

Abstract
AIM: To investigate the roles of PLCγ2 and PKCα in TPA-induced apoptosis of gastric cancer cells.
METHODS: Human gastric cancer cell line MGC80-3 was used. Protein expression levels of PLCγ2 and PKCα were detected by Western blot. Protein localization of PLCγ2 and PKCα was shown by immunofluoscence analysis under laser-scanning confocal microscope. Apoptotic morphology was observed by DAPI fluorescence staining, and apoptotic index was counted among 1000 cells randomly.
RESULTS: Treatment of gastric cancer cells MGC80-3 with TPA not only up-regulated expression of PLC-γ2 protein, but also induced PLC-γ2 translocation from the cytoplasm to the nucleus. However, this process was not directly associated with apoptosis induction. Further investigation showed that PKCα translocation from the cytoplasm to the nucleus was correlated with initiation of apoptosis. To explore the inevitable linkage between PLC-γ2 and PKCα during apoptosis induction, PLC inhibitor U73122 was used to block PLC-γ2 translocation, in which neither stimulating PKCα translocation nor inducing apoptosis occurred in MGC80-3 cells. However, when U73122-treated cells were exposed to TPA, not only PLC-γ2, but also PKCα was redistributed. On the other hand, when cells were treated with PKC inhibitor alone, PLC-γ2 protein was still located in the cytoplasm. However, redistribution of PLC-γ2 protein occurred in the presence of TPA, no matter whether PKC inhibitor existed or not.
CONCLUSION: PLC-γ2 translocation is critical in transmitting TPA signal to its downstream molecule PKCα. As an effector, PKCα directly promotes apoptosis of MGC80-3 cells. Therefore, protein translocation of PLCγ2 and PKCα is critical event in the process of apoptosis induction.
INTRODUCTION
Once binding to the cell surface receptors, many extracellular signaling molecules could elicit intracellular responses by activating inositol phospholipid-specific phopholipase C (PLC)[1,2]. Activated PLC could catalyze the hydrolysis of phosphatidylinositol 4, 5-biphosphate (PIP2) to diacylglycerol (DAG) and inositol 1, 4, 5-triphosphate (IP3). DAG and IP3 are second messengers, the former could function to activate protein kinase C (PKC), and the latter could stimulate release of Ca2+ from internal stores[1,3-5]. This bifurcating pathway constitutes the cornerstone of a transmembrane signal transductionmechanism that was known to regulate a large array of cellular processes[5-9].
Ten types of PLC isoform have been divided into three types, β, γ and δ[4-6,10,11]. One PLC isoform, PLC-γ, is a substrate of epidermal growth factor (EGF) receptor, and its catalytic activity could be stimulated by tyrosine phosphorylation[12]. PLC-γhas been implicated in mitogenic signaling by platelet-derived growth factor (PDGF) receptor. Through the pleiotropic actions of IP3 and DAG, PLC-γ could participate in regulation of cell proliferation and differentiation[13]. PLC-γ overexpression could favor cell survival in response to acute oxidative stress[14,15]. In rat pheochromocytoma PC12 cells, overexpression of PLC-γcould inhibit apoptosis induced by short wave length ultraviolet radiation[16].
Protein kinase C (PKC) could play a variety of regulatory roles in proliferation, differentiation, apoptosis, gene expression, membrane transportation, and signal transduction[17-19]. There have been at least 11 distinct PKC isoforms[20]. In many tissues, both PKC activation and Ca2+ mobilization could act synergistically to evoke some cellular responses[21-24]. For example, activation of PKC and increase in Ca2+ were the "on" signals for T-cell activation[27,28]. By contrast, this PKC activator could also act as a negative regulator of T-cell activation[27,28]. Thus, PKC activation in T-cells has bidirectional effects on the cellular activation process.
Several evidences suggest that activation of PKC could attenuate receptor-coupled PLC activity in certain types of cell, providing a negative feedback signal to limit the magnitude and duration of receptor signaling[3,5]. Although such a regulatory mechanism has not been fully understood, it is possible that the targets might include receptors, G protein, PTK, protein tyrosine phosphatase and PLC itself[5]. Phosphorylation of Thr-654 of the EGF receptor by PKC reduced the ability of the receptor tyrosine kinase to phosphorylate PLC-γ, thereby preventing PLC-γ activation[29]. A decrease in the extent of tyrosine phosphorylation of PLC-γ has also been proved to be the mechanism by which the PKC activator, TPA and cAMP could attenuate the PIP2 hydrolysis induced by TCR[30]. Thus, interaction between PLC-γ and PKC is related to PLC-γphosphorylation.
To date, little is known about the molecular event of PLC-γ translocation, and the functional consequences of PLC-γ in response to extracellular signal stimulation. It is generally accepted that PLC-γ localizes and functions in cytosol. EGF or PDGF treatment of cells could induce translocation of PLC-γ1 from a predominantly cytosolic localization to membrane fraction[31], showing a preliminary clue that the function of PLC-γ may be in relation with its intracellular changes. In this study, we found that 12-O-tetradecanoylphorbol-1, 3-acetate (TPA) not only up-regulated the expression level of PLC-γ2, but also induced PLC-γ2 translocation from the cytoplasm to the nucleus in gastric cancer cells MGC80-3. In addition, according to our previous studies concerning the critical role of PKC-associated signaling pathway in promoting apoptosis of gastric cancer cell[32], we hypothesized that PLC-γ2 may play an important role in transmitting TPA signal to PKCα, which finally lead to apoptosis induction in gastric cancer cells. This notion may provide a novel strategy for exploring the cross-talk between PLC-γ2 and PKCα in apoptosis.
MATERIALS AND METHODS
Cell line and culture condition
Human gastric cancer cell line, MGC80-3 was established by Cancer Research Center, Xiamen University[33]. The cells were maintained in RPMI-1640 medium, supplemented with 10% FCS, 1 mmol/L glutamine, and 100 U/mL penicillin.
Western blot analysis[34]
The cells were harvested, and suspended in RIPA buffer (10 mmol/L Tris (pH7.4), 150 mmol/L NaCl, 1% Triton X-100, 1% deoxycholic acid, 0.1% SDS, 5 mmol/L EDTA (pH8.0), 1 mmol/L PMSF). Protein concentration was determined using the Bio-Rad protein assay system according to the manufacturer's instructions (Bio-Rad Hereules, CA). 50 μg protein was subjected to SDS-PAGE and transferred to nitrocellulose membrane for Western blot analysis. The membrane was subsequently blocked with 5% dry milk in TBS-T and then immunnoblotted with the responding antibody. Binding was detected by using the ECL kit (Pierce) according to the manufacturer's instructions.
Apoptosis analysis[32]
The cells were trypsinized, and washed in PBS. The harvested cells were fixed in 3.7% paraformaldehyde on ice, washed in PBS and stained with 50 μg/mL of 4, 6-diamidino-2-phenylindole (DAPI, Sigma) containing 100 μg/mL of DNase-free RNase A per mL. The cells were observed under fluorescence microscope. Apoptotic cells were counted among 1000 cells randomly. The apoptotic index was the mean of three independent experiments.
Immunofluorescence analysis[35]
The cells were cultured on a cover glass overnight. After washed in PBS, the cells were fixed in 4% paraformaldehyde. To display PLC-γ2 or PKCα protein, the cells were incubated first with anti-PLC-γ2 or anti-PKCα antibody (Santa Cruz), and then reacted with their corresponding FITC-conjugated anti-IgG (Pharmingen) as secondary antibodies. To visualize the nuclei, the cells were stained with propidium iodine (PI, 50 μg/mL) containing 100 μg of DNase-free RNase A per mL. Fluorescent image was observed under laser-scanning confocal microscope (Bio-Rad MRC-1024ES).
RESULTS
Expression and translocation of PLC-γ2 in response to TPA
To determine the effect of TPA on PLC-γ2 protein expression, Western blot analysis was carried out. As shown in Figure 1A, PLC-γ2 protein was normally expressed in MGC80-3 cells. When exposed to TPA for different time, expression level of PLC-γ2 protein was increased in a time-dependent manner (Figure 1A), implying that PLC-γ2 might play a role in response to TPA. To examine whether redistribution of PLC-γ2 protein also occurred in response to TPA, immunofluorescent localization of PLC-γ2 protein was conducted and observed by laser-scanning confocal microscope. The result illustrated that in intact MGC80-3 cells, PLC-γ2 protein was more abundant in cytoplasm, and formed a much brighter circle a round the nuclear membrane (Figure 1B). After treatment of TPA for different time, PLC-γ2 protein was translocated from the cytoplasm to the nucleus by degrees (Figure 1B). To further confirm this translocation, cytoplasmic and nuclear protein fractions were prepared, and expression level of PLC-γ2 protein was analyzed by Western blot. Figure 1C shows that PLC-γ2 protein in intact cells mainly appears in the cytoplasmic fraction, and only a trace amount in the nuclear fraction. However, TPA-treatment changed the proportion of PLC-γ2 protein between the two fractions and such a change was time-dependent. After 48 h TPA treatment, PLC-γ2 protein could only be detected in the nuclear fraction (Figure 1C). Clearly, this result was in accordance with that of Figure 1B, indicating that transloation of PLC-γ2 protein by TPA occurred in MGC80-3 cells.
Figure 1.
