

Abstract
AIM: Our previous studies showed increased sensitivity to 5-FU in colon cancer cell lines with microsatellite instability, and considered that mutations of TGFβ-R II, IGF IIR, RIZ gene might enhance the potentials of cell growth and proliferation, which increased the sensitivity to 5-FU. Here we compared the distribution of cell cycle and P53 status between two human colon cancer cell lines with different sensitivity to 5-FU. Because mechanistic differences exist between 5-FU and CDDP, we also analyzed the efficacy of CDDP and combination therapy on two human colon cancer cell lines.
METHODS: We compared the sensitivity to CDDP of these two cell lines by MTT assay. Distribution of cell cycle under treatment of 5-FU, CDDP alone or both was analyzed by Flow Cytometry, and expression of P53 was detected by immunocytochemical staining.
RESULTS: SW480 cells were more sensitive to CDDP than LoVo cells at the concentrations above 16 μmol/L (Ratio of absorption is 0.64 and 0.79 at 16 μmol/L, respectively; P < 0.01). Efficacy of combination therapy was conversely lower than that of single-therapy of 5-FU (Ratio of absorption in LoVo + 5-FU, SW480 + 5-FU, LoVo + 5-FU + CDDP and SW480 + 5-FU + CDDP is 0.53, 0.54, 0.72, 0.78, respectively; P < 0.01). LoVo cells were negative whereas SW480 cells positive in P53 expression. 5-FU induced G1-phase arrest in both cell lines, but LoVo cells peaked 24 h earlier than SW480 cells, and 48 h earlier for an apparent hypodiploid DNA. However, CDDP showed the contrary, inducing S-phase arrest, and SW480 cells peaking 36 h earlier. Both cell lines showed hypodipliod nuclei 48 h after CDDP treatment. Percentage of cells in G1-phase and S-phase dominated alternatively under combination therapy in both cell lines.
CONCLUSION: These results suggest that colon cancer cells with microsatellite instability are more sensitive to 5-FU, whereas more resistant to CDDP. Combination therapy of 5-FU and CDDP shows fewer efficacies than 5-FU single-therapy, although it can render a cell cycle arrest. P53 may be involved in the shift of G1-phase to S-phase, but inessentially.
INTRODUCTION
5-FU is currently the first-line agent for colorectal cancer after surgical cytoreduction with an overall response rate of less than 15%[1,2], this has stimulated intensive effort in the development of novel compounds with improved pharmacological properties and new regimens for colorectal cancer patients. Efforts have been made in combination of 5-FU with several second-line agents, such as paclitaxel, mitomycin, calcium folinate, INF-α, irinotecan, leucovorin, suramin and tegafur, and so on, unfortunately, improvement is far from satisfaction. Our studies previously demonstrated that colorectal cancer cell lines with microsatellite instability showed increased sensitivity to 5-FU, and that mutations were found in 8 loci from different genes, among which 3 loci harbored in the exon of TGF-RII, IGFIIR, and RIZ, respectively. All these three genes are closely associated with cell growth and proliferation. On basis of these results, we proposed that these mutations may enhance the proliferative potentials of cancer cells and increase chemosensitivity to 5-FU.
In this study, We explored the differences of cell cycle arrest and apoptosis between two cell lines under 5-FU treatment, and found that in G1-phase arrest and presence of hypodiploid DNA in LoVo cells happened 48 h earlier than that in SW480 cells. Because the options available to colorectal cancer patients for second-line therapy were limited, and mechanistic differences existed between 5-FU and CDDP, we analyzed the efficacies of CDDP and combination therapy on these two cell lines. Results indicated that SW480 cells were more sensitive to CDDP than LoVo cells, and that combination therapy of 5-FU and CDDP showed less efficacy than single-therapy of 5-FU in both cell lines, although it can render a cell cycle arrest. P53 may be involved in the entry of G1-phase to S-phase, but inessentially.
MATERIALS AND METHODS
Cell lines
LoVo, a human colon adenocarcinoma cell line, was purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences, Shanghai, China; SW480, a human colon adenocarcinoma cell lines, was donated by the Cancer Institute, Zhe Jiang University. They were maintained in RPMI1640 supplemented with 10% fetal bovine serum in a humidified 5% CO2 atmosphere at 37 °C.
Drugs and agents
5-fluorouracil (5-FU), [cis-diamminedichloroplatinum (II)] (cisplatin/CDDP), 3-[4-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), dimethylsulfoxide (DMSO) and propidium iodide (PI), were all purchased from the Sigma Chemical Co. The primary mouse antibody of P53 (DO-7), biotinylated anti-mouse immunoglobulin, horseradish peroxidase-conjugated streptavidin, and the chromogenic substrate solution 3,3-diaminobenzidine were bought from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
MTT assay
MTT assay was performed as described by Lu et al[3]. Briefly, logarithmically growing cells were seeded in the 96-well plate at a concentration of 1 × 104 per well and incubated for 12 h. Then medium with various concentrations of drugs was added in quadruplicate and exposed for 72 h. The culture medium was then removed and about 300 μL fresh medium containing 0.5 mg/mL MTT was added to each well. Four hours later, the medium was replaced with 100 μL DMSO and vortexed for 10 min. Absorbance (A) was then recorded at 570 nm using an Enzyme-linked Immunosorbent Assay device DG3022A. Cell viability was assessed as follows: Viability [%] = Atreat/Acontrol × 100%.
Immunocytochemical staining
Immunocytochemical staining was performed using DO-7 anti-P53 on logarithmically growing cell lines, LoVo and SW480, on coverslip. Firstly, cells were plated onto coverslips, adhered overnight. Then, rinsed three times with PBS, cells were fixed in cold acetone for 8-10 min. Endogenous peroxidase was blocked with 1% hydrogen peroxide in absolute methanol for 30 min. The primary antibodies were applied for 2 h at 37 °C at 1:300 dilution in a humidified chamber. Then the typical SP strategy followed.
Flow Cytