

Abstract
AIM: JNK cascade plays an important role in cell proliferation, differentiation and apoptosis. However, the exact function of JNK cascade for apoptosis induction remains largely unknown. In this study, the role of JNK activation stimulated by TPA in the process of apoptosis induction and its signaling transduction pathway in gastric cancer cells were investigated and determined.
METHODS: Expressions of mRNA and protein were detected by Northern blot and Western blot. Transcription activity was measured by transient transfection and CAT assay. Apoptotic cells were displayed through staining the nucleus with DAPI and were observed under fluorescence microscope. The apoptotic index was determined by counting 1000 cells randomly.
RESULTS: JNK protein was stimulated rapidly by TPA, and reached its highest peak within 3 hr, then decreased in a time-dependent manner, but the expression level of JNK protein induced by TPA was always keeping higher than that in untreated cells. Similar pattern was seen in c-jun mRNA level induced by TPA. TPA significantly activated the transcriptional activity of activator protein-1 with a TPA-dose-dependent manner. Furthermore, activation of JNK was mediated through PKC pathway. Treatment of cells with PKC specific inhibitor, Wortmannin, led to repression of JNK even in the presence of TPA. More importantly, all these effects were associated with induction of apoptosis in gastric cancer cells. TPA inducted apoptosis obviously in gastric cancer cells. The apoptotic cells became smaller and rounded, and their nuclei became condensation and fragmentation with brightly stained chromatin. However, suppression of JNK by PKC specific inhibitor, Wortmannin, resulted in the decrease of apoptosis induced by TPA in a time-dependent manner, apoptotic index dramatically decreased from 32.56% to 8.71%.
CONCLUSION: TPA stimulates JNK cascade, including up-regulation of JNK protein expression level and c-jun mRNA expression level, and activation of activator protein-1 transcriptional activity. Activation of JNK is mediated through PKC pathway, which has an association with induction of apoptosis by TPA. Thus, activation of JNK via PKC pathway may represent one of important mechanisms for TPA to induce apoptosis in gastric cancer cells.
INTRODUCTION
Mitogen-activated protein kinase (MAPK) pathway is an important signal transduction pathway that transduces extracellular signals in intracellular responses and has been implicated in a wide array of physiological processes including cell growth, differentiation, and apoptosis[1-6]. The extracellular signal-regulated kinase (ERK), the c-jun N-terminal kinase (JNK) and the p38 MAPK are the members of MAPK family[7-9]. Among them, JNK is one of the key mediators of stress signal and inflammatory response evoked by a variety of agents such as UV-irradiation, γ-irradiation, heat shock, and inflammatory cytokinase[10,11]. Once activated by specific kinases, JNK mediates the phosphorylation and activation of the transcription factors, such as c-Jun, ATF2, and ElK1[12,13].
JNK can be strongly activated through stress signal that ultimately leads to apoptosis. Accumulating reports have recently been focused on the potential role of JNK in apoptotic signaling. In PC12 cells, JNK plays a critical role in mediating apoptosis caused by nerve growth factor withdrawal[14]. The JNK pathway is also required for the induction of apoptosis by ceramide, γ- and UV-irradiation, some chemotherapeutic drugs[15-17]. Inversely, other reports have shown evidences that activation of JNK is not mechanistically implicated in the apoptotic process. activation of JNK does not correlate with apoptosis induced by the detachment of epithelial cells[18]. Inhibition of JNK activity by the expression of c-jun mutant does not prevent TNF-mediated cell death[19,20].
The present study investigates the effects of 12-O-tetradecanoyl-Phorbol-13-acetate (TPA) on activation of JNK and its role in apoptosis induction in gastric cancer cells. Our results indicated that TPA activated the expressions of JNK protein, which led to induction of c-jun mRNA expression, and was associated with activation of activator protein-1 transcriptional activity. Furthermore, activation of JNK cascade was mediated through protein kinase C (PKC) pathway. All these effects had an association with induction of apoptosis by TPA. Thus, activation of JNK via PKC pathway might represent one of mechanisms for TPA to induce apoptosis in gastric cancer cells.
MATERIALS AND METHODS
Cell line and culture condition
Human gastric cancer cell line, MGC80-3 was established by Cancer Research Center, Xiamen University[21]. Cells were maintained in RPMI-1640 medium, supplemented with 10% FCS, 1 m mol/L glutamine, and 100 µ/mL penicillin. The concentration of TPA was 100 ng/mL.
Western blot analysis[22]
Cells treated by TPA were harvested, and suspended in RIPA buffer (10 m mol/L Tris (pH7.4), 150 m mol/L NaCl, 1% Triton X-100, 1% Deoxycholic Acid, 0.1% SDS, 5 m mol/L EDTA (pH8.0), 1 m mol/L PMSF). Protein concentration was determined by using the Bio-Rad protein assay system according to the manufacturer’s direction (Bio-Rad Hereules, CA). Total protein (50 μg) was subjected to SDS-PAGE and transferred to nitrocellulose membrane for western blot analysis. The membrane was subsequently blocked with 5% dry milk in TBS-T and then immunnoblotted with the corresponding antibody. Protein was detected by using the ECL kit (Amersham Inc.) according to the manufacturer’s direction. Before incubation with antibody, the same membrane was stained by ponceau S. (Sigma) to show the amount of protein used in each well. The intensities of bands indicated by Western blot were quantified by densitometer.
RNA preparation and northern blotting
Extraction of total RNA was performed by the guanidine hydrochloride/ultracentrifugation method. Total RNA (20 μg) was separated on 1% agarose gel containing formaldehyde, and then transferred to nylon membrane, hybridized with α-32p-dATP and α-32p-dCTP labeled c-jun probe (Zhong Shang Biotec. CO., China), washed, and autoradiographed as described previously[23]. The same membrane was probed again with β-actin cDNA to show the amount of RNA used in each well. The intensities of bands indicated by Northern blot were quantified by densitometer.
Transient transfection and CAT assay[24]
Cells were seeded in a six-well plate and were approximately 70% confluent at the time of transfection.Cells were transfected by CeLLFECTINtm (Gibco/BRI Life Technologies) by the following procedure: 6 μL CeLLFECTINtm in 1.0 mL standard medium was added to each well containing 1.0 mL of standard medium supplemented with the -73Col-CAT reporter plasmid (100 ng), β-galactosidase expression vector (400 ng, pCH110, pharmacia). CAT activity was determined using 3H-acetyl-CoA as substrate. β-galactosidase activity was measured to normalize transfection efficiency.
Apoptosis analysis[25]
Cells were treated with TPA, trypsinized, washed with PBS. The harvested cells were fixed in 3.7% paraformaldehyde on ice, washed with PBS, then stained with 50 μg/mL of 4,6-diamidino-2-phenylindole (DAPI, Sigma) containing 100 μg/mL of DNase-free RNase A to facilitate the examination of nuclei under fluorescence microscope. Apoptotic cells were investigated and counted among 1000 cells randomly. The apoptotic index was a mean of three independent experiments.
RESULTS
Stimulation of JNK expression by TPA
To determine whether TPA activates the JNK expression in gastric cancer cells, MGC80-3 cells were treated with TPA in a time course, and the expression level of JNK protein was detected by western blot analysis. As shown in Figure 1, JNK expressed in MGC80-3 cells, after cells were treated with TPA, the JNK expression was stimulated markedly. It was noted simultaneously that JNK expression was stimulated rapidly by TPA and reached a highest peak within 3 hr. After 3 hr treatment of TPA, although TPA-stimulated JNK expression decreased in a time course manner, the level of JNK protein was always keeping higher than that in MGC80-3 cells (Figure 1). Thus, the result clearly indicates the activation of JNK by TPA.
Figure 1.
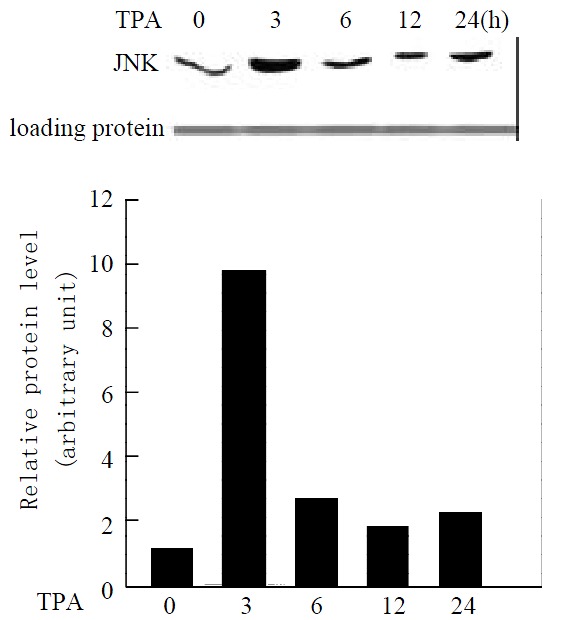
Expression of JNK protein in response to TPA induction. Cells treated with TPA for different time indicated. Expression of JNK protein was detected by Western blot. Amount of protein used in each lane was indicated by stain-ing with ponceau S. The intensity of each band was quantified by densitometer.