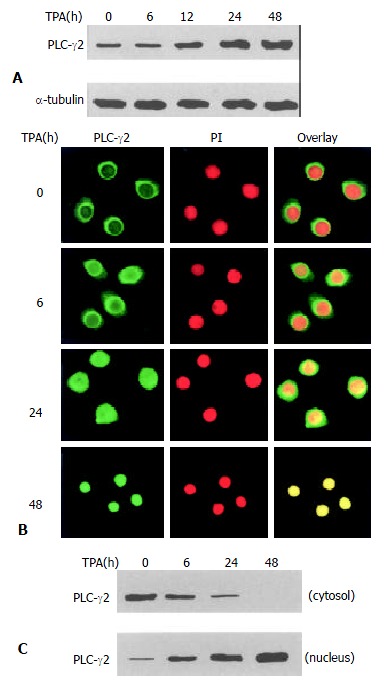
Expression and redistribution of PLC-γ2 in response to TPA. A: Effect of TPA on PLC-γ2 protein expression. Cells were treated with TPA (100 ng/mL) for indicated time, and expression level of PLC-γ2 protein was analyzed by Western blot. α-tubulin was used as a control to quantify the amount of protein used in each lane; B: Translocation of PLC-γ2 in response to TPA. Cells were treated with TPA for indicated time, then immunostained with corresponding antibodies or dye as described in Materials and Methods. The fluorescent images were observed under laser-scanning confocal microscope; C: Redistribution of PLC-γ2 protein in response to TPA. The nuclear and cytosolic fractions were prepared as described in Materials and Methods.
PLC-γ2 expression was not required for apoptosis induction by TPA
Since TPA could induce apoptosis in many types of cancer cell lines[32,35-37], it would be interesting to know whether activation of PLC-γ2 was a necessary link for apoptosis induction by TPA. A PLC-specific inhibitor U73122[38] was thus used upon gastric cancer cells. Prior to be stimulated with TPA, U73122 partially repressed the expression level of PLC-γ2 protein (Figure 2A). In the presence of U73122, TPA-induced increase of PLC-γ2 expression was also blocked. Even after TPA treatment was prolonged for 48 h, the expression level of PLC-γ2 was always lower than the control or TPA treatment alone (Figure 2A). In parallel, apoptosis of MGC80-3 cells was examined by DAPI staining. When the cells were treated with TPA alone, apoptotic cells became smaller, the nuclei became condensed and fragmented with bright chromatins. Importantly, an increased apoptotic index was clearly shown with the extension of TPA treatment (Figure 2B: TPA). However, no such a change was observed in cells treated with U73122 alone (Figure 2B: U73122). By contrast, when cells pretreated with U73122, followed by TPA stimulation, many apoptotic cells were still seen, which not only showed morphological changes similar to those treated with TPA alone, but also had an increased apoptotic index with the extension of TPA treatment (Figure 2B: U73122 + TPA). Therefore, these results showed that additional expression of PLC-γ2 protein was not required for TPA-induced apoptosis in MGC80-3 cells.
Figure 2.
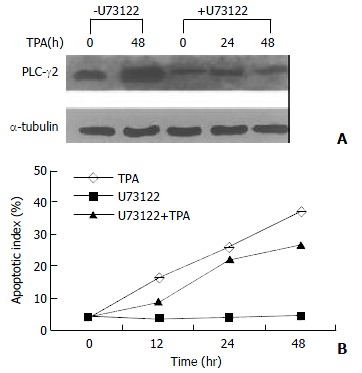
Effect of PLC-γ2 protein expression on apoptosis inducted by TPA. A: Effect of PLC-specific inhibitor U73122 on PLC-γ2 protein expression detected by Western blot. Cells were pretreated with or without U73122 (10-3 mmol/L) for 3 h, and followed by TPA for indicated time; B: Analysis of apoptotic index. Cells were pretreated with or without U73122 for 3 h, and then exposed to TPA for indicated time. Apoptotic cells were counted among 1000 cells randomly.
PKCα translocation was directly associated with induction of apoptosis by TPA
The consequent question was that since TPA could induce rapid expression of PLC-γ2 protein which seemed to be not critical in TPA-induced apoptosis in MGC80-3 cells, then, what was the role of rapidly expressed PLC-γ2 protein in TPA-induced event As PKC was a downstream molecule of PLC[3,4], it was inferred that PLC-γ2 might indirectly play its final effect through the downstream PKC signaling pathway in relation to TPA-induced apoptosis. To test this possibility, the effects of TPA on PKCαwere investigated. Firstly, the expression of PKCα protein was determined by Western blot analysis. The result showed that PKCα was intrinsically expressed in MGC80-3 cells, but its expression was not changeable by TPA (data not shown). Secondly, the possible TPA-induced translocation of PKC in MGC80-3 cells was observed. PKCα protein was shown to localize in cytoplasm (Figure 3A). However, TPA treatment stimulated the translocation of PKCα protein from cytoplasm to nucleus in a time-dependent manner. After 48 h of TPA treatment, cellular PKCα protein was completely translocated into the nucleus, showing a unique yellow color (Figure 3A). Finally, to investigate the behavior of PKCα translocation, PKC-specific inhibitor, a PKC inhibitor peptide (PIP)[39], was used. As expected, PKC inhibitor peptide could not induce PKCα protein translocated to the nucleus (Figure 3B, PIP.), even plus TPA for 48 h it no longer initiated PKCα protein translocation (Figure 3B, PIP + TPA).
