

Abstract
AIM: To explore the mode of inheritance of hepatocellular carcinoma (HCC) in a moderately high-incidence area of East China.
METHODS: A pedigree survey was conducted in 210 families (3315 individuals) ascertained through 210 HCC probands in Haimen, Jiangsu Province. Simple segregation analysis was conducted using SEGRANB software. The probability of ascertainment (π), segregation ratio (p), and the proportion of sporadic cases (x) were estimated. Complex segregation analysis was performed using the REGTL program of S.A.G.E. Models were fitted on the data of 3212 individuals that allowed for personal HBsAg status and variable age of onset in REGTL program.
RESULTS: The estimate of segregation ratio was 0.191 by SEGRANB. The probability of ascertainment was 0.0266, and the proportion of sporadic cases was 0.465. The results of complex segregation analysis showed that Mendelian autosomal recessive inheritance of a major gene that influenced the age of onset distribution of HCC, provided the best fit to the data. In the best-fitting recessive model, the frequency of the disease allele was 0.11138. HBsAg seropositive status would significantly increase the risk of developing HCC.
CONCLUSION: These results suggest that at least one major gene is involved in the genetic predisposition to develop HCC at an earlier age of onset. The seropositive HBsAg status can significantly increase the risk of developing HCC, which provides strong support for the interaction between genetic and environmental risk factors.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common types of malignant tumor in the world, the estimated number of new cases worldwide annually is over 500000[1], ranking third in cause of cancer deaths in China. An estimated 26000 deaths are from HCC annually worldwide, among which about 40% are in China[2]. Classical epidemiological studies have shown that certain risk factors are associated with HCC, though the odds ratios (ORs) vary considerably from study to study[3-5]. On a worldwide basis, chronic infection with hepatitis B virus (HBV) appears to be the most important risk factor for HCC. About 80% of the patients of HCC in China are seropositive for hepatitis B surface antigen (HBsAg)[6]. An IARC working group in 1993 found that the evidence for a causal association of HBV with HCC was pretty strong. HCC is common in cirrhotic patients when cirrhosis is secondary to chronic viral hepatitis[1]. In addition to viral factors, environmental exposures and genetic susceptibility are clearly involved. Dietary aflatoxin exposure is an important codeterminant of HCC risk in Africa and parts of Asia[7]. Aflatoxins together with chronic hepatitis B virus (HBV) infection contribute to the high incidence of hepatocellular carcinoma in developing countries[8-10]. Contaminated drinking water and chemical carcinogens are also associated with the development of HCC[3].
Many familial clusterings of HCC have been reported[11,12]. The prevalence of HCC among the first-degree relatives is significantly higher than that among the second-degree and third-degree relatives, which suggests that genetic mechanisms may be responsible for familial HCC. Haimen city is a HCC high-incidence area of East China, however, only a few studies have been performed to explore the genetic mode of HCC in this area. Consequently, in this study, data of 210 pedigrees ascertained through HCC probands were collected and segregation analysis of HCC was performed.
MATERIALS AND METHODS
Materials and data collection
Probands were 210 HCC patients from an eight-year follow-up of a 90000-person Haimen City cohort. All the patients were pathologically diagnosed in hospitals of counties and cities. Information on four-generation pedigrees was obtained for 210 families. Data on 3315 individuals in the 210 families were collected primarily by face-to-face interviews or by checking the medical records and according to the recall of their relatives if the proband patients were dead. Data on 3212 individuals (97%) were used to fit models in the REGTL program of S.A.G.E. The remaining individuals were excluded because of little information available.
Family history interview and data management
A family history of HCC was collected as part of an interview to gather information on the medical history of probands and their relatives. The questionnaire included questions on age, occupation, tobacco use, drinking water source by decades (60 s, 70 s, 80 s, 90 s), staple food consumption by decades, result of HBsAg test, history of chronic hepatitis B, history of other chronic diseases, family history of HCC and relationship of family member with HCC to proband cases. All the questionnaires were reviewed and checked for quality control. Pedigree information database was developed using Epi-Info v5.0.
