration on glass wool and glass powder and detection by RT-PCR. J Virol Methods. 1997;65:265–271. doi: 10.1016/s0166-0934(97)02193-9. [DOI] [PubMed] [Google Scholar]
- 6.Tsai YL, Tran B, Sangermano LR, Palmer CJ. Detection of poliovirus, hepatitis A virus, and rotavirus from sewage and ocean water by triplex reverse transcriptase PCR. Appl Environ Microbiol. 1994;60:2400–2407. doi: 10.1128/aem.60.7.2400-2407.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zoll GJ, Melchers WJ, Kopecka H, Jambroes G, van der Poel HJ, Galama JM. General primer-mediated polymerase chain reaction for detection of enteroviruses: application for diagnostic routine and persistent infections. J Clin Microbiol. 1992;30:160–165. doi: 10.1128/jcm.30.1.160-165.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li JW, Wang XW, Rui QY, Song N, Zhang FG, Ou YC, Chao FH. A new and simple method for concentration of enteric viruses from water. J Virol Methods. 1998;74:99–108. doi: 10.1016/s0166-0934(98)00078-0. [DOI] [PubMed] [Google Scholar]
- 9.Sheng DL. Current Protocols for Molecular Biology (1st editation) Beijin: Senior Education Press; 1993. pp. 147–149. [Google Scholar]
- 10.Sellner LN, Coelen RJ, Mackenzie JS. A one-tube, one manipulation RT-PCR reaction for detection of Ross River virus. J Virol Methods. 1992;40:255–263. doi: 10.1016/0166-0934(92)90084-q. [DOI] [PubMed] [Google Scholar]
- 11.Abraham R, Chonmaitree T, McCombs J, Prabhakar B, Lo Verde PT, Ogra PL. Rapid detection of poliovirus by reverse transcription and polymerase chain amplification: application for differentiation between poliovirus and nonpoliovirus enteroviruses. J Clin Microbiol. 1993;31:395–399. doi: 10.1128/jcm.31.2.395-399.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu QF. Modern Stanitary Microbiology (1st editation) Beijin: People Medical Press; 1995. pp. 54–66. [Google Scholar]
- 13.Li JW, Zhang FG, Chao FH, Wang XW, Song N, Wang FY. Detection of enteroviruses by semi-nested RT-PCR. Zhongguo Weisheng Jianyan Zazhi. 1996;6:125–127. [Google Scholar]
- 14.Yang DL, Wang BC. DNA Amplification and Applification in Medicine. Jinan: Shangdong Science and Technological Press; 1992. pp. 74–79. [Google Scholar]
- 15.Seah CL, Chow VT, Tan HC, Can YC. Rapid, single-step RT-PCR typing of dengue viruses using five NS3 gene primers. J Virol Methods. 1995;51:193–200. doi: 10.1016/0166-0934(94)00104-o. [DOI] [PubMed] [Google Scholar]
- 16.Straub TM, Pepper IL, Gerba CP. Comparison of PCR and cell culture for detection of enteroviruses in sludge-amended field soils and determination of their transport. Appl Environ Microbiol. 1995;61:2066–2068. doi: 10.1128/aem.61.5.2066-2068.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shieh YS, Wait D, Tai L, Sobsey MD. Methods to remove inhibitors in sewage and other fecal wastes for enterovirus detection by the polymerase chain reaction. J Virol Methods. 1995;54:51–66. doi: 10.1016/0166-0934(95)00025-p. [DOI] [PubMed] [Google Scholar]
- 18.Moore NJ, Margolin AB. Evaluation of radioactive and nonradioactive gene probes and cell culture for detection of poliovirus in water samples. Appl Environ Microbiol. 1993;59:3145–3146. doi: 10.1128/aem.59.9.3145-3146.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams SJ, Schwer C, Krishnarao AS, Heid C, Karger BL, Williams PM. Quantitative competitive polymerase chain reaction: analysis of amplified products of the HIV-1 gag gene by capillary electrophoresis with laser-induced fluorescence detection. Anal Biochem. 1996;236:146–152. doi: 10.1006/abio.1996.0143. [DOI] [PubMed] [Google Scholar]
- 20.Doktycz MJ, Hurst GB, Habibi-Goudarzi S, McLuckey SA, Tang K, Chen CH, Uziel M, Jacobson KB, Woychik RP, Buchanan MV. Analysis of polymerase chain reaction-amplified DNA products by mass spectrometry using matrix-assisted laser desorption and electrospray: current status. Anal Biochem. 1995;230:205–214. doi: 10.1006/abio.1995.1465. [DOI] [PubMed] [Google Scholar]
- 21.Li JW, Zhang FG, Chao FH, Wang XW, Song N. Detection of HAV by one-step, one-tube RT-PCR. Zhongguo Wei sheng Jianyan Zazhi. 1996;6:63–65. [Google Scholar]
- 22.Ma JF, Gerba CP, Pepper IL. Increased sensitivity of poliovirus detection in tap water concentrates by reverse transcriptase-polymerase chain reaction. J Virol Methods. 1995;55:295–302. doi: 10.1016/0166-0934(95)00065-6. [DOI] [PubMed] [Google Scholar]
- 23.Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xun ZX. Current Biochemical Theory and Research Technology (1st editation) Beijin: Military Medical Sciences Press; 1995. pp. 422–424. [Google Scholar]
- 25.Tilston P, Corbitt G. A single tube nested PCR for the detection of hepatitis C virus RNA. J Virol Methods. 1995;53:121–129. doi: 10.1016/0166-0934(95)00006-g. [DOI] [PubMed] [Google Scholar]