

Abstract
AIM: Both Hepatitis B virus (HBV) and Hepatitis C virus (HCV) are major causative agents of transfusion-associated and community-acquired hepatitis worldwide. Development of a HCV vaccine as well as more effective HBV vaccines is an urgent task. DNA immunization provides a promising approach to elicit protective humoral and cellular immune responses against viral infection. The aim of this study is to achieve immune responses against both HCV and HBV by DNA immunization with fusion constructs comprising various HCV E2 gene fragments fused to HBsAg gene of HBV.
METHODS: C57BL/6 mice were immunized with plasmid DNA expressing five fragments of HCV E2 fused to the gene for HBsAg respectively. After one primary and one boosting immunizations, antibodies against HCV E2 and HBsAg were tested and subtyped in ELISA. Splenic cytokine expression of IFN-γ and IL-10 was analyzed using an RT-PCR assay. Post-immune mouse antisera also were tested for their ability to capture HCV viruses in the serum of a hepatitis C patient in vitro.
RESULTS: After immunization, antibodies against both HBsAg and HCV E2 were detected in mouse sera, with IgG2a being the dominant immunoglobulin sub-class. High-level expression of INF-γ was detected in cultured splenic cells. Mouse antisera against three of the five fusion constructs were able to capture HCV viruses in an in vitro assay.
CONCLUSION: The results indicate that these fusion constructs could efficiently elicit humoral and Th1 dominant cellular immune responses against both HBV S and HCV E2 antigens in DNA-immunized mice. They thus could serve as candidates for a bivalent vaccine against HBV and HCV infection. In addition, the capacity of mouse antisera against three of the five fusion constructs to capture HCV viruses in vitro suggested that neutralizing epitopes may be present in other regions of E2 besides the hypervariable region 1.
INTRODUCTION
Both Hepatitis B virus (HBV) and Hepatitis C virus (HCV) are major causative agents of transfusion-associated and community-acquired hepatitis worldwide[1,2]. It is estimated that there are 250 million HBV carriers in the world and more than 10% of chronically infected HBV patients eventually develop cirrhosis and hepatocellular carcinoma[3]. About 2%-3% of the world population are HCV carriers. More than 70% of HCV infections become chronic, among which 5%-20% progress to liver cirrhosis and hepatocellular carcinoma[4,5]. Available HBV vaccines have proven to be safe and effective in preventing HBV infection. However, high costs, exclusion of some escape mutants and neonatal intolerance are elimilating their wide use[6]. So far, no vaccine is available against HCV infection. IFN-γ treatment is the only useful therapy available. However, only 20%-30% of treated patients develop long-term responses[7]. Therefore, HBV and HCV infections pose a worldwide health threat and the development of uniformly effective vaccines of affordable prices is an urgent task.
DNA immunization, which allows the de novo synthesis of antigens in host’s cells, is able to elicit protective humoral and cellular immune responses in several animal models of viral infection[8-10]. The cellular context for de novo synthesized proteins to achieve proper maturation is a particularly important advantage for proteins such as those constituting viral envelopes whose maturation requires the help of additional cellular factors. Increasing data showed that DNA immunization against HBsAg elicited strong humoral and cellular immune responses that protect chimpanzees against the challenge with HBV. Moreover, DNA immunization in transgenic mice expressing HBsAg in the liver resulted in the clearance of HBsAg and long-term control of transgene expression, suggesting that DNA immunization is a potential tool in the treatment of HBV chronic carriers[11-15]. DNA immunization with HCV E2 protein that was believed to carry the major neutralization epitopes of HCV[16] also was studied in several animal models including primates[17-21]. These studies demonstrated that DNA immunization with HCV E2 elicited strong humoral and cellular immune responses in various animals, though it did not elicit sterilizing immunity in chimpanzees against the challenge with a monoclonal homologous virus. The DNA immunization did appear to modify the infection and might have prevented the progression to chronicity, suggesting that DNA vaccine could be a promising approach for HCV treatment.
The objective of this research was to simultaneously stimulate immune responses against both HBV and HCV by DNA immunization with fusion constructs comprising of various HCV E2 gene fragments fused to the HBsAg gene of HBV. HBsAg carries all the information required for membrane translocation, particle assembly and secretion from mammalian cells. We have previously shown that HBsAg carrying HBV preS1 (21-47) at its truncated carboxyl terminal end could present the preS1 epitope on the surface of the chimeric particle and induce preS1 specific antibodies in mice[22-24]. Moreover, humoral and cellular immune responses were successfully induced via direct injection of the plasmid containing the HBsAg-preS1 fusion gene[15]. These data indicated that gene fragments of proper size could be fused to the C-terminal of HBsAg without affecting particle assembly and secretion, and were capable of inducing immune responses against both HBsAg and the fused epitope. Although the epitopes on envelope protein E2 are not very clear yet, there have been some successful experiments to determine the immune determinants[25-27]. Based on these previous findings, five fragments of HCV-E2 were selected in the hydrophilic region of E2 protein and fused to the truncated 3’ end of HBsAg gene. The humoral and cellular immune responses of the plasmids expressing fusion proteins were evaluated. Furthermore, virus-capture ability of antibodies against those HCV-E2 fragments was also examined. The results are opening a new approach to the development of a bivalent vaccine against HBV and HCV, and lead to a better understanding of the immunological property of HCV-E2.
MATERIALS AND METHODS
Expression plasmids
Five HCV E2 gene fragments (A to E) were amplified from pUC18/E (E1-E2 gene of HCV genome type 1b inserted in pUC18)[28] with following primers:
Math 1
Math 1.

Math(A1).
Acc I and EcoR I sites were introduced in sense and antisense primers, respectively. After digestion with Acc I and EcoR I, PCR products were inserted downstream from the HBV S gene in pcDNA3S[29] to obtain pcDNA3SE2-A, pcDNA3SE2-B, pcDNA3SE2-C, pcDNA3SE2-D and pcDNA3SE2-E (Figure 1). A fragment coding for amino acids 384 to 649 of HCV-E2 was also amplified from pUC18/E with primers P11 (5’-CCGGCGGATCCATGAACACC-3’) and P12 (5’-GCGAATTCATCCTCGAGTCCAG-3’). PcDNA3E2 was constructed by inserting this PCR product digested with BamH I and EcoR I into the MCS of pcDNA3 (Figure 1).