Statistical methods
Simple segregation analysis by SEGRANB program In SEGRANB, models were fitted to the data and maximum likelihood scores of parameters were estimated. Hypothesis tests were performed to determine which set of parameter was most consistent with the observed data. The involved parameters were probability of ascertainment (π) (the probability for a patient to be identified as a proband), segregation ratio (p), and the proportion of sporadic cases (x). S is the sibship size, r is the total number of the affected siblings and a is the total number of the probands. The function for the probability of ascertainment is:
Math 1
Math 1.
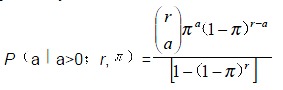
Math(A1).
The function for the estimate of the segregation ratio in the pedigrees with more than one patient is:
Math 2
Math 2.
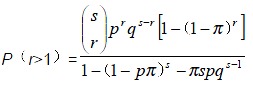
Math(A1).
The functions for estimate of p and x are:
Math 3
Math 3.
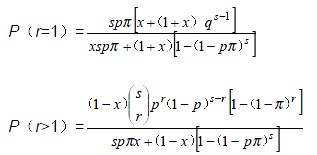
Math(A1).
Uπ, Up, and Ux are the maximum likelihood scores for π, p and x, respectively. If the value of U was negative, the initial estimated value of the parameter should be reduced to fit the observed data, and vice versa.
Complex segregation analysis using REGTL program of S.A.G.E. (Statistical analysis for genetic epidemiology, v3.1. Case Western Reserve University, Cleveland, OH) The REGTL program of S.A.G.E. (1997) (release 3.1) under a Window's 9x operating system was used to perform complex segregation analysis. This program uses maximum-likelihood methods to estimate parameters of mathematical models of disease occurrence in families. It assumes that under a class A regressive model, a censored trait, such as age of onset to a disease or of the disease susceptibilities, follows a logistic distribution. Sibs are dependent on one another only because of common parentage. Mendelian inheritance, if present, is presumed to be through a single autosomal locus with two alleles, A and B, A being associated with the affected state. The "type" was used to describe the discrete factors that affect a person's phenotype. The same concept was denoted as "ousiotype". Genotypes are the special case of types, or ousiotypes, that transmit to offspring in Mendelian fashion. Thus we should use the term "type" to allow for any kind of discrete transmission, whether Mendelian or not. Two general models can be assumed. In model 1, the segregation of a possible major locus is allowed for by letting the baseline and age coefficient parameters of the age of onset distribution, but not the susceptibility, depends on an unobserved qualitative factor u = AA, AB, BB. Susceptibility depends solely on randomly distributed environmental characteristics of the population studied. In model 2, type is presumed to influence susceptibility to the affected state, but not the parameters of the age-at-onset distribution, and groups of individuals of different types have the same mean age at onset. Analysis was performed under the model 1 in this study. HCC was represented by a dichotomous variable y, in which y = 1 for affected and 0 for unaffected. The following parameters were estimated: type frequencies ψu (u = AA, AB, BB. If the type frequencies are in Hardy-Weinberg equilibrium proportions, then they are defined in terms of qA = frequency of allele A), transmission probabilities τu (the probability that a parent of type u transmits allele A to an offspring, under Mendelian transmission, τAA = 1, τAB = 0.5, τBB = 0), baseline parameter β which can be sex-dependent and/or type-dependent, covariate coefficient ζHbsAg which is coded 1 if the individual is HBsAg positive and is coded 0 if negative or 9 if unknown. Thus, the coefficient ζHBsAg is the change in the logit (the risk of HCC) according to the HBsAg status, age adjustment coefficient α, and susceptibility parameter γ which can be sex-dependent. Susceptibility is the probability that an individual is susceptible.
To correct for ascertainment bias, the likelihood of each pedigree is conditioned on the proband's HCC status by age at exam or death and his or her actual age of onset. This assumes single ascertainment, which is a reasonable approximation since only 1 (0.5%) of 210 families has more than one patient eligible to be a proband. Under single ascertainment, the probability that any one family will be ascertained is small and proportional to the number of affected children in the family.