Figure 3.
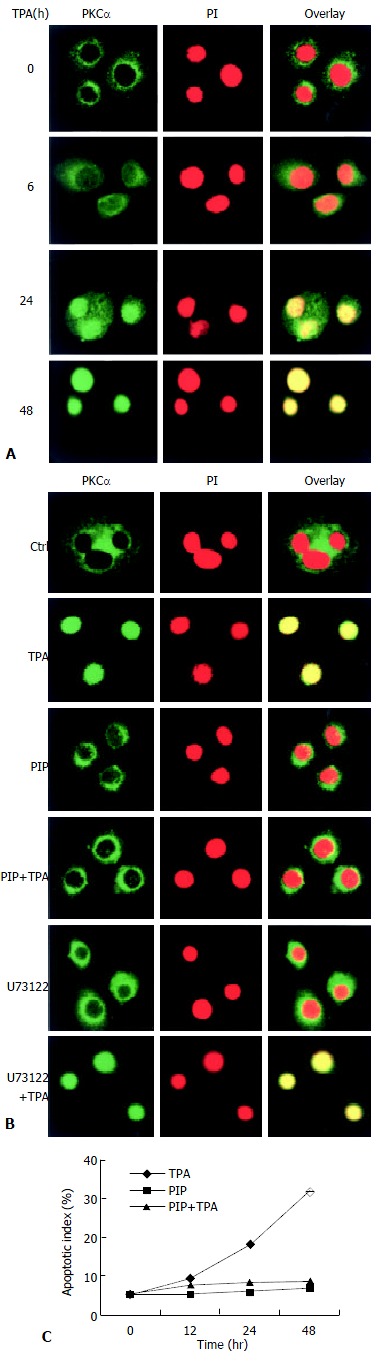
Translocation of PKCα protein in the process of apoptosis induced by TPA. A: Redistribution of PKCα protein. Cells were treated with TPA for indicated time, then immunostained with corresponding antibodies or dye as described in Materials and Methods. The images were observed under laser-scanning confocal microscope; B: Effects of various inhibitors, including PKC-specific inhibitor (PIP., 12 μmol/L) and PLC-specific inhibitor (U73122), on redistribution of PKCα. The detecting method was the same as described in Figure 3A; C: Analysis of apoptotic index. The method was similar as in Figure 2B.
Since TPA could regulate redistribution, but not expression of PKCα protein, it would be attractive to ask whether redistribution of PKCα was in action in TPA-induced apoptosis. The results showed that TPA could no longer induce apoptosis when translocation of PKCα was blocked by PKC inhibitor peptide. Even though these cells were subsequently treated by TPA for 48 h, the apoptotic index was obviously reduced reduced as compared with the TPA treatment alone (Figure 3C), suggesting that translocation of PKCα protein into the nucleus might be intrinsic in the mechanism of TPA-induced apoptosis.
PKCα was a downstream factor of PLC-γ2
It was then necessary to probe into the intrinsic mechanism of whether there was some inevitable linkage between PLC-γ2 and PKCα in the process of TPA-induced apoptosis. Since enhanced expression of PLCγ2 protein was not required for apoptosis induction (Figure 2), we therefore focused on the translocation behavior of PLC-γ2 with regard to the TPA-induced apoptosis effect. Out of our expectation, when cells were pre-treated with PLC-specific inhibitor U73122 for 3 h alone, both PLC-γ2 and PKCα proteins did not redistribute (Figure 3B and Figure 4A, U73122). However, when cells were exposing to TPA for 48 h, not only PLC-γ2 protein but also PKCα protein were translocated into the nucleus (Figure 3B and Figure 4A, U73122+TPA), although expression level of PKCα protein was not changed (Figure 4B). Together with the results in Figure 2, it was suggested that PLC-γ2 might function in passing TPA message to its downstream molecule PKCαin gastric cancer cells.
Figure 4.