Figure 1.

Schematic diagram of the expression plasmids. The coordinates below the bars refer to the corresponding amino acid residues of HBV surface antigen (HBsAg) and HCV E2 protein. The fusion genes were under the control of CMV immediate early promoter in pcDNA3. Bovine growth hormone (bGH) 3’-untranslated sequences were used as polyadenylation signals.
ELISA for transiently expressed HBV surface antigen
COS-M6 cells were maintained in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 5% fetal calf serum (Gibco, BRL, USA). 10 µg of plasmid DNA and 5 µg of an internal control pSK110, which expresses β-galactosidase, were transfected into 5 × 105 COS-M6 cells by calcium phosphate co-precipitation method. Forty-eight hours after transfection, cells and media were collected. The activity of β-galactosidase in cell lysate obtained by cycles of freezing and thawing was analyzed to normalize the variation of transfection efficiency among different samples. HBsAg in cell lysates and media was determined with a commercial ELISA kit (Sino-American Co., Shanghai, China). The kit was adapted for quantitative measurements of HBsAg using purified yeast derived HBsAg (National Vaccine and Serum Institute, Beijing, China) as potency standard. Results were presented as means of three independent experiments[23,30].
Western Blotting of transiently expressed products
HeLa cells were maintained in DMEM supplemented with 5% fetal calf serum. To enhance the expression level of fusion genes, cells were first infected with vaccinia virus vTT7 that expresses T7 RNA polymerase at a M.O.I. of 10 pfu per cell[31]. Two hours post-infection, 1-2 × 105 cells were transfected with 10 µg of plasmid DNA by calcium phosphate method. Eighteen hours after transfection, cells were collected and cell lysate was electrophoresed on a 12.5% SDS-PAGE and subsequently electro-blotted onto nitrocellulose membrane. After blocking for 1 h with 5% powdered lipid-free milk in PBST (PBS containing 0.1% Tween 20), the membrane was incubated with anti-HBs McAb H116 (kindly provided by Dr. E. Hildt, Munich, Germany) or anti-HCV E2 (450-565) polyclonal antiserum RE2-116[32], followed by incubation with HRP conjugated secondary antibody (Dako Co., Denmark). Signals were detected by enhanced chemi-luminescence (ECL) blotting analysis system (Amersham-Pharmacia Co., UK).
DNA immunization of mice
C57BL/6 (H-2b) mice were purchased from Shanghai Laboratory Animal Center. Groups of five female mice, 6-8 weeks old, were injected with 100 µg of expression plasmids into regenerated tibialis anterior (TA) muscles 5 days after treatment of 100 µL 10-5 mol/l cardiotoxin (Sigma, USA)[15,29]. Four weeks later, the mice were boosted in the same way.
Serologic tests
Sera were collected by tail bleeding at different time points pre-and post-injection and assayed for anti-HBs and anti-HCV-E2 by ELISA. Microwell plates were coated with serum derived HBsAg particles[29] (Kehua Co., Shanghai, China), or 0.1 µg/well HCV-E2 (385-565) expressed in E. coli[32]. After blocking with 5% powdered lipid-free milk in PBST (PBS containing 0.1% Tween 20), serial two-fold dilutions of sera were added. The bound antibodies were detected with HRP-conjugated rabbit anti-mouse IgG (1:1000) (Dako Co., Denmark) after extensive washing with PBST. Sera from mice immunized with pcDNA3 vector at 1 in 20 dilution were used as negative controls. A positive result was defined as an absorbance value of great than twice the absorbance of negative control with a cutoff of 0.050. Seroconversion was defined as giving a positive reading at 1:20 dilution. Antibody titres of pooled seroconverted animal sera were set as the highest dilution giving a positive reading (end-point dilution method)[22,32].
To type the subclasses of IgG, mouse sera of each group were collected 6 weeks after the first immunization, and the titers of IgG1 and IgG2a antibodies assayed individually by a similar end-point dilution method, using the HRP-conjugated rabbit anti-mouse IgG1 and IgG2a (Serotec Co., Oxford, UK) for detection.
Splenic cytokine expression test
Expression of IFN-γ and IL-10 were assayed semi-quantitatively as described[33,34]. Mice were killed 10 weeks after the first injection and spleens were removed aseptically. The single cell suspensions of the splenocytes were prepared and maintained in DMEM supplemented with 5% fetal calf serum and 5 × 10-5 M β-ME at a density of 107 cells per well. Cells were stimulated with 0.5 µg/ml rDNA yeast derived HBsAg (National Vaccine and Serum Institute, Beijing, China), followed by culture at 37 °C. At 0, 24, 48 hr post-stimulation, splenic cells were sampled. Total RNA was extracted with Trizol (Gibcol, BRL, USA) and reverse transcribed with random hexamer primers (New England Biolabs Inc, Beverly, USA.). Messenger RNAs for IFN-γ and IL-10 were semi-quantitated by competitive RT-PCR[33,34]. Splenic cells from mice immunized with pcDNA3 and mitogen ConA (5 µg/ml) served as a negative and a positive control respectively.
Analysis of the virus capture capability of immune sera
The virus capture capability of antibodies in immune sera was assessed according to the method of affinity capture RT-PCR (AC-RT-PCR) detection of HEV in patient stool specimens[35] with a few modifications. Briefly, 0.5 mL microcentrifuge tubes were coated with 50 µL goat anti-mouse IgG F (ab’)ª-2 (0.02 µg/µL, Jackson ImmunoResearch Laboratories, Inc., West Grove, USA), capped and incubated at room temperature for 1 hr. After blocking with 5% powdered lipid-free milk in PBST for 15 min at 37 °C and overnight at 4 °C, tubes were aspirated and washed with 150 µL PBST. The mouse sera were adjusted to same titre of 1:80 by dilution with PBS, to avoid the variations of the results caused by different antibody titers in different samples. Tubes were subsequently loaded with 25 µL properly diluted mouse immune sera and 25 µL HCV-RNA-PCR-positive patient’s serum (1.6 × 107 HCV genome Equivalent [Geq] per mL, provided by Dr. X. Zhang, Ruijin Hospital, Shanghai, China) diluted 1:50 in PBS. The mixtures were mixed gently and incubated for 1 hr at room temperature and overnight at 4 °C. Tubes were then washed three times with 150 µL wash buffer (25 mM Tris-HCl pH8.0, 75 mM KCl, 2.5 mM MgCl2). Captured virus was detected with a commercial HCV PCR testing kit (Sino-American Co., Shanghai, China) which specifically amplifies a 225 bp fragment in the 5’ UTR of HCV RNA. HCV which was captured directly by a conformation specific monoclonal antibody (McAb) against HCV-E2, 219A2, (kindly provided by Dr. S. Abrignani, IRIS, Siena, Italy) was used as positive control.