As the process of parameter estimate is complex and time consuming when complex models are fitted, a program written in Perl program was used to facilitate the estimate of the initial value of parameters. This program was used to generate random values for the initial estimate of parameters and to call modules in the REGTL program of the S.A.G.E. package to perform segregation analysis. For each model, thirty converged results were saved, among which the best-fitted result of each model was chosen.
Under the model 1, six hypotheses were tested against the likelihood of a general (unrestricted) model, in which all parameters were unrestricted and allowed to fit the empirical data. Thus this general model would give the best fit to the data. The six hypotheses of transmission are as follows: major gene type, Mendelian dominant, Mendelian recessive, Mendelian additive, purely environmental effect and no transmission. Twice the differences between the natural log likelihood (InL) for the data under the hypothesis of interest and that under the unrestricted model is in accordance with χ² distribution, thus it is used to assess the degree of departure from expectation statistically. The degree of freedom (df) for the χ² statistic is given by the differences in the number of estimated parameters between the hypothesis and the unrestricted model. If one or more parameters are fixed at a bound at the end of the estimation process, a range of the df and P values are given when appropriate. A non-significant χ² indicates that the hypothetical model cannot be rejected. When covariates and major gene effects are considered simultaneously, the number of potential hypothesis tests is large and the natural hierarchy of the models may not be clear. In this case, models can also be compared with use of Akaike's (1974) information criterion (AIC), which is defined as AIC = -2lnL + 2 × (Number of parameters estimated). A lower value of AIC represents a better fitting model.
RESULTS
Distribution of HCC and HBsAg status among relatives
Of 3315 individuals in the 210 extended pedigrees, 330 (10.0%) including probands were affected with HCC. The distribution of HCC and HBsAg status among the relatives is summarized in Table 1. Among the first-degree relatives, the prevalence of HCC was 6.6% (100/1511). Among the second-degree relatives, it was 2.1% (20/945). The prevalence of HCC was 23.1% (30/130) among the HBsAg positive first-degree relatives, it was 19.2 (5/26) among the HBsAg positive second-degree relative. As shown in Table 2, among the fathers of the probands, the prevalence of HCC was 5.5% (11/200). Among siblings, it was 11.1% (74/664) (16.1% for brothers, 5.0% for sisters). Among offsprings, it was 0.2% (1/448), and among spouses of the probands, it was 1.0% (2/202).
Table 1.
HBsAg status of HCC cases among first-degree and second-degree relatives
| HBsAg status |
First-degree relatives |
Second-degree relatives |
||
| No. of relatives | No. (%) of HCC | No. of relatives | No. (%) of HCC | |
| + | 130 | 30 (23.1%) | 26 | 5 (19.2%) |
| - | 1104 | 47 (4.3%) | 711 | 4 (0.6%) |
| Unknown | 277 | 23 (8.3%) | 208 | 11 (5.3%) |
| Total | 1511 | 100 (6.6%) | 945 | 20 (2.1%) |
Table 2.
Distribution of HCC among the relatives of 210 probands
| Characteristic |
Relatives |
||||||
| Father | Mother | Brother | Sister | Son | Daughter | Spouse | |
| No. (%) of HCC | 11 (5.5%) | 14 (7.0%) | 59 (16.1%) | 15 (5.0%) | 1 (0.5%) | 0 (0%) | 2 (1.0%) |
| Total | 200 | 199 | 366 | 298 | 221 | 227 | 202 |
Parameter estimate in simple segregation analysis by SEGRANB software
The distribution of probands and their affected siblings among the 210 families is shown in Table 3. There was one family with two patients eligible to be a proband. The best fitted probability of ascertainment π was 0.026556, Uπ was 6.500244 × 10-3, χ² was 2.848217 × 10-8. The estimated sets of parameters are shown in Table 4. The best fitted probability of ascertainment π was 0.0266, the segregation ratio p was 0.191, and the proportion of sporadic cases x was 0.465.
Table 3.