Effect of TPA on c-jun mRNA expression
Since activation of the JNK cascade is know to induce c-jun expression in several cell systems[26,27], we examine whether c-jun expression in mRNA level is also induced by TPA in gastric cancer cells. Northern blot analysis, using c-jun as a probe, revealed that MGC80-3 cells expressed c-jun mRNA at lower level, after treatment of TPA, expression level of c-jun mRNA was upregulated in a time-dependent manner (Figure 2). More interestingly, the induction patterns between JNK and c-jun by TPA were very similar. c-jun mRNA also reached its maximal peak induced by TPA within 3 hr, followed by a decrease with extension of TPA treatment in a time-course. Taken together, the data with Figure1 demonstrate that JNK cascade indeed plays a role in TPA signaling in gastric cancer cells.
Figure 2.
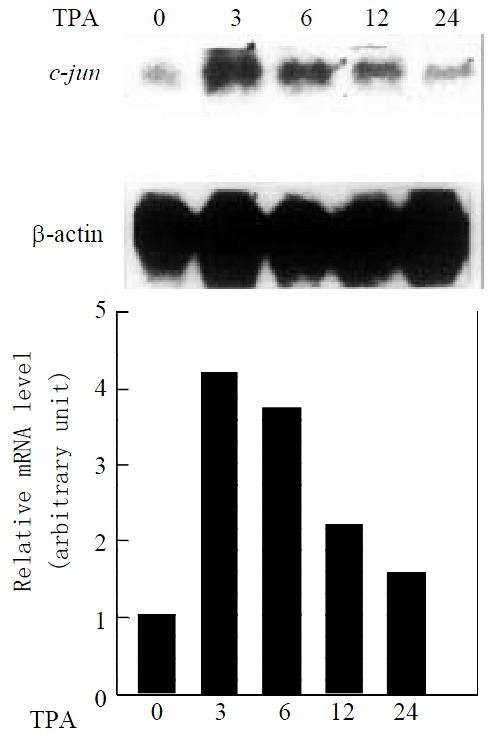
Expression of c-jun mRNA in response to TPA induction. Cells treated with TPA for different time indicated. Expression of c-jun mRNA was revealed by Northern blot. The same membrane was probed again with β-actin cDNA to show the amount of RNA used in each well. The intensity of each band was quantified by densitometer.
Transcriptional activity of activator protein-1 in response to TPA
Activator protein-1 is composed of products of the proto-oncogenes c-jun and c-fos[28]. The result mentioned above implies that TPA may affect transcriptional activity of activator protein-1 due to the activation of c-jun. To further determine whether TPA activates the transcriptional activity of activator protein-1, we measured transcriptional activity of activator protein-1 in MGC80-3 cells treated with various concentrations of TPA by the methods of transient transfection and CAT assay. When the reporter -73Col-CAT expression vector that contains an AP-1 binding site[29] was transfected into MGC80-3 cells, a strong induction of reporter transcriptional activity was observed when cells were treated by TPA. This induction of reporter transcriptional activity was TPA-dose-dependent obviously (Figure 3). Thereby, TPA had a capability of activating the transcriptional activity of activator protein-1.
Figure 3.
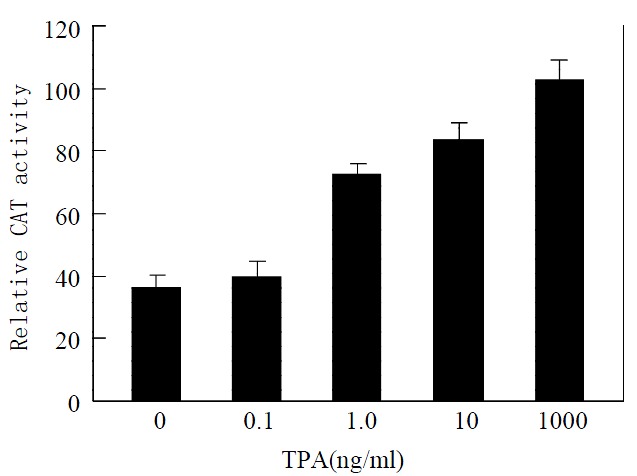
Effect of TPA on transcriptional activity of activator protein-1. Cells were treated with various concentrations of TPA indicated. Various expression vectors were transfected into cells as described in materials and methods. Transcrip-tional activity was measured by the method of transient trans-fection and CAT assay. Data shown represent mean of dupli-cate experiments (± SE).