RESULTS
Transient expression of HBsAg-HCV E2 fusion proteinsFive expression plasmids containing the HCV-E2 coding sequences (corresponding to amino acids 384-413, 414-443, 460-489, 490-519, 529-549) fused to the 3’ end of HBV-S gene were constructed as described in "Materials and Methods". COS-M6 cells were then transfected with these plasmids respectively.
Forty-eight hours post-transfection, the presence of HBsAg fusion protein in the cell lysates and culture media was measured with HBsAg ELISA. HBsAg was detected in both the cell lysates and culture media, indicating that the fusion constructs were able to express and secrete HBsAg efficiently (Figure 2).
Figure 2.
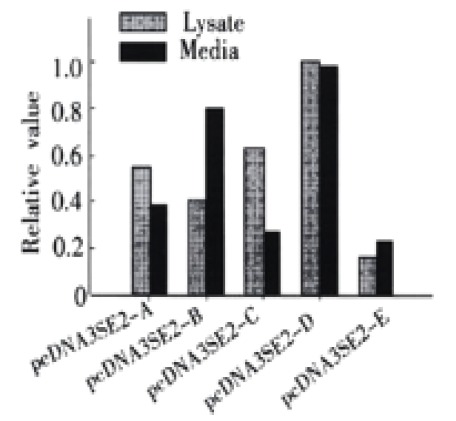
Comparison of the expression and secretion level of fusion proteins in COS cells. pcDNA3SE2-A, pcDNA3SE2-B, pcDNA3SE2-C, pcDNA3SE2-D, pcDNA3SE2-E are the plasmids expressing HBsAg fused with different HCV E2 fragments. The HBsAg in the culture medium (gray) and cell lysates (black) was measured by a commercial ELISA kit (Sino-American Co., Shanghai, China). Results were presented as means of three independent experiments (see Materials and Methods). The amount of HBsAg expressed by pcDNA3SE2-D in cell lysates was arbitrarily taken as 1.0.
Expressed HBsAg and HCV-E2 fusion proteins were also evaluated by Western Blot. They were separated by SDS-PAGE gel, followed by incubation with HBsAg specific McAb H116 (Figure 3A). Compared with HBsAg expressed by pcDNA3S (Figure 1), which contained the major surface gene of HBV only and displayed 24000 and 27000 protein bands for the unglycosylated and glycosylated forms respectively (Figure 3A, lane1)[29], all the fusion antigens showed slower migratory bands (27000 and 29000), indicating successful expression of the fusion proteins. Two fusion constructs SE2-B (414-443) and SE2-D (490-519) displayed additional bands at 31000, which might result from multi-glycosylation. Expressed fusion proteins SE2-C (460-489), SE2-D (490-519) and SE2-E (529-549) were also incubated with polyclonal antibody RE2-116. The band pattern in the Western Blots (Figure 3B) was consistent with that in Figure 3A, suggesting that these fusion proteins possess both HBV-S and HCV-E2 antigenicity. SE2-A (384-413) and SE2-B (414-443) were not assayed for HCV-E2, since they did not locate in the reactive region (450-565) of RE2-116. By contrast, the non-fused truncated E2 (384-649) (Figure 1) displayed only a single band as large as the expected unglycosylated product (Figure 3B, lane 1).
Figure 3.
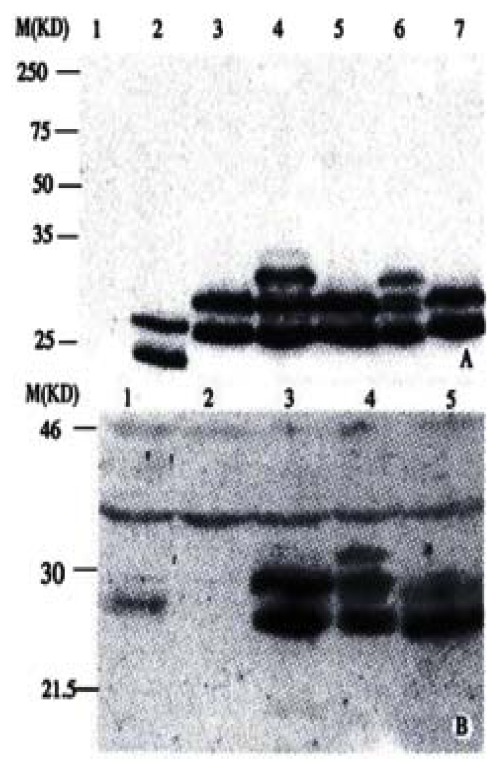
Characterization of the fusion antigens transiently expressed in HeLa cells by Western blotting. After separation by a 12.5% SDS-PAGE, the proteins were blotted onto a nitro-cellulose membrane and incubated with (A) anti-S McAb (H166, 1:300 diluted), (B) anti-HCV E2 (450-565) polyclonal antiserum RE2-116 (1:500 diluted) respectively. (A) lane 1-7, fusion proteins expressed by pcDNA3, pcDNA3S, pcDNA3SE2-A, pcDNA3SE2-B, pcDNA3SE2-C, pcDNA3SE2-D and pcDNA3SE2-E respectively. (B) lane 1-5, fusion proteins expressed by pcDNA3E2-pcDNA3, pcDNA3SE2-C, pcDNA3SE2-D, pcDNA3SE2-E respectively.