Distribution of probands and affected siblings among families
| Number of affected siblings | 1 | 2 | 3 | 4 | 4 | 5 |
| Number of probands in family | 1 | 1 | 1 | 1 | 2 | 1 |
| Number of families | 162 | 32 | 7 | 7 | 1 | 1 |
Table 4.
Maximum likelihood estimates of parameters of segregation models for HCC by SEGRANB
| π | p | x | Uπ | Up | Ux | χ²π | χ²p | χ²x | ¯π | ¯p | ¯x | P value |
| 0.026556 | 0.50 | 0.00 | 116.922 | -929.338 | 2482.747 | 1069.75 | 1074.303 | 1712.618 | 9.176 | -0.655 98 | 0.689 81 | < 0.05 |
| 0.026556 | 0.25 | 0.00 | 50.272 | -530.853 | 230.405 | 134.657 | 134.794 | 144.777 | 2.705 | -0.003 92 | 0.628 36 | < 0.05 |
| 0.026556 | 0.20 | 0.60 | -7.363 | 50.253 | -34.966 | 4.140 | 4.069 | 4.090 | -0.536 | 0.280 97 | 0.483 03 | < 0.05 |
| 0.026556 | 0.280 97 | 0.483 | 8.856 | -41.824 | 36.699 | 3.566 | 3.806 | 3.817 | 0.429 | 0.189 96 | 0.587 05 | < 0.05 |
| 0.026556 | 0.189 96 | 0.483 | -0.055 | 0.200 | 0.464 | 0.097 | 0.098 | 0.09 | -0.055 | 0.200 11 | 0.463 91 | > 0.05 |
| 0.026556 | 0.189 96 | 0.464 | -0.054 | 0.559 | 0.007 | 1.976E-04 | 3.118E-04 | 2.146E-07 | 0.029 | 0.190 52 | 0.463 94 | > 0.05 |
| 0.026556 | 0.190 52 | 0.464 | 0.013 | -0.003 | 0.276 | 1.097E-05 | 8.709E-09 | 3.252E-04 | 0.027 | 0.190 52 | 0.465 08 | > 0.05 |
| 0.026 6 | 0.190 52 | 0.465 | -0.054 | 0.529 | 0.016 | 1.909E-04 | 2.822E-04 | 1.064E-06 | 0.030 | 0.191 06 | 0.465 07 | > 0.05 |
| 0.026 6 | 0.191 | 0.465 | 0.005 | 0.059 | 0.244 | 1.517E-06 | 3.486E-06 | 2.505E-04 | 0.027 | 0.191 06 | 0.466 03 | > 0.05 |
π, p and x: initially estimated values for π, p and x, respectively. Uπ, Up and Ux: maximum likelihood scores for π, p and x, respectively. χ²π, χ²p and χ²x: χ² of the estimated π, p and x, respectively. π, p, and x: corrected value for ¯π, ¯p and ¯x, respectively.
Results of complex segregation analysis by REGTL program of S.A.G.E.
Of the 210 families, 48 (22.9%) had two or more HCC patients. The mean age at onset was 50 (range 21-83). Complex segregation analyses were performed by REGTL under model 1 that did not include regressive familial effects in the models and in which age at onset and susceptibility were not sex-dependent. One environmental covariate, HBsAg status, as a dichotomous variable was included in the models.
In Table 5, the best fitted parameter estimates for general and hypothetical models were reported. As a result, the Mendelian recessive and additive (codominant) hypotheses were not rejected (P > 0.1) and the major gene model was marginally not rejected (0.01 < P < 0.03). All other models were rejected at a 0.001 significance level. According to AIC, Mendelian recessive inheritance was the best-fitted hypothesis, but the AICs for recessive (811.68716) and major gene (812.20661) models were very close. The recessive model suggested that approximately 11.1% of the population could be expected to carry the candidate gene (or gene pattern). The coefficient of the covariate HBsAg was positive. If its standard deviation is to do the Wald's test, we got |βHBsAg|/|SHBsAg| = 1.36380/0.40661 = 3.354 > 1.96, P < 0.05, which suggested that HBsAg seropositive status would increase the risk of developing HCC.
Table 5.
Complex segregation analysis on HCC among the 210 extended pedigrees (SAGE-REGTL, model 1)
| Parametera |
Hypothesis |
||||||
|
Mendelian |
Majorgene | Environmental | Notransmission | General | |||
| Dominant | Recessive | Additive | |||||
| qA | 0.00304 | 0.11138 | 0.11451 | 0.87964 | 0.02229 | 0b | 0.00861 |
| τAA | 1.0b | 1.0b | 1.0b | 1.0b | 1.0c | 0b | 1.0c |
| τAB | 0.5b | 0.5b | 0.5b | 0.5b | 1.0c | 0b | 0.0c |
| τBB | 0b | 0b | 0b | 0b | 1.0c | 0b | 1.0c |
| βAA | -5.74896 | -6.86219 | -7.30081 | -13.664 | -7.27445 | -6.74403 | -9.22295 |
| βAB | -5.74896d | -14.22974 | -14.45292 | -22.64140 | -4.14230 | -6.74403d | -6.19460 |
| βBB | -11.62577 | -14.22974d | -21.60502 | -6.83546 | -9.94049 | -6.74403d | -20.47273 |
| α | 0.10039 | 0.12335 | 0.12867 | 0.12385 | 0.07499 | 0.05710 | 0.08464 |
| γ | 0.72125 | 1.000c | 0.86915 | 1.000c | 0.89922 | 1.000c | 1.000c |
| ζHBsAg | 0.93155 | 1.36380 | 1.84008 | 1.20200 | 2.72339 | 2.70874 | 1.47535 |
| -2lnL | 831.07918 | 799.68716 | 805.17010 | 798.20661 | 822.20467 | 842.79594 | 803.24635 |
| AIC | 843.07918 | 811.68716 | 817.17010 | 812.20661 | 838.20467 | 850.79594 | 825.24635 |
| χ² | 27.83283 | 3.55919 | 1.92375 | 5.03974 | 18.95832 | 39.54959 | |
| dfe | 1-5 | 2-5 | 1-5 | 1-4 | 2-3 | 4-7 | |
| Pvalue | P < 0.001 | P > 0.1 | P > 0.1 | 0.01 < P < 0.03 | P < 0.001 | P < 0.001 | |
a: See Materials and Methods for definitions of the parameters. b: Parameter was fixed at this value and estimation was not carried out. c: Parameter estimate reached its bound. d: Parameter was constrained to equal the preceding one and was not estimated. e: Range in degrees of freedom was given since parameters in models reached their bounds.
DISCUSSION
In this study, we explored the inheritance mode of HCC in the 210 extended pedigrees in Haimen City by simple and complex segregation analyses. This study used SEGRANB software and REGTL program of S.A.G.E. analyzing a population-based family data set to investigate the presence of a major gene effect for HCC. In simple segregation analysis procedure, as the estimate of segregation ratio p could be substantially affected by the estimated value of the probability of ascertainment π[13,14], especially on complex traits, therefore π was calculated using maximum likelihood method by SEGRANB program in this study. The estimated value of π was 0.0266, which suggested a single ascertainment for this study. Then the segregation ratio and the proportion of sporadic cases were also estimated by maximum likelihood method using SEGRANB. The best-fitted parameter set is shown in Table 4. The segregation ratio was 0.191 and the proportion of sporadic cases was 0.465 for this data, which suggested that HCC did not follow a mono-gene inheritance pattern in this population and environmental factors appeared to influence the development of HCC since 46.5% of the cases were sporadic.
Due to the modest sample size and relatively high proportion of missed information for the elder generations of the probands in the studied pedigrees, gender and familial residual effects were not estimated simultaneously with other parameters of the corresponding models in complex segregation analysis. The results showed that although both the recessive and additive (codominant) hypotheses could not be rejected, Mendelian autosomal recessive inheritance of a major gene that influenced baseline and age of onset of HCC provided the best fit to the data. The estimated gene frequency under the recessive hypothesis was 0.11138. The result in this study was consistent with the result of an earlier complex segregation analysis by Shen et al[3] (1991) using the POINTER program in which an autosomal recessive major gene yielding lifetime risk of PHC (primary hepatocellular carcinoma) was suggested. In this study, the information of serum HBsAg status for relatives was obtained through the questionnaire and sometimes the report of laboratory test was unavailable. Although this situation could cause miss of information for many individuals in analysis, which might ruin the statistical power, the significance of seropositive HBsAg effect could still be observed. It strongly suggested that chronic HBV infection was a risk factor in developing HCC in this population.