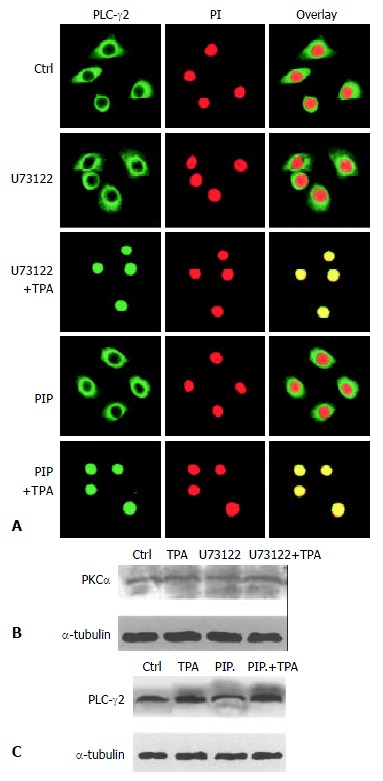
Correlation between PLC-γ2 and PKa in their redistribution. A: Effects of PKC- and PLC-specific inhibitors on PLC-γs redistribution. The Method was as described in Figure 3B; B: Effects of PKC- and PLC-specific inhibitors on PKCαexpression detected by Western blot; C: Effects of PKC- and PLC-specific inhibitors on PLC-γ2 expression.
On the other hand, PKC-specific inhibitor was also used to test its role in the expression of PLC-γ2 protein. When cells were treated with PKC inhibitor peptide (PIP) for 2 h, PLC-γ2 protein was still located in the cytoplasm (Figure 4A, PIP). However, followed by TPA treatment for 48 h, translocation of PLC-γ2 protein into the nucleus was not blocked by this inhibitor (Figure 4A: PIP + TPA). Similar result was observed in Western blot analysis, in which the expression level of PLC-γ2 protein was enhanced by TPA, no matter whether PKC inhibitor existed or not (Figure 4C). Taken together, all these findings above convincingly indicated that PKC inhibitor-induced inhibition of PKCα protein (mainly its translocation) and TPA-induced PLC-γ2 protein expression and translocation were two separate events. It also suggested that induction of PLC-γ2 protein translocation was a critical event in signal transmission between TPA and PKCα.
DISCUSSION
PLC-γ has been reported to be activated and up-regulated in response to external signals[13,15,40-44]. Most of these previous studies focused on PLC-γ2 phosphorylation and the relevant growth factor receptor (s), but its physiologic function and signaling pathway were rarely concerned. In the present study, we found that the enhanced expression of PLC-γ2 by TPA was not directly correlated to apoptosis induction. PLC-γ exerted its influence on intracellular process largely through the initiation of second messengers IP3 and DAG, and the subsequent Ca2+ mobilization and PKC activation. More importantly, these PLC-γ-induced chain reactions could be stimulated by some external stimuli[40,41]. Taking this fact into account, we therefore turned to investigate the possible regulatory mechanism of PLC-γ2 not correlated with its expression level.
PLC-specific inhibitor U73122 alone could partially suppress the expression of PLC-γ2 protein, but not induce apoptosis in MGC80-3 cells. However, TPA could still induce apoptosis in U73122-pretreated MGC80-3 cells, even though the expression of PLC-γ2 was in its supressed state, indicating that up-regulation of PLCγ2 expression was not required for apoptosis induced by TPA. However, TPA-induced apoptosis in MGC80-3 cells depended on PKCα protein translocation from cytoplasm into nucleus. When translocation of PKCα protein was blocked by its specific inhibitor PKC inhibitor peptide, the apoptosis decreased dramatically even in the presence of TPA. Therefore, these evidences strongly suggest that PLC-γ2 and PKCα can exert distinct effects in response to TPA, and regulation of PKCα protein translocation is closely associated with apoptosis induction.
We have indicated that TPA could promote PLC-γ2 translocation from cytoplasm to nucleus in a time-dependent manner, so it is interesting to figure out why PLC-γ2 is regulated (particularly its translocation) in response to TPA, and what the underlying functional implication is. Recently, some important evidences revealed that PLC-γ2 was critical for transmission of the B-cell antigen receptor complex (BCR)-dependent signals that led to the nuclear translocation of NF-κB[45], and PLC-γ1 was important for transducing survival signals against the cytotoxic effect of oxidant exposure[15]. These studies strongly imply that PLC-γ is capable of transducting signal, leading to translocation of its downstream molecule. To clarify whether TPA-induced apoptosis via PKC pathway was correlated with PLC-γ2, we investigated the effect of PLC-γ2 protein translocation on promoting PKCα translocation and initiating apoptosis. PLC-inhibitor U73122 alone could block the expression of PLC-γ2, but did not initiate the translocation of PLC-γ2 protein and induce PKCα expression, in which neither PKCα translocation nor apoptotic process could be detected. It is thus conceivable that PKCα translocation and cell apoptosis induced by TPA are PLC-γ2 translocation dependent in gastric cancer cells. Furthermore, our present study strongly supports the view that protein redistribution is an important event, which is critical for the function of protein.