Humoral responses to fusion antigens in DNA-immunized mice
C57BL/6 mice were injected with HCV-E2 and HBV-S fusion constructs. As shown in Table 1 and Table 2, the immunized mice produced antibodies against both HBsAg and HCV-E2 after the first injection, and the seroconversion rate increased quickly after the boost, reaching 100% at week 6 or 8. Antibodies were still detectable after 24 weeks. Impressively, the anti-HCV E2 antibody titers elicited in mice immunized with fusion constructs were much higher than that induced in mice injected with pcDNA3E2 (up to 40 fold difference), which contained non-fused HCV E2 (384-649). Compared with the humoral response induced by pcDNA3S, all the fusion constructs were able to induce equivalent or slightly smaller anti-HBs antibody responses.
Table 1.
Sero-conversion Ratios and Titres of Anti-HBsAg in DNA Immunized Mice
| Weeks |
DNA Immunogen |
|||||
| pcDNA3S | pcDNA3SE2-A | pcDNA3SE2-B | pcDNA3SE2-C | pcDNA3SE2-D | pcDNA3SE2-E | |
| 0a | (0/5) | (0/5) | (0/5) | (0/5) | (0/5) | (0/5) |
| 2 | (5/5) | (1/5) | (1/5) | (1/5) | (1/5) | (2/5) |
| 4b | 1280d (5/5c) | 1280 (4/5) | 640 (2/5) | 320 (2/5) | 320 (2/5) | 320 (4/5) |
| 6 | 10240 (5/5) | 10240 (5/5) | 5120 (4/5) | 5120 (4/5) | 5120 (5/5) | 2560 (5/5) |
| 8 | 5120 (5/5) | 5120 (5/5) | 5120 (5/5) | 5120 (5/5) | 5120 (5/5) | 5120 (5/5) |
| 10 | 5120 (5/5) | 5120 (5/5) | 2560 (4/5) | 2560 (5/5) | 2560 (5/5) | 5120 (5/5) |
| 16 | 1280 (5/5) | 1280 (5/5) | 640 (4/5) | 320 (5/5) | 640 (5/5) | 80 (5/5) |
| 24 | 1280 (5/5) | 1280 (5/5) | 640 (4/5) | 320 (5/5) | 640 (5/5) | 80 (5/5) |
Note: Groups of mice (n = 5) were injected i.m. with the respective plasmids, and anti-HBsAg antibody titers were measured at different time points by ELISA. Sera from mice immunized with pcDNA3 vector were used as negative controls and showed no reactivity with any coated antigens (data not shown). For sero-conversion, 1:20 starting dilution were assayed and the sera giving positive reading were taken as converted one. To deter-mine antibody titers, serial two-fold dilutions of pooled sero-converted immune sera were incubated in antigen-coated wells. A positive result was defined as an absorbance value of greater than twice the absorbance of the negative control with a cutoff of 0.050 (also see Materials and Methods). At week 2 only sero-conversion testing was performed at 1:20 dilution.
the first immunization;
the second immunization (boost);
(n/n); sero-conversion ratio, numbers of sero-converted mice relative to the total number tested;
titer; reciprocal of the two-fold dilution factor at end point; pools of sero-converted sera from test groups were tested.
Table 2.
Sero-conversion Ratios and Titres of Anti-HCV-E2 in DNA Immunized Mice
| Weeks |
DNA Immunogen |
|||||
| pcDNA3S | pcDNA3SE2-A | pcDNA3SE2-B | pcDNA3SE2-C | pcDNA3SE2-D | pcDNA3SE2-E | |
| 0a | (0/5) | (0/5) | (0/5) | (0/5) | (0/5) | (0/5) |
| 2 | (1/5) | (2/5) | (1/5) | (1/5) | (1/5) | (1/5) |
| 4b | 40d (2/5c) | 320 (3/5) | 160 (1/5) | 320 (3/5) | 320 (2/5) | 320 (1/5) |
| 6 | 160 (5/5) | 640 (5/5) | 640 (5/5) | 2560 (5/5) | 2560 (5/5) | 5120 (4/5) |
| 8 | 880 (5/5) | 640 (5/5) | 320 (5/5) | 5120 (5/5) | 5120 (5/5) | 5120 (4/5) |
| 10 | 80 (5/5) | 160 (5/5) | 320 (5/5) | 5120 (5/5) | 5120 (5/5) | 5120 (4/5) |
| 16 | 80 (5/5) | 80 (5/5) | 160 (5/5) | 160 (5/5) | 320 (5/5) | 80 (4/5) |
| 24 | 40 (5/5) | 40 (5/5) | 80 (5/5) | 80 (5/5) | 160 (5/5) | 40 (4/5) |
Note: Groups of mice (n = 5) were injected i.m. with the respective plasmids, and sero-conversion ratio and anti-HCV E2 antibody titers were measured at different time points by ELISA. With the exception of coating antigen (E2 (385-565)), all experimental procedures were the same as described in Table 1.
the first immunization;
the second immunization (boost);
(n/n); sero-conversion ratio, numbers of sero-converted mice relative to the total number tested;
titer; reciprocal of the two-fold dilution factor at end point, pools of positive sera from test groups were tested.
Subsets of IgG against both HBsAg and HCV-E2 were also determined. The IgG profiles specific to HBsAg indicated the obvious predominance of IgG2a (Figure 4A), while IgG profiles specific to HCV-E2 varied among different constructs (Figure 4B). Only pcDNA3SE2-A and pcDNA3SE2-D induced predominant IgG2a against HCV-E2, suggesting that the T helper responses elicited against these two fragments were Th1 dominant.
Figure 4.
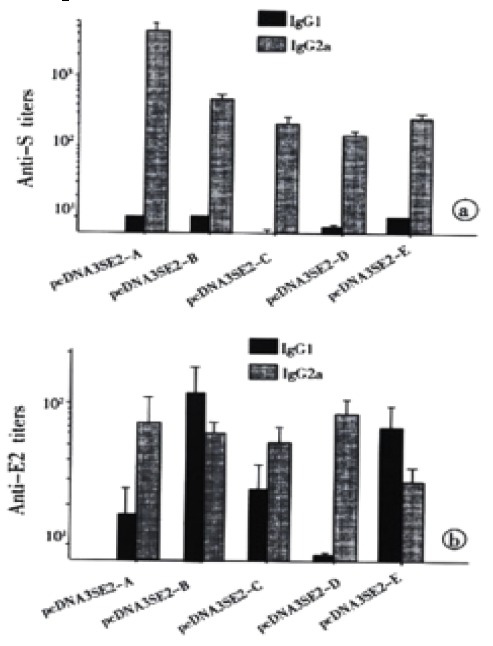
IgG subtypes profile of antibodies against HBsAg and HCV E2. Sera collected from mice 6 weeks after boosting were assayed for the IgG1 and IgG2a antibodies against HBsAg and HCV E2. For each group of 5 mice, titers of IgG1 and IgG2a antibodies were determined individually by serial two-fold dilution titration methods using HRP-conjugated rabbit anti-mouse IgG1 and IgG2a (Serotec Co., Oxford, UK) for detection. (as described in Materials and Methods). The arithmetic mean ± standard deviation (SD) (n = 5) is shown. (A) Anti-HBs, (B) Anti-HCV E2.
Expression of cytokines in the cultured splenic cells from DNA-immunized mice
After the in vitro stimulation of splenic cells derived from immunized mice, expressions of cytokines were analyzed by competitive RT-PCR. 24 hrs post-stimulation with rDNA yeast derived HBsAg (National Vaccine and Serum Institute, Beijing, China), high levels of IFN-γ were detected in the splenic cells from the mice immunized with pcDNA3SE2-A, pcDNA3SE2-B, pcDNA3SE2-C and pcDNA3SE2-E (Figure 5). The IL-10 expression was also detectable, but weaker than IFN-γ expression.
Figure 5.
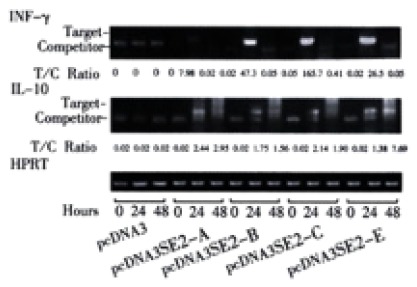
IFN-γ and IL-10 mRNA expression. mRNA was isolated from splenic cells of immunized mice after stimulation with HBsAg for 0, 24, 48 hours. IFN-γ and IL-10 mRNA expression was determined by semi-quantitative RT-PCR after standardization of the cDNA concentration by amplification of HPRT. Level of expression is indicated as ratio of target to competitor, calculated from densitometric analysis of their PCR products. (High ratio indicates high expression of target mRNA).
Virus-capture ability of antisera from DNA-immunized mice
To test the virus-capture capability of antibodies elicited in response to DNA immunization, affinity-capture-RT-PCR (AC-RT-PCR) was performed as described in "Materials and Methods". The HCV virions bound to anti-HCV-E2 were detected by nested RT-PCR to amplify a specific fragment in the 5’-UTR of HCV genome. The results indicated that immune sera of mice immunized with pcDNA3SE2-A, pcDNA3SE2-B and pcDNA3SE2-D were able to capture HCV virions in the serum of a hepatitis C patient in vitro (Figure 6).
Figure 6.
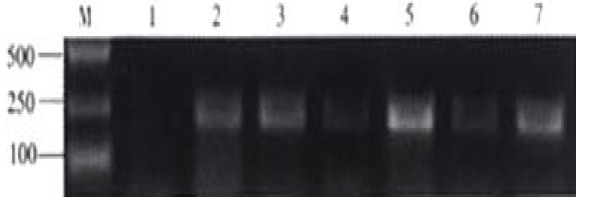
Detection of HCV captured by mouse antisera by nested RT-PCR. After immuno capture of HCV from hepatitis C patient serum (1.6 × 107 HCV GEq per mL) by antisera from groups of mice immunized with pcDNA3, pcDNA3SE2-A, pcDNA3SE2-B, pcDNA3SE2-C, pcDNA3SE2-D and pcDNA3SE2-E (lane 1 to lane 6), viruses were detect1ed by nested RT-PCR using the HCV RT-PCR testing kit (Sino-America Co., Shanghai, China). Lane 7, the positive control using McAb against HCV E2 to capture HCV. Lane M, molecular markers.
DISCUSSION
In this report, five fusion constructs comprising various fragments within the hydrophilic region of HCV-E2 fused to the 3’ end of the S gene of HBV were constructed. The immune responses of mice to immunization with these fusion construct s indicated that all five constructs were able to elicit strong antibody responses against both HBsAg and HCV-E2. Importantly, antibody typing showed the HBsAg specific Th1 like responses, which were consistent with the high levels of IFN-γ expression in cultured spleen cells from the immunized mice after stimulation with HBsAg. The IgG subtype responses to HCV-E2 also showed E2 specific Th1 like responses to E2-A and E2-D. Since Th1 like response is essential for triggering a wide range of cellular responses against infectious agents, including NK cell responses, CTL responses and inflammatory reactions, the results of the DNA immunization with our fusion plasmids suggest a promising strategy to clear both HBV and HCV in the host.
The excellent vaccine potential of our constructs may be ascribed to the use of HBsAg as a carrier. HBsAg has the advantage of assembling into large secretable particles, which form a virus-like polymeric structure that enhances antigenic stability and provides a high-density presentation to antigen-presenting cells (APC). HBsAg has been used successfully as vaccine carrier to present various antigens[15,23,29,36,37]. The successful immune responses to both HBV S and HCV E2 in these reports support our prelimilary observation that antigens of a proper size fused to C-terminal of HBsAg could be successfully presented without affecting the immunogenicity of HBsAg. Moreover, our fusion constructs were able to elicit higher levels of antibody responses to HCV E2, compared to pcDNA3E2 that encodes only a truncated HCV-E2 (384-649). This of course suggests that fusion to HBsAg may help these fragments to be presented more effectively to APCs, and hence induce stronger humoral responses against HCV-E2. These fusion constructs may turn out to be promising candidates for the vaccines against HCV and HBV.
It is well established that the intact E2 glycoproteins expressed alone or together with E1 (E1-E2 heterodimer) are retained in the endoplasmic reticulum[38] and that E2 proteins expressed in mammalian cells without the signal sequence at the C-terminus of E1 are not glycosylated[39]. Consistently, we also observed that the truncated E2 (384-649) was expressed in unglycosylated form in mammalian cells. However, the use of HBsAg as carrier seems to give rise to expression of fusion proteins carrying HCV-E2 epitopes in glycosylated and secretable form, which emphasizes the advantage of HBsAg as a carrier. Notably, two products, SE2-B and SE2-D expressed by pcDNA2SE2-B and pcDNA3SE2-D, produced an additional 31000 glycosylated form, suggesting that in addition to the glycosylation site in HBsAg, some sites in fragment B and D also may be glycosylated. Since glycosylation is a decisive factor for biological molecules to carry out correct biological functions, the additional glycosylation of the fusion constructs may elicit immune responses similar to that of natural infections by the HCV. This may be an additional reason that our constructs are able to elicit strong immune responses.