The ultimate test of the utility of segregation analysis is whether the resulting models can be used in linkage analysis to identify disease-susceptibility loci. Although the recessive hypothesis provided the best fit to the data, segregation analysis had its limitation that it could not distinguish the effect of a single locus that underlied a trait from the effects of two or more independently acting loci with similar transmission patterns[15]. Thus, more than one gene, with different penetrances and modes of inheritance, may be involved. Even under oligogenic inheritance, the parameters derived from a single locus segregation analysis can provide power for detecting the susceptibility loci. Many studies have been performed to search for susceptibility genes for HCC[16-29], and several novel genes have been suggested to play a role in HCC development[30-32]. The HCC mortality differs in different areas in China and there are some environmental factors (e.g. local economic status, geographical factors, habit of food consumption and viral infection) that might differ in different areas. With the same environmental exposures, HCC occurrence differs a great deal from family to family, which is typified by the existence of highly aggregated "cancer families". It is hard to say which factor (environmental or genetic) is more important. All can play important roles in the etiology of HCC. The mutation of HCC gene (s) may act as a trigger for HCC onset, or people with the gene (s) may tend to be more susceptible to HCC under the influence of certain environmental risk factors.
In conclusion, our study showed that a Mendelian autosomal recessive major gene might play an important role in the etiology of HCC in a moderately high-incidence area of East China. This study provides some modeling parameters of HCC (in this area) for further linkage studies. The identification of the putative gene detected by the study is warranted.
ACKNOWLEDGEMENTS
This work was supported in part by the National Natural Science Foundation of China (No: 39930160). Some of the results in this paper were obtained by using the program package S.A.G.E., which was supported by a U.S. Public Health Service Resource Grant (1 P41 RR03655) from the National Center for Research Resources. Prof. Fu-Min Shen donated the version of the program package S.A.G.E. used in this study. We thank Dr. Yin Hu of the NCI Center for Bio-informatics for his help on compiling Perl script and on tackling some technical problems.
Footnotes
Edited by Wu XN and Wang XL
Supported by the National Natural Science Foundation of China, No. 39930160
References
- 1.Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13–20. doi: 10.1111/j.1749-6632.2002.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 2.Zhao ZT, Jia CX. Epidemiology of liver cancer. In: Liu Q, Wang WQ, editors. Cancer. Beijing: People's Health Press; 2000. pp. 221–239. [Google Scholar]
- 3.Evans AA, Chen G, Ross EA, Shen FM, Lin WY, London WT. Eight-year follow-up of the 90, 000-person Haimen City cohort: I. Hepatocellular carcinoma mortality, risk factors, and gender differences. Cancer Epidemiol Biomarkers Prev. 2002;11:369–376. [PubMed] [Google Scholar]
- 4.Yoshizawa H. Hepatocellular carcinoma associated with hepatitis C virus infection in Japan: projection to other countries in the foreseeable future. Oncology. 2002;62 Suppl 1:8–17. doi: 10.1159/000048270. [DOI] [PubMed] [Google Scholar]