TPA activates JNK cascade through PKC pathway
Since the target of TPA is protein kinase C (PKC)[30,31], we examine whether stimulation of JNK cascade by TPA is through PKC pathway in gastric cancer cells. When MGC80-3 cells were pretreated with PKC specific inhibitor, Wortmannin[32,33] for 2 hr, and then treated with TPA for another 3 hr, there was a significant inhibition observed in TPA-stimulated JNK protein expression (Figure 4, lane 4). As controls, JNK expression was clearly detected in MGC80-3 cells (Figure 4, lane 1) and TPA- stimulated JNK expression was seen obviously in the TPA-treated cells (Figure 4, lane 3), and Wortmannin alone slightly repressed JNK expression (Figure 4, lane 2), respectively. The similar result was observed in MGC80-3 cells treated with another PKC inhibitor, PKC inhibitor peptide[34] (data not shown). These results suggest that activation of JNK by TPA is mediated through PKC pathway.
Figure 4.
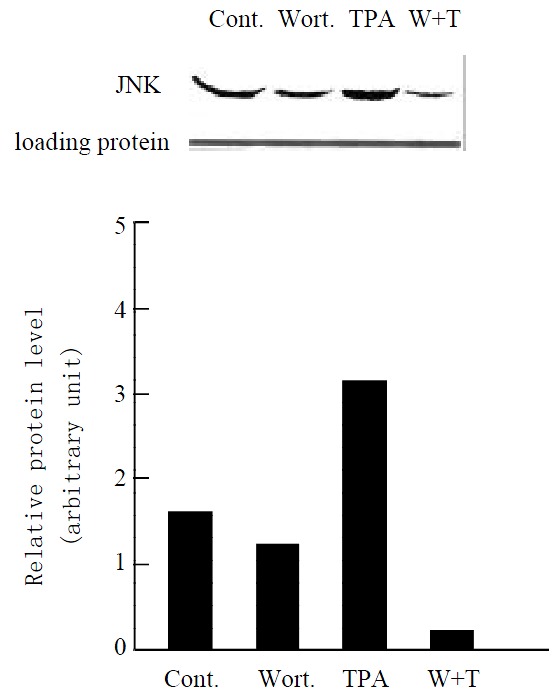
Effect of various agents, including Wortmannin (shown by Wort. or W), a PKC specific inhibitor, on expres-sion of JNK protein. Cells were treated with Wortmannin for 2 hr, following by TPA induction for another 3 hr. Protein was detected by Western blot. Amount of protein used in each lane was indicated by staining with ponceau S. The intensity of each band was quantified by densitometer.
TPA-induced apoptosis is correlated with JNK expression through PKC pathway
To determine the effects of TPA on JNK and PKC functions, and their relationship with induction of apoptosis in gastric cancer cells, we analyzed apoptotic rate in MGC80-3 cells treated with relevant agents. Compared to the untreated MGC80-3 cells, treatment of cells with TPA caused the classical morphological characteristics of apoptosis, cells became smaller and rounded, and their nuclei became condensation and fragmentation with brightly stained chromatin (Figure 5A). However, when treatment of cells with PKC specific inhibitor, Wortmannin, subsequently with TPA, the apoptotic cells were decreased obviously, cells displayed a normal nuclear morphology similar to that of untreated cells (Figure 5A). In addition, the number of apoptotic cells was increased by the induction of TPA in a time-dependent manner. The highest apoptotic rate reached 32.56% after treating cells with TPA for 48 hr, but only 5.46% in untreated MGC80-3 cells (Figure 5B).
Figure 5.