The results presented here also suggest that the DNA immunization with different HCV E2 fragments fused to HBsAg may be a useful approach to screen for epitopes or immune determinants on HCV-E2. Nakano et al[18] inserted different regions of HCV E2 into the preS2 region of HBV surface protein, and evaluated the humoral immune responses of these fusion constructs via DNA immunization. Their study also suggest that fusion constructs of E2 with HBsAg might be an efficient way to identify restricted immunogenic domains within E2. In our study, each of the chosen E2 fragments contained only 30 amino acids, it provides a possibility to study the antigenic domains of HCV E2 more precisely. Antibody responses and IgG subtype typing against these E2 fragments suggest that B- and T-epitopes were present in fragment D (490-519). Furthermore, the results of virus-capture assay showed that HCV virions in the serum of a hepatitis C patient could be captured by immune sera against SE2-A, SE2-B and SE2-D in vitro. In particular, antibodies against fragment D exhibited the strongest ability to capture HCV virions. Because the ability of antibodies to bind viruses is a prerequisite for neutralizing viruses in vivo and preventing bodies from being infected, our data suggest that besides HVR1-containing fragment A, fragments B and D probably also harbour neutralizing epitopes. So far, only HVR1 has been confirmed to be an important epitope[40-48] whose corresponding antibody can neutralize the binding of HCV-E2 to susceptible cells in vitro, and prevent HCV infection after in vitro neutralization[42,43]. Recently, correlation between circulating antibodies against HVR1 and resolution of chronic hepatitis C has been confirmed[49]. However, the high variability of HVR1 interferes with the development of HVR1-based vaccines. Some researchers suggested that there may be B-cell epitopes located downstream from HVR1[17,18,50]. Rosa et al[43] speculated that there may be at least one additional neutralizing epitope located somewhere other than HVR1 of HCV E2, this was based on the finding that HVR-1 peptide failed to completely block the binding of HCV to susceptible cells. Our results suggest that potential neutralizing epitopes may be present within fragments B (414-443) and D (490-519) that are downstream from HVR1. Since fragments B and D are relatively more conservative than HVR1, it would seem that vaccines containing fragments B and/or D may be more promising than that containing only HVR1. Further studies on these neutralizing epitopes are under way, which hopefully will lead to a better understanding of the immunological property of HCV E2, and facilitate the design of efficient HCV vaccines.
ACKNOWLEDGMENTS
The authors are grateful to Dr. E. Hildt for providing monoclonal antibody (McAb) H166, Dr. S. Abrignani for McAb 219A2, Dr. Yuan ZH for HRP-Rabbit antibody against mouse IgG1 and IgG2a, Dr. Zhang XX for HCV patient serum and Qu D for her help in cytokine test.
Footnotes
Supported by the National High-Technology Program of China, No. 863-102-07-02-02
Edited by Pang LH
References
- 1.Farci P. Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome [Science 1989; 244: 359-362] J Hepatol. 2002;36:582–585. doi: 10.1016/s0168-8278(02)00051-x. [DOI] [PubMed] [Google Scholar]
- 2.Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 3.Tiollais P, Buendia MA. Hepatitis B virus. Sci Am. 1991;264:116–123. doi: 10.1038/scientificamerican0491-116. [DOI] [PubMed] [Google Scholar]
- 4.Alter MJ, Margolis HS, Krawczynski K, Judson FN, Mares A, Alexander WJ, Hu PY, Miller JK, Gerber MA, Sampliner RE. The natural history of community-acquired hepatitis C in the United States. The Sentinel Counties Chronic non-A, non-B Hepatitis Study Team. N Engl J Med. 1992;327:1899–1905. doi: 10.1056/NEJM199212313272702. [DOI] [PubMed] [Google Scholar]
- 5.Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe Y, Koi S, Onji M, Ohta Y. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87:6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thanavala Y. Novel approaches to vaccine development against HBV. J Biotechnol. 1996;44:67–73. doi: 10.1016/0168-1656(95)00117-4. [DOI] [PubMed] [Google Scholar]
- 7.Fried MW, Hoofnagle JH. Therapy of hepatitis C. Semin Liver Dis. 1995;15:82–91. doi: 10.1055/s-2007-1007265. [DOI] [PubMed] [Google Scholar]
- 8.Lai WC, Bennett M. DNA vaccines. Crit Rev Immunol. 1998;18:449–484. doi: 10.1615/critrevimmunol.v18.i5.30. [DOI] [PubMed] [Google Scholar]
- 9.Donnelly JJ, Ulmer JB, Liu MA. Immunization with DNA. J Immunol Methods. 1994;176:145–152. doi: 10.1016/0022-1759(94)90308-5. [DOI] [PubMed] [Google Scholar]
- 10.McDonnell WM, Askari FK. DNA vaccines. N Engl J Med. 1996;334:42–45. doi: 10.1056/NEJM199601043340110. [DOI] [PubMed] [Google Scholar]
- 11.Davis HL, Michel ML, Whalen RG. DNA-based immunization induces continuous secretion of hepatitis B surface antigen and high levels of circulating antibody. Hum Mol Genet. 1993;2:1847–1851. doi: 10.1093/hmg/2.11.1847. [DOI] [PubMed] [Google Scholar]