- 5.Donato MF, Arosio E, Del Ninno E, Ronchi G, Lampertico P, Morabito A, Balestrieri MR, Colombo M. High rates of hepatocellular carcinoma in cirrhotic patients with high liver cell proliferative activity. Hepatology. 2001;34:523–528. doi: 10.1053/jhep.2001.26820. [DOI] [PubMed] [Google Scholar]
- 6.Wang YL, Zhou HG, Gu GW. Advances in liver cancer research. Shanghai: Shanghai Science and Technology Literature Press; 1999. pp. 13–27. [Google Scholar]
- 7.Ming L, Thorgeirsson SS, Gail MH, Lu P, Harris CC, Wang N, Shao Y, Wu Z, Liu G, Wang X, et al. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology. 2002;36:1214–1220. doi: 10.1053/jhep.2002.36366. [DOI] [PubMed] [Google Scholar]
- 8.Wild CP, Yin F, Turner PC, Chemin I, Chapot B, Mendy M, Whittle H, Kirk GD, Hall AJ. Environmental and genetic determinants of aflatoxin-albumin adducts in the Gambia. Int J Cancer. 2000;86:1–7. doi: 10.1002/(sici)1097-0215(20000401)86:1<1::aid-ijc1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.Chen SY, Chen CJ, Tsai WY, Ahsan H, Liu TY, Lin JT, Santella RM. Associations of plasma aflatoxin B1-albumin adduct level with plasma selenium level and genetic polymorphisms of glutathione S-transferase M1 and T1. Nutr Cancer. 2000;38:179–185. doi: 10.1207/S15327914NC382_6. [DOI] [PubMed] [Google Scholar]
- 10.Wang JS, Huang T, Su J, Liang F, Wei Z, Liang Y, Luo H, Kuang SY, Qian GS, Sun G, et al. Hepatocellular carcinoma and aflatoxin exposure in Zhuqing Village, Fusui County, People's Republic of China. Cancer Epidemiol Biomarkers Prev. 2001;10:143–146. [PubMed] [Google Scholar]
- 11.Tai DI, Changchien CS, Hung CS, Chen CJ. Replication of hepatitis B virus in first-degree relatives of patients with hepatocellular carcinoma. Am J Trop Med Hyg. 1999;61:716–719. doi: 10.4269/ajtmh.1999.61.716. [DOI] [PubMed] [Google Scholar]
- 12.Yu MW, Chang HC, Liaw YF, Lin SM, Lee SD, Liu CJ, Chen PJ, Hsiao TJ, Lee PH, Chen CJ. Familial risk of hepatocellular carcinoma among chronic hepatitis B carriers and their relatives. J Natl Cancer Inst. 2000;92:1159–1164. doi: 10.1093/jnci/92.14.1159. [DOI] [PubMed] [Google Scholar]
- 13.Tai JJ, Hsiao CK. Effects of implicit parameters in segregation analysis. Hum Hered. 2001;51:192–198. doi: 10.1159/000053342. [DOI] [PubMed] [Google Scholar]
- 14.Haghighi F, Hodge SE. Likelihood formulation of parent-of-origin effects on segregation analysis, including ascertainment. Am J Hum Genet. 2002;70:142–156. doi: 10.1086/324709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarvik GP. Complex segregation analyses: uses and limitations. Am J Hum Genet. 1998;63:942–946. doi: 10.1086/302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tufan NL, Lian Z, Liu J, Pan J, Arbuthnot P, Kew M, Clayton MM, Zhu M, Feitelson MA. Hepatitis Bx antigen stimulates expression of a novel cellular gene, URG4, that promotes hepatocellular growth and survival. Neoplasia. 2002;4:355–368. doi: 10.1038/sj.neo.7900241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu MW, Yang YC, Yang SY, Chang HC, Liaw YF, Lin SM, Liu CJ, Lee SD, Lin CL, Chen PJ, et al. Androgen receptor exon 1 CAG repeat length and risk of hepatocellular carcinoma in women. Hepatology. 2002;36:156–163. doi: 10.1053/jhep.2002.33897. [DOI] [PubMed] [Google Scholar]
- 18.Kinoshita M, Miyata M. Underexpression of mRNA in human hepatocellular carcinoma focusing on eight loci. Hepatology. 2002;36:433–438. doi: 10.1053/jhep.2002.34851. [DOI] [PubMed] [Google Scholar]