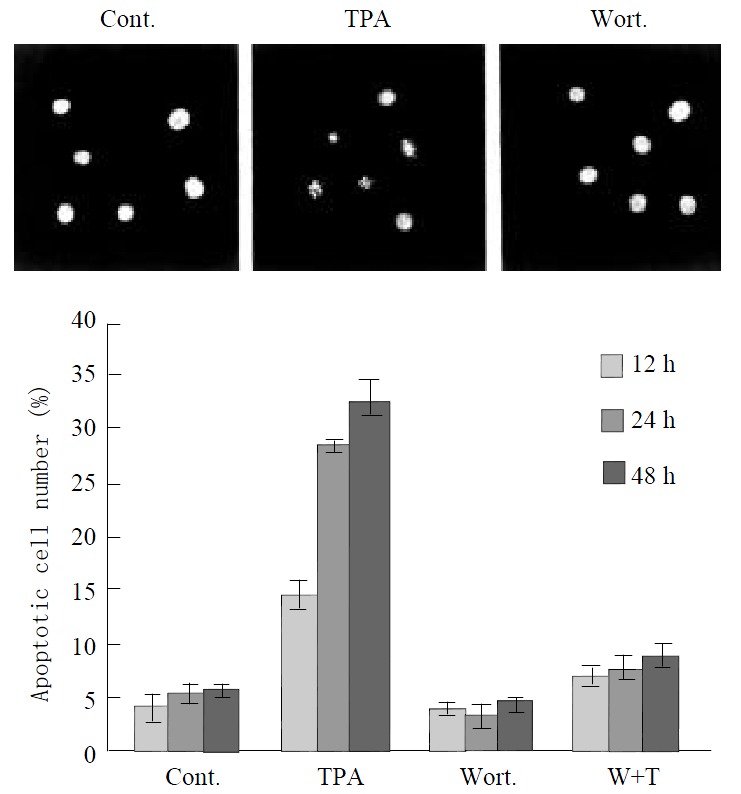
Effect of various agents, including TPA and Wortmannin, on apoptosis induction. Cells were treated with TPA and/or Wortmannin for different time indicated, and then the apoptotic rate was analyzed as described in material and methods. (A) The morphological changes in nucleus in re-sponse to different agents. (B) Apoptosis index.
Since TPA-activated JNK cascade was mediated through PKC pathway in MGC80-3 cells (Figure 4), we analyzed whether this pathway was an association with apoptosis induced by TPA. Compared to TPA that induced apoptosis obviously, pretreated cells with PKC specific inhibitor, Wortmannin, significantly inhibited apoptosis induced by TPA, apoptotic rate was rather low, the highest apoptotic rate was only 8.71%, although those cells were treated subsequently by TPA for another 48 h (Figure 5B). Accordingly, all these data further suggest that induction of apoptosis by TPA is through PKC pathway, which may associate with the activation of JNK cascade.
DISCUSSION
The dynamic balance among the extracellular signal-regulated kinase (ERK), the c-jun N-terminal kinase (JNK) and the p38 Mitogen-activated protein kinase (p38 MAPK) activities critically determines the cellular fate in response to stimuli of proliferation, differentiation and apoptosis[35,36]. The role of ERK signaling in the processes of cell differentiation and proliferation has been clearly recognized[37,38], while requirement of JNK and p38 MAPK activations for apoptosis remains controversy. In some cases, JNK and p38 MAPK activity is strictly required for apoptosis[39-41]. However, in other circumstances, they are not associated with the induction of apoptosis[42]. In this study, we demonstrate that TPA stimulates JNK cascade through PKC pathway, which is associated with induction of apoptosis in gastric cancer cells.
JNK cascade plays an important role in cell proliferation, differentiation and apoptosis[43,44]. In HeLa cells, the activation of JNK was very rapid and sustained for at least 24 hr post-irradiation[45]. We also found that JNK activity was activated by TPA in gastric cancer cells, MGC80-3. This activation occurred very rapidly within 3 hr of the TPA addition (Figure 1), suggesting that JNK protein may be one of the TPA primary response targets. More importantly, JNK expression level activated by TPA was always keeping higher than that in untreated MGC80-3 cells. This evidence also suggests that the JNK pathway is a major one for mediating TPA effect, which should be functional in gastric cancer cells.