Some literatures pointed out that down regulation of surface receptor expression represented an obvious mechanism by which PKC might block PLC activation[29,30]. For example, in Jurkat T cells, activation of PKC by PKC-stimulating agonists resulted in a decrease in its tyrosine phosphorylation, which was responsible for the apparently decreased PLC activity[5]. By contrast, in the present study, the relation between PLC-γ2 and PKCα seemed to be inter-dependent and synergistic. Our results showed a definite linkage between PLC-γ2 and PKCα translocation, and the synergistic effect of PLC-γ2 and PKCα in the initiation of apoptosis in MGC80-3 cells induced by TPA. In addition, the fact that pretreatment of cells with PKC inhibitor did not affect PLC-γ2 activation and translocation by TPA was consistent with the observation in which pretreatment of 293 cells for 30 min with PKC inhibitor Gö6976 did not stimulate PLC activation by EGF[46], demonstrating that PKC is a downstream molecule of PLCa pathway. Taken together, PLC-γ2 functions in signal transmission to initiate TPA-induced apoptosis via PKCαpathway.
In summary, PLC-γ2 and its downstream molecule PKCα are essential for initiating TPA-induced apoptosis in gastric cancer cells. Translocation of PLC-γ2 and PKCα proteins is critical events in the process of apoptosis. In the cross-talk between TPA, PLC-γ2 and PKCα, PLCγ2 receives TPA message, then transmits it to PKCα. PKCα functions as an effector, directly promoting apoptosis of MGC80-3 cells. This is a novel concept for PLC-γ2 and PKCα functions, which will help us to get further insights into the relationship between PLC-γ2/PKCα and the subsequent cellular events.
Footnotes
Edited by Zhang JZ and Wang XL
Supported by the National Natural Science Foundation of China (No. 30170477), the National Outstanding Youth Science Foundation of China (No.39825502), and the Natural Science Foundation of Fujian Province (C0110004)
References
- 1.Rana RS, Hokin LE. Role of phosphoinositides in transmembrane signaling. Physiol Rev. 1990;70:115–164. doi: 10.1152/physrev.1990.70.1.115. [DOI] [PubMed] [Google Scholar]
- 2.Majerus PW. Inositol phosphate biochemistry. Annu Rev Biochem. 1992;61:225–250. doi: 10.1146/annurev.bi.61.070192.001301. [DOI] [PubMed] [Google Scholar]
- 3.Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272:15045–15048. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- 4.Noh DY, Shin SH, Rhee SG. Phosphoinositide-specific phospholipase C and mitogenic signaling. Biochim Biophys Acta. 1995;1242:99–113. doi: 10.1016/0304-419x(95)00006-0. [DOI] [PubMed] [Google Scholar]
- 5.Rhee SG, Choi KD. Regulation of inositol phospholipid-specific phospholipase C isozymes. J Biol Chem. 1992;267:12393–12396. [PubMed] [Google Scholar]
- 6.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 7.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 8.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 9.Ji QS, Winnier GE, Niswender KD, Horstman D, Wisdom R, Magnuson MA, Carpenter G. Essential role of the tyrosine kinase substrate phospholipase C-gamma1 in mammalian growth and development. Proc Natl Acad Sci USA. 1997;94:2999–3003. doi: 10.1073/pnas.94.7.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cockcroft S, Thomas GM. Inositol-lipid-specific phospholipase C isoenzymes and their differential regulation by receptors. Biochem J. 1992;288(Pt 1):1–14. doi: 10.1042/bj2880001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SB, Rhee SG. Significance of PIP2 hydrolysis and regulation of phospholipase C isozymes. Curr Opin Cell Biol. 1995;7:183–189. doi: 10.1016/0955-0674(95)80026-3. [DOI] [PubMed] [Google Scholar]
- 12.Nishibe S, Wahl MI, Wedegaertner PB, Kim JW, Rhee SG, Carpenter G. Selectivity of phospholipase C phosphorylation by the epidermal growth factor receptor, the insulin receptor, and their cytoplasmic domains. Proc Natl Acad Sci USA. 1990;87:424–428. doi: 10.1073/pnas.87.1.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kayali AG, Eichhorn J, Haruta T, Morris AJ, Nelson JG, Vollenweider P, Olefsky JM, Webster NJ. Association of the insulin receptor with phospholipase C-gamma (PLCgamma) in 3T3-L1 adipocytes suggests a role for PLCgamma in metabolic signaling by insulin. J Biol Chem. 1998;273:13808–13818. doi: 10.1074/jbc.273.22.13808. [DOI] [PubMed] [Google Scholar]