- 19.Fu XY, Wang HY, Tan L, Liu SQ, Cao HF, Wu MC. Overexpression of p28/gankyrin in human hepatocellular carcinoma and its clinical significance. World J Gastroenterol. 2002;8:638–643. doi: 10.3748/wjg.v8.i4.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito Y, Miyoshi E, Takeda T, Nagano H, Sakon M, Noda K, Tsujimoto M, Monden M, Matsuura N. Linkage of elevated ets-2 expression to hepatocarcinogenesis. Anticancer Res. 2002;22:2385–2389. [PubMed] [Google Scholar]
- 21.Nakau M, Miyoshi H, Seldin MF, Imamura M, Oshima M, Taketo MM. Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res. 2002;62:4549–4553. [PubMed] [Google Scholar]
- 22.Wei Y, Van Nhieu JT, Prigent S, Srivatanakul P, Tiollais P, Buendia MA. Altered expression of E-cadherin in hepatocellular carcinoma: correlations with genetic alterations, beta-catenin expression, and clinical features. Hepatology. 2002;36:692–701. doi: 10.1053/jhep.2002.35342. [DOI] [PubMed] [Google Scholar]
- 23.Chiao PJ, Na R, Niu J, Sclabas GM, Dong Q, Curley SA. Role of Rel/NF-kappaB transcription factors in apoptosis of human hepatocellular carcinoma cells. Cancer. 2002;95:1696–1705. doi: 10.1002/cncr.10829. [DOI] [PubMed] [Google Scholar]
- 24.Jiang Y, Zhou XD, Liu YK, Wu X, Huang XW. Association of hTcf-4 gene expression and mutation with clinicopathological characteristics of hepatocellular carcinoma. World J Gastroenterol. 2002;8:804–807. doi: 10.3748/wjg.v8.i5.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikeguchi M, Hirooka Y, Kaibara N. Quantitative analysis of apoptosis-related gene expression in hepatocellular carcinoma. Cancer. 2002;95:1938–1945. doi: 10.1002/cncr.10898. [DOI] [PubMed] [Google Scholar]
- 26.Guo LL, Guo Y, Cao CA. [Relationship between hepatitis C virus infection and expression of apoptosis-related gene bcl-2, bax and ICH-1 in hepatocellular carcinoma tissues] Diyi Junyi Daxue Xuebao. 2002;22:797–799, 805. [PubMed] [Google Scholar]
- 27.Lévy L, Renard CA, Wei Y, Buendia MA. Genetic alterations and oncogenic pathways in hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:21–36. doi: 10.1111/j.1749-6632.2002.tb04091.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Wu MC, Sham JS, Tai LS, Fang Y, Wu WQ, Xie D, Guan XY. Different expression of hepatitis B surface antigen between hepatocellular carcinoma and its surrounding liver tissue, studied using a tissue microarray. J Pathol. 2002;197:610–616. doi: 10.1002/path.1150. [DOI] [PubMed] [Google Scholar]
- 29.Joo M, Kang YK, Kim MR, Lee HK, Jang JJ. Cyclin D1 overexpression in hepatocellular carcinoma. Liver. 2001;21:89–95. doi: 10.1034/j.1600-0676.2001.021002089.x. [DOI] [PubMed] [Google Scholar]
- 30.Rahman MA, Kohno H, Nagasue N. COX-2 - a target for preventing hepatic carcinoma. Expert Opin Ther Targets. 2002;6:483–490. doi: 10.1517/14728222.6.4.483. [DOI] [PubMed] [Google Scholar]
- 31.Zeng JZ, Wang HY, Chen ZJ, Ullrich A, Wu MC. Molecular cloning and characterization of a novel gene which is highly expressed in hepatocellular carcinoma. Oncogene. 2002;21:4932–4943. doi: 10.1038/sj.onc.1205652. [DOI] [PubMed] [Google Scholar]
- 32.Harada H, Nagai H, Ezura Y, Yokota T, Ohsawa I, Yamaguchi K, Ohue C, Tsuneizumi M, Mikami I, Terada Y, et al. Down-regulation of a novel gene, DRLM, in human liver malignancy from 4q22 that encodes a NAP-like protein. Gene. 2002;296:171–177. doi: 10.1016/s0378-1119(02)00855-7. [DOI] [PubMed] [Google Scholar]