JNK cascade involves many factors. Activated transcription factor 2 (ATF-2) and c-jun are activated mainly by JNK[43]. Transcriptional regulation of c-jun expression is mainly mediated by a TPA-response element in its promoter, which binds to c-jun/ATF-2 heterodimers[46]. Increase of c-jun mRNA by various stimuli is highly regulated by phosphorylation of c-jun or ATF-2 that bind to the activator protein-1 binding sites present in the c-jun promoter[47]. Our observation showed a possible interrelation between JNK and c-jun, which were activated rapidly by TPA and reach its maximal peak within 3 hr, and then decreased with time-dependent manner (Figures 1 and 2). Activator protein-1 is a transcriptional factor mainly composed of the c-jun/c-jun homodimer or c-jun/c-fos heterodimer. The fact that TPA also activated the transcriptional activity of activator protein-1 significantly in MGC80-3 cells (Figure 3) suggests that signaling that causes the transcriptional activity of activator protein-1 is functional in gastric cancer cells. Thus, induction of c-jun by TPA in MGC80-3 cells is likely due to the activation of JNK, which led to the activation of activator protein-1 transcriptional activity. This is a JNK cascade stimulated by TPA indeed.
How to function for JNK activation by TPA in gastric cancer cells is still largely unknown. MGC80-3 cells underwent apoptosis in response to TPA, as evidenced by typical morphological changes (Figure 5A, B), which is consistent with our previous observation[48,49]. Our results demonstrated that apoptosis induction by TPA was associated with JNK activition via PKC pathway. When PKC specific inhibitor, Wortmannin was used to treat MGC80-3 cells prior to TPA treatment, it would prevent TPA further to activate JNK expression (Figure 4). In this case, apoptotic cells were much fewer than that in TPA-treated cells, apoptotic index dramatically decreased from 32. 56% to 8.71% (Figure 5A, B). Thus, these data not only confirm that TPA-induced apoptosis is associated with JNK activation through PKC pathway, but also indicate that JNK cascade is required for apoptosis induction by TPA, which may be one mechanism for TPA to inhibit the growth of gastric cancer cells.
In summary, the present study demonstrates that the activation of JNK cascade is mediated through a signaling transduction pathway that involves PKC-dependent pathway, which leads to activation of JNK, induction of the c-jun expression and activation of activator protein-1 transcriptional activity in response to TPA. The activation of JNK cascade is closely associated with induction of apoptosis by TPA in gastric cancer cells.
ACKNOWLEDGEMENTS
We acknowledge the kind gift of reporter plasmid from Dr. Xiao-Kun Zhang (The Burnham Institute, CA, USA).
Footnotes
Supported by the National Outstanding Youth Science foundation of China, No. 39825502; the National Natural Science Foundation of China, No.39880015, 30170477; the Natural Science Foundation of Fujian Province, No.C0110004
Editted by Zhao M
References
- 1.Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouysségur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng DY, Zheng H, Tan Y, Cheng RX. Effect of phosphorylation of MAPK and Stat3 and expression of c-fos and c-jun proteins on hepatocarcinogenesis and their clinical significance. World J Gastroenterol. 2001;7:33–36. doi: 10.3748/wjg.v7.i1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 4.Nishida E, Gotoh Y. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem Sci. 1993;18:128–131. doi: 10.1016/0968-0004(93)90019-j. [DOI] [PubMed] [Google Scholar]
- 5.Mao H, Yuan AL, Zhao MF, Lai ZS, Zhang YL, Zhou DY. Effect of p38MAPK signal pathway on ultrastructural change of liver cancer cells induced by VEGF. Shijie Huaren Xiaohua Zazhi. 2000;8:536–538. [Google Scholar]
- 6.Fleischer F, Dabew R, Göke B, Wagner AC. Stress kinase inhibition modulates acute experimental pancreatitis. World J Gastroenterol. 2001;7:259–265. doi: 10.3748/wjg.v7.i2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frost JA, Xu S, Hutchison MR, Marcus S, Cobb MH. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol Cell Biol. 1996;16:3707–3713. doi: 10.1128/mcb.16.7.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vrana JA, Grant S. Synergistic induction of apoptosis in human leukemia cells (U937) exposed to bryostatin 1 and the proteasome inhibitor lactacystin involves dysregulation of the PKC/MAPK cascade. Blood. 2001;97:2105–2114. doi: 10.1182/blood.v97.7.2105. [DOI] [PubMed] [Google Scholar]