- 14.Lee YH, Kim SY, Kim JR, Yoh KT, Baek SH, Kim MJ, Ryu SH, Suh PG, Kim JH. Overexpression of phospholipase Cbeta-1 protects NIH3T3 cells from oxidative stress-induced cell death. Life Sci. 2000;67:827–837. doi: 10.1016/s0024-3205(00)00677-9. [DOI] [PubMed] [Google Scholar]
- 15.Wang XT, McCullough KD, Wang XJ, Carpenter G, Holbrook NJ. Oxidative stress-induced phospholipase C-gamma 1 activation enhances cell survival. J Biol Chem. 2001;276:28364–28371. doi: 10.1074/jbc.M102693200. [DOI] [PubMed] [Google Scholar]
- 16.Lee YH, Kim S, Kim J, Young Kim K, Kim MJ, Ryu SH, Suh P. Overexpression of phospholipase C-gamma1 suppresses UVC-induced apoptosis through inhibition of c-fos accumulation and c-Jun N-terminal kinase activation in PC12 cells. Biochim Biophys Acta. 1999;1440:235–243. doi: 10.1016/s1388-1981(99)00128-6. [DOI] [PubMed] [Google Scholar]
- 17.Han Y, Han ZY, Zhou XM, Shi R, Zheng Y, Shi YQ, Miao JY, Pan BR, Fan DM. Expression and function of classical protein kinase C isoenzymes in gastric cancer cell line and its drug-resistant sublines. World J Gastroenterol. 2002;8:441–445. doi: 10.3748/wjg.v8.i3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Wu Q, Song SY, Su WJ. Activation of JNK by TPA promotes apoptosis via PKC pathway in gastric cancer cells. World J Gastroenterol. 2002;8:1014–1018. doi: 10.3748/wjg.v8.i6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodnight JA, Mischak H, Kolch W, Mushinski JF. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J Biol Chem. 1995;270:9991–10001. doi: 10.1074/jbc.270.17.9991. [DOI] [PubMed] [Google Scholar]
- 20.Shao RG, Cao CX, Pommier Y. Activation of PKCalpha downstream from caspases during apoptosis induced by 7-hydroxystaurosporine or the topoisomerase inhibitors, camptothecin and etoposide, in human myeloid leukemia HL60 cells. J Biol Chem. 1997;272:31321–31325. doi: 10.1074/jbc.272.50.31321. [DOI] [PubMed] [Google Scholar]
- 21.Nishizuka Y. Turnover of inositol phospholipids and signal transduction. Science. 1984;225:1365–1370. doi: 10.1126/science.6147898. [DOI] [PubMed] [Google Scholar]
- 22.Majerus PW, Connolly TM, Deckmyn H, Ross TS, Bross TE, Ishii H, Bansal VS, Wilson DB. The metabolism of phosphoinositide-derived messenger molecules. Science. 1986;234:1519–1526. doi: 10.1126/science.3024320. [DOI] [PubMed] [Google Scholar]
- 23.Isakov N, Mally MI, Scholz W, Altman A. T-lymphocyte activation: the role of protein kinase C and the bifurcating inositol phospholipid signal transduction pathway. Immunol Rev. 1987;95:89–111. doi: 10.1111/j.1600-065x.1987.tb00501.x. [DOI] [PubMed] [Google Scholar]
- 24.Kaibuchi K, Takai Y, Nishizuka Y. Protein kinase C and calcium ion in mitogenic response of macrophage-depleted human peripheral lymphocytes. J Biol Chem. 1985;260:1366–1369. [PubMed] [Google Scholar]
- 25.Albert F, Hua C, Truneh A, Pierres M, Schmitt-Verhulst AM. Distinction between antigen receptor and IL 2 receptor triggering events in the activation of alloreactive T cell clones with calcium ionophore and phorbol ester. J immunol. 1985;134:3649–3655. [PubMed] [Google Scholar]
- 26.Isakov N, Altman A. Human T lymphocyte activation by tumor promoters: role of protein kinase C. J immunol. 1987;138:3100–3107. [PubMed] [Google Scholar]
- 27.Abraham RT, Ho SN, Barna TJ, Rusovick KM, McKean DJ. Inhibition of T-cell antigen receptor-mediated transmembrane signaling by protein kinase C activation. Mol Cell Biol. 1988;8:5448–5458. doi: 10.1128/mcb.8.12.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills GB, May C, Hill M, Ebanks R, Roifman C, Mellors A, Gelfand EW. Physiologic activation of protein kinase C limits IL-2 secretion. J immunol. 1989;142:1995–2003. [PubMed] [Google Scholar]