- 12.Michel ML, Davis HL, Schleef M, Mancini M, Tiollais P, Whalen RG. DNA-mediated immunization to the hepatitis B surface antigen in mice: aspects of the humoral response mimic hepatitis B viral infection in humans. Proc Natl Acad Sci USA. 1995;92:5307–5311. doi: 10.1073/pnas.92.12.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mancini M, Davis H, Tiollais P, Michel ML. DNA-based immunization against the envelope proteins of the hepatitis B virus. J Biotechnol. 1996;44:47–57. doi: 10.1016/0168-1656(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 14.Mancini M, Hadchouel M, Davis HL, Whalen RG, Tiollais P, Michel ML. DNA-mediated immunization in a transgenic mouse model of the hepatitis B surface antigen chronic carrier state. Proc Natl Acad Sci USA. 1996;93:12496–12501. doi: 10.1073/pnas.93.22.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hui J, Mancini M, Li G, Wang Y, Tiollais P, Michel ML. Immunization with a plasmid encoding a modified hepatitis B surface antigen carrying the receptor binding site for hepatocytes. Vaccine. 1999;17:1711–1718. doi: 10.1016/s0264-410x(98)00430-7. [DOI] [PubMed] [Google Scholar]
- 16.Choo QL, Kuo G, Ralston R, Weiner A, Chien D, Van Nest G, Han J, Berger K, Thudium K, Kuo C. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc Natl Acad Sci USA. 1994;91:1294–1298. doi: 10.1073/pnas.91.4.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tedeschi V, Akatsuka T, Shih JW, Battegay M, Feinstone SM. A specific antibody response to HCV E2 elicited in mice by intramuscular inoculation of plasmid DNA containing coding sequences for E2. Hepatology. 1997;25:459–462. doi: 10.1002/hep.510250234. [DOI] [PubMed] [Google Scholar]
- 18.Nakano I, Maertens G, Major ME, Vitvitski L, Dubuisson J, Fournillier A, De Martynoff G, Trepo C, Inchauspe G. Immunization with plasmid DNA encoding hepatitis C virus envelope E2 antigenic domains induces antibodies whose immune reactivity is linked to the injection mode. J Virol. 1997;71:7101–7109. doi: 10.1128/jvi.71.9.7101-7109.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forns X, Emerson SU, Tobin GJ, Mushahwar IK, Purcell RH, Bukh J. DNA immunization of mice and macaques with plasmids encoding hepatitis C virus envelope E2 protein expressed intracellularly and on the cell surface. Vaccine. 1999;17:1992–2002. doi: 10.1016/s0264-410x(98)00448-4. [DOI] [PubMed] [Google Scholar]
- 20.Fournillier A, Depla E, Karayiannis P, Vidalin O, Maertens G, Trépo C, Inchauspé G. Expression of noncovalent hepatitis C virus envelope E1-E2 complexes is not required for the induction of antibodies with neutralizing properties following DNA immunization. J Virol. 1999;73:7497–7504. doi: 10.1128/jvi.73.9.7497-7504.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forns X, Payette PJ, Ma X, Satterfield W, Eder G, Mushahwar IK, Govindarajan S, Davis HL, Emerson SU, Purcell RH, et al. Vaccination of chimpanzees with plasmid DNA encoding the hepatitis C virus (HCV) envelope E2 protein modified the infection after challenge with homologous monoclonal HCV. Hepatology. 2000;32:618–625. doi: 10.1053/jhep.2000.9877. [DOI] [PubMed] [Google Scholar]
- 22.Xu X, Li GD, Kong YY, Yang HL, Zhang Z, Cao HT, Wang Y. A modified hepatitis B virus surface antigen with the receptor-binding site for hepatocytes at its C terminus: expression, antigenicity and immunogenicity. J Gen Virol. 1994;75(Pt 12):3673–3677. doi: 10.1099/0022-1317-75-12-3673. [DOI] [PubMed] [Google Scholar]
- 23.Hui J, Li G, Kong Y, Wang Y. Expression and characterization of chimeric hepatitis B surface antigen particles carrying preS epitopes. J Biotechnol. 1999;72:49–59. doi: 10.1016/s0168-1656(99)00049-8. [DOI] [PubMed] [Google Scholar]
- 24.Yang JY, Jin J, Kong YY, Wei J, Zhang ZC, Li GD, Wang Y, Yuan HY, Li YY. Purification and Characterization of Recombinant Hepatitis B Virus Surface Antigen SS1 Expressed in Pichia pastoris. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2000;32:503–508. [PubMed] [Google Scholar]
- 25.Koshy R, Inchauspé G. Evaluation of hepatitis C virus protein epitopes for vaccine development. Trends Biotechnol. 1996;14:364–369. doi: 10.1016/0167-7799(96)10049-4. [DOI] [PubMed] [Google Scholar]
- 26.Fournillier A, Nakano I, Vitvitski L, Depla E, Vidalin O, Maertens G, Trépo C, Inchauspé G. Modulation of immune responses to hepatitis C virus envelope E2 protein following injection of plasmid DNA using single or combined delivery routes. Hepatology. 1998;28:237–244. doi: 10.1002/hep.510280131. [DOI] [PubMed] [Google Scholar]
- 27.Lee JW, Kim Km, Jung SH, Lee KJ, Choi EC, Sung YC, Kang CY. Identification of a domain containing B-cell epitopes in hepatitis C virus E2 glycoprotein by using mouse monoclonal antibodies. J Virol. 1999;73:11–18. doi: 10.1128/jvi.73.1.11-18.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Okamoto H, Tsuda F, Nagayama R, Tao QM, Mishiro S. Prevalence, genotypes, and an isolate (HC-C2) of hepatitis C virus in Chinese patients with liver disease. J Med Virol. 1993;40:254–260. doi: 10.1002/jmv.1890400316. [DOI] [PubMed] [Google Scholar]
- 29.Hui JY, Li GD, Kong YY, Yuan Wang. DNA-based immunization against hepatitis B surface antigen carrying preS epitopes. Chinese Sci Bull. 1999;44:620–623. [Google Scholar]
- 30.Feng Y, Kong YY, Wang Y, Qi GR. Intracellular inhibition of the replication of hepatitis B virus by hammerhead ribozymes. J Gastroenterol Hepatol. 2001;16:1125–1130. doi: 10.1046/j.1440-1746.2001.02548.x. [DOI] [PubMed] [Google Scholar]
- 31.Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Zhu L, Zhang X, Lu M, Kong Y, Wang Y, Li G. Expression, purification, immunological characterization and application of Escherichia coli-derived hepatitis C virus E2 proteins. Biotechnol Appl Biochem. 2001;34:109–119. doi: 10.1042/ba20010036. [DOI] [PubMed] [Google Scholar]