- 9.Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- 10.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 11.Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol. 2000;60:1015–1021. doi: 10.1016/s0006-2952(00)00372-5. [DOI] [PubMed] [Google Scholar]
- 12.Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Campbell D, Dérijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 14.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 15.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 16.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 17.Osborn MT, Chambers TC. Role of the stress-activated/c-Jun NH2-terminal protein kinase pathway in the cellular response to adriamycin and other chemotherapeutic drugs. J Biol Chem. 1996;271:30950–30955. doi: 10.1074/jbc.271.48.30950. [DOI] [PubMed] [Google Scholar]
- 18.Khwaja A, Downward J. Lack of correlation between activation of Jun-NH2-terminal kinase and induction of apoptosis after detachment of epithelial cells. J Cell Biol. 1997;139:1017–1023. doi: 10.1083/jcb.139.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 20.Lenczowski JM, Dominguez L, Eder AM, King LB, Zacharchuk CM, Ashwell JD. Lack of a role for Jun kinase and AP-1 in Fas-induced apoptosis. Mol Cell Biol. 1997;17:170–181. doi: 10.1128/mcb.17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang KH. An in vitro cell line (MGc80-3) of a pooly differenti-ated mucoid adenocarcinoma of human stomach. Shiyan Shengwu Xiebao. 1983;16:257–267. [Google Scholar]
- 22.Wu Q, Chen Z, Su W. Growth inhibition of gastric cancer cells by all-trans retinoic acid through arresting cell cycle progression. Chin Med J (Engl) 2001;114:958–961. [PubMed] [Google Scholar]
- 23.Wu Q, Chen Z, Su W. Mechanism of inhibition on activator protein-1 activity by all-trans retinoic acid in gastric cancer cells. Chin Med J (Engl) 2000;113:972–976. [PubMed] [Google Scholar]
- 24.Wu Q, Li Y, Liu R, Agadir A, Lee MO, Liu Y, Zhang X. Modulation of retinoic acid sensitivity in lung cancer cells through dynamic balance of orphan receptors nur77 and COUP-TF and their heterodimerization. EMBO J. 1997;16:1656–1669. doi: 10.1093/emboj/16.7.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Lee MO, Wang HG, Li Y, Hashimoto Y, Klaus M, Reed JC, Zhang X. Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol Cell Biol. 1996;16:1138–1149. doi: 10.1128/mcb.16.3.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Lin B, Agadir A, Liu R, Dawson MI, Reed JC, Fontana JA, Bost F, Hobbs PD, Zheng Y, et al. Molecular determinants of AHPN (CD437)-induced growth arrest and apoptosis in human lung cancer cell lines. Mol Cell Biol. 1998;18:4719–4731. doi: 10.1128/mcb.18.8.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 28.Konta T, Xu Q, Furusu A, Nakayama K, Kitamura M. Selective roles of retinoic acid receptor and retinoid x receptor in the suppression of apoptosis by all-trans-retinoic acid. J Biol Chem. 2001;276:12697–12701. doi: 10.1074/jbc.M011000200. [DOI] [PubMed] [Google Scholar]
- 29.Yang-Yen HF, Zhang XK, Graupner G, Tzukerman M, Sakamoto B, Karin M, Pfahl M. Antagonism between retinoic acid receptors and AP-1: implications for tumor promotion and inflammation. New Biol. 1991;3:1206–1219. [PubMed] [Google Scholar]
- 30.Goodnight JA, Mischak H, Kolch W, Mushinski JF. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J Biol Chem. 1995;270:9991–10001. doi: 10.1074/jbc.270.17.9991. [DOI] [PubMed] [Google Scholar]
- 31.Mischak H, Goodnight JA, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH, Mushinski JF. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268:6090–6096. [PubMed] [Google Scholar]
- 32.Cross MJ, Stewart A, Hodgkin MN, Kerr DJ, Wakelam MJ. Wortmannin and its structural analogue demethoxyviridin inhibit stimulated phospholipase A2 activity in Swiss 3T3 cells. Wortmannin is not a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1995;270:25352–25355. doi: 10.1074/jbc.270.43.25352. [DOI] [PubMed] [Google Scholar]