- 29.Decker SJ, Ellis C, Pawson T, Velu T. Effects of substitution of threonine 654 of the epidermal growth factor receptor on epidermal growth factor-mediated activation of phospholipase C. J Biol Chem. 1990;265:7009–7015. [PubMed] [Google Scholar]
- 30.Park DJ, Min HK, Rhee SG. Inhibition of CD3-linked phospholipase C by phorbol ester and by cAMP is associated with decreased phosphotyrosine and increased phosphoserine contents of PLC-gamma 1. J Biol Chem. 1992;267:1496–1501. [PubMed] [Google Scholar]
- 31.Todderud G, Wahl MI, Rhee SG, Carpenter G. Stimulation of phospholipase C-gamma 1 membrane association by epidermal growth factor. Science. 1990;249:296–298. doi: 10.1126/science.2374928. [DOI] [PubMed] [Google Scholar]
- 32.Wu Q, Liu S, Ding L, Ye X, Su W. PKCalpha translocation from mitochondria to nucleus is closely related to induction of apoptosis in gastric cancer cells. Sci China C Life Sci. 2002;45:237–244. doi: 10.1360/02yc9026. [DOI] [PubMed] [Google Scholar]
- 33.Wang KH. An in vitro cell line (MGc80-3) of a pooly differentiated mucoid adenocarcinoma of human stomach. Shiyan Shengwu Xiebao. 1983;16:257–267. [Google Scholar]
- 34.Wu Q, Chen Z, Su W. Growth inhibition of gastric cancer cells by all-trans retinoic acid through arresting cell cycle progression. Chin Med J ( Engl) 2001;114:958–961. [PubMed] [Google Scholar]
- 35.Wu Q, Liu S, Ye XF, Huang ZW, Su WJ. Dual roles of Nur77 in selective regulation of apoptosis and cell cycle by TPA and ATRA in gastric cancer cells. Carcinogenesis. 2002;23:1583–1592. doi: 10.1093/carcin/23.10.1583. [DOI] [PubMed] [Google Scholar]
- 36.Champelovier P, Richard MJ, Seigneurin D. Autocrine regulation of TPA-induced apoptosis in monoblastic cell-line U-937: role for TNF-alpha, MnSOD and IL-6. Anticancer Res. 2000;20:451–458. [PubMed] [Google Scholar]
- 37.Li Y, Bhuiyan M, Mohammad RM, Sarkar FH. Induction of apoptosis in breast cancer cells by TPA. Oncogene. 1998;17:2915–2920. doi: 10.1038/sj.onc.1202218. [DOI] [PubMed] [Google Scholar]
- 38.Bleasdale JE, Bundy GL, Bunting S, Fitzpatrick FA, Huff RM, Sun FF, Pike JE. Inhibition of phospholipase C dependent processes by U-73, 122. Adv Prostaglandin Thromboxane Leukot Res. 1989;19:590–593. [PubMed] [Google Scholar]
- 39.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 40.McLaughlin AP, De Vries GW. Role of PLCgamma and Ca (2+) in VEGF- and FGF-induced choroidal endothelial cell proliferation. Am J Physiol Cell Physiol. 2001;281:C1448–C1456. doi: 10.1152/ajpcell.2001.281.5.C1448. [DOI] [PubMed] [Google Scholar]
- 41.Doong H, Price J, Kim YS, Gasbarre C, Probst J, Liotta LA, Blanchette J, Rizzo K, Kohn E. CAIR-1/BAG-3 forms an EGF-regulated ternary complex with phospholipase C-gamma and Hsp70/Hsc70. Oncogene. 2000;19:4385–4395. doi: 10.1038/sj.onc.1203797. [DOI] [PubMed] [Google Scholar]
- 42.Jílek F, Hüttelová R, Petr J, Holubová M, Rozinek J. Activation of pig oocytes using calcium ionophore: effect of protein synthesis inhibitor cycloheximide. Anim Reprod Sci. 2000;63:101–111. doi: 10.1016/s0378-4320(00)00150-0. [DOI] [PubMed] [Google Scholar]
- 43.Kurosaki T, Maeda A, Ishiai M, Hashimoto A, Inabe K, Takata M. Regulation of the phospholipase C-gamma2 pathway in B cells. Immunol Rev. 2000;176:19–29. doi: 10.1034/j.1600-065x.2000.00605.x. [DOI] [PubMed] [Google Scholar]
- 44.Carpenter G, Ji Qs. Phospholipase C-gamma as a signal-transducing element. Exp Cell Res. 1999;253:15–24. doi: 10.1006/excr.1999.4671. [DOI] [PubMed] [Google Scholar]
- 45.Petro JB, Khan WN. Phospholipase C-gamma 2 couples Bruton's tyrosine kinase to the NF-kappaB signaling pathway in B lymphocytes. J Biol Chem. 2001;276:1715–1719. doi: 10.1074/jbc.M009137200. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt M, Frings M, Mono ML, Guo Y, Weernink PA, Evellin S, Han L, Jakobs KH. G protein-coupled receptor-induced sensitization of phospholipase C stimulation by receptor tyrosine kinases. J Biol Chem. 2000;275:32603–32610. doi: 10.1074/jbc.M004784200. [DOI] [PubMed] [Google Scholar]