- 33.Sun B, Wells J, Goldmuntz E, Silver P, Remmers EF, Wilder RL, Caspi RR. A simplified, competitive RT-PCR method for measuring rat IFN-gamma mRNA expression. J Immunol Methods. 1996;195:139–148. doi: 10.1016/0022-1759(96)00099-3. [DOI] [PubMed] [Google Scholar]
- 34.Sun B, Rizzo LV, Sun SH, Chan CC, Wiggert B, Wilder RL, Caspi RR. Genetic susceptibility to experimental autoimmune uveitis involves more than a predisposition to generate a T helper-1-like or a T helper-2-like response. J Immunol. 1997;159:1004–1011. [PubMed] [Google Scholar]
- 35.He J, Binn LN, Caudill JD, Asher LV, Longer CF, Innis BL. Antiserum generated by DNA vaccine binds to hepatitis E virus (HEV) as determined by PCR and immune electron microscopy (IEM): application for HEV detection by affinity-capture RT-PCR. Virus Res. 1999;62:59–65. doi: 10.1016/s0168-1702(99)00047-7. [DOI] [PubMed] [Google Scholar]
- 36.Prange R, Werr M, Birkner M, Hilfrich R, Streeck RE. Properties of modified hepatitis B virus surface antigen particles carrying preS epitopes. J Gen Virol. 1995;76(Pt 9):2131–2140. doi: 10.1099/0022-1317-76-9-2131. [DOI] [PubMed] [Google Scholar]
- 37.Major ME, Vitvitski L, Mink MA, Schleef M, Whalen RG, Trépo C, Inchauspé G. DNA-based immunization with chimeric vectors for the induction of immune responses against the hepatitis C virus nucleocapsid. J Virol. 1995;69:5798–5805. doi: 10.1128/jvi.69.9.5798-5805.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duvet S, Cocquerel L, Pillez A, Cacan R, Verbert A, Moradpour D, Wychowski C, Dubuisson J. Hepatitis C virus glycoprotein complex localization in the endoplasmic reticulum involves a determinant for retention and not retrieval. J Biol Chem. 1998;273:32088–32095. doi: 10.1074/jbc.273.48.32088. [DOI] [PubMed] [Google Scholar]
- 39.Saito T, Sherman GJ, Kurokohchi K, Guo ZP, Donets M, Yu MY, Berzofsky JA, Akatsuka T, Feinstone SM. Plasmid DNA-based immunization for hepatitis C virus structural proteins: immune responses in mice. Gastroenterology. 1997;112:1321–1330. doi: 10.1016/s0016-5085(97)70146-x. [DOI] [PubMed] [Google Scholar]
- 40.Weiner AJ, Geysen HM, Christopherson C, Hall JE, Mason TJ, Saracco G, Bonino F, Crawford K, Marion CD, Crawford KA. Evidence for immune selection of hepatitis C virus (HCV) putative envelope glycoprotein variants: potential role in chronic HCV infections. Proc Natl Acad Sci USA. 1992;89:3468–3472. doi: 10.1073/pnas.89.8.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zibert A, Schreier E, Roggendorf M. Antibodies in human sera specific to hypervariable region 1 of hepatitis C virus can block viral attachment. Virology. 1995;208:653–661. doi: 10.1006/viro.1995.1196. [DOI] [PubMed] [Google Scholar]
- 42.Scarselli E, Cerino A, Esposito G, Silini E, Mondelli MU, Traboni C. Occurrence of antibodies reactive with more than one variant of the putative envelope glycoprotein (gp70) hypervariable region 1 in viremic hepatitis C virus-infected patients. J Virol. 1995;69:4407–4412. doi: 10.1128/jvi.69.7.4407-4412.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosa D, Campagnoli S, Moretto C, Guenzi E, Cousens L, Chin M, Dong C, Weiner AJ, Lau JY, Choo QL, et al. A quantitative test to estimate neutralizing antibodies to the hepatitis C virus: cytofluorimetric assessment of envelope glycoprotein 2 binding to target cells. Proc Natl Acad Sci USA. 1996;93:1759–1763. doi: 10.1073/pnas.93.5.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, Shapiro M, Alter HJ, Purcell RH. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci USA. 1996;93:15394–15399. doi: 10.1073/pnas.93.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zibert A, Dudziak P, Schreier E, Roggendorf M. Characterization of antibody response to hepatitis C virus protein E2 and significance of hypervariable region 1-specific antibodies in viral neutralization. Arch Virol. 1997;142:523–534. doi: 10.1007/s007050050098. [DOI] [PubMed] [Google Scholar]
- 46.Esumi M, Ahmed M, Zhou YH, Takahashi H, Shikata T. Murine antibodies against E2 and hypervariable region 1 cross-reactively capture hepatitis C virus. Virology. 1998;251:158–164. doi: 10.1006/viro.1998.9393. [DOI] [PubMed] [Google Scholar]
- 47.Watanabe K, Yoshioka K, Ito H, Ishigami M, Takagi K, Utsunomiya S, Kobayashi M, Kishimoto H, Yano M, Kakumu S. The hypervariable region 1 protein of hepatitis C virus broadly reactive with sera of patients with chronic hepatitis C has a similar amino acid sequence with the consensus sequence. Virology. 1999;264:153–158. doi: 10.1006/viro.1999.0004. [DOI] [PubMed] [Google Scholar]
- 48.Shang D, Zhai W, Allain JP. Broadly cross-reactive, high-affinity antibody to hypervariable region 1 of the hepatitis C virus in rabbits. Virology. 1999;258:396–405. doi: 10.1006/viro.1999.9730. [DOI] [PubMed] [Google Scholar]
- 49.Ishii K, Rosa D, Watanabe Y, Katayama T, Harada H, Wyatt C, Kiyosawa K, Aizaki H, Matsuura Y, Houghton M, et al. High titers of antibodies inhibiting the binding of envelope to human cells correlate with natural resolution of chronic hepatitis C. Hepatology. 1998;28:1117–1120. doi: 10.1002/hep.510280429. [DOI] [PubMed] [Google Scholar]
- 50.Mink MA, Benichou S, Madaule P, Tiollais P, Prince AM, Inchauspe G. Characterization and mapping of a B-cell immunogenic domain in hepatitis C virus E2 glycoprotein using a yeast peptide library. Virology. 1994;200:246–255. doi: 10.1006/viro.1994.1182. [DOI] [PubMed] [Google Scholar]