- 33.Bandyopadhyay G, Standaert ML, Galloway L, Moscat J, Farese RV. Evidence for involvement of protein kinase C (PKC)-zeta and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology. 1997;138:4721–4731. doi: 10.1210/endo.138.11.5473. [DOI] [PubMed] [Google Scholar]
- 34.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 35.Cambien B, Pomeranz M, Schmid-Antomarchi H, Millet MA, Breittmayer V, Rossi B, Schmid-Alliana A. Signal transduction pathways involved in soluble fractalkine-induced monocytic cell adhesion. Blood. 2001;97:2031–2037. doi: 10.1182/blood.v97.7.2031. [DOI] [PubMed] [Google Scholar]
- 36.Zhou H, Lin A, Gu Z, Chen S, Park NH, Chiu R. 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced c-Jun N-terminal kinase (JNK) phosphatase renders immortalized or transformed epithelial cells refractory to TPA-inducible JNK activity. J Biol Chem. 2000;275:22868–22875. doi: 10.1074/jbc.M909273199. [DOI] [PubMed] [Google Scholar]
- 37.Rovida E, Marra F, Baccarini M, Dello Sbarba P. Constitutive activation of the MAPK pathway mediates v-fes-induced mitogenesis in murine macrophages. Blood. 2000;95:3959–3963. [PubMed] [Google Scholar]
- 38.Olson NE, Kozlowski J, Reidy MA. Proliferation of intimal smooth muscle cells. Attenuation of basic fibroblast growth factor 2-stimulated proliferation is associated with increased expression of cell cycle inhibitors. J Biol Chem. 2000;275:11270–11277. doi: 10.1074/jbc.275.15.11270. [DOI] [PubMed] [Google Scholar]
- 39.Sluss HK, Barrett T, Dérijard B, Davis RJ. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol Cell Biol. 1994;14:8376–8384. doi: 10.1128/mcb.14.12.8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butterfield L, Storey B, Maas L, Heasley LE. c-Jun NH2-terminal kinase regulation of the apoptotic response of small cell lung cancer cells to ultraviolet radiation. J Biol Chem. 1997;272:10110–10116. doi: 10.1074/jbc.272.15.10110. [DOI] [PubMed] [Google Scholar]
- 41.Moreno-Manzano V, Ishikawa Y, Lucio-Cazana J, Kitamura M. Suppression of apoptosis by all-trans-retinoic acid. Dual intervention in the c-Jun n-terminal kinase-AP-1 pathway. J Biol Chem. 1999;274:20251–20258. doi: 10.1074/jbc.274.29.20251. [DOI] [PubMed] [Google Scholar]
- 42.Sakata N, Patel HR, Terada N, Aruffo A, Johnson GL, Gelfand EW. Selective activation of c-Jun kinase mitogen-activated protein kinase by CD40 on human B cells. J Biol Chem. 1995;270:30823–30828. doi: 10.1074/jbc.270.51.30823. [DOI] [PubMed] [Google Scholar]
- 43.Agadir A, Chen Gq, Bost F, Li Y, Mercola D, Zhang X. Differential effect of retinoic acid on growth regulation by phorbol ester in human cancer cell lines. J Biol Chem. 1999;274:29779–29785. doi: 10.1074/jbc.274.42.29779. [DOI] [PubMed] [Google Scholar]
- 44.Huang C, Li J, Ma WY, Dong Z. JNK activation is required for JB6 cell transformation induced by tumor necrosis factor-alpha but not by 12-O-tetradecanoylphorbol-13-acetate. J Biol Chem. 1999;274:29672–29676. doi: 10.1074/jbc.274.42.29672. [DOI] [PubMed] [Google Scholar]
- 45.Assefa Z, Vantieghem A, Declercq W, Vandenabeele P, Vandenheede JR, Merlevede W, de Witte P, Agostinis P. The activation of the c-Jun N-terminal kinase and p38 mitogen-activated protein kinase signaling pathways protects HeLa cells from apoptosis following photodynamic therapy with hypericin. J Biol Chem. 1999;274:8788–8796. doi: 10.1074/jbc.274.13.8788. [DOI] [PubMed] [Google Scholar]
- 46.Karin M. Signal transduction from the cell surface to the nucleus through the phosphorylation of transcription factors. Curr Opin Cell Biol. 1994;6:415–424. doi: 10.1016/0955-0674(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 47.van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Q, Liu S, Ding L, Ye X, Su W. PKCalpha translocation from mitochondria to nucleus is closely related to induction of apoptosis in gastric cancer cells. Sci China C Life Sci. 2002;45:237–244. doi: 10.1360/02yc9026. [DOI] [PubMed] [Google Scholar]
- 49.Liu S, Wu Q, Ye XF, Cai JH, Huang ZW, Su WJ. Induction of apoptosis by TPA and VP-16 is through translocation of TR3. World J Gastroenterol. 2002;8:446–450. doi: 10.3748/wjg.v8.i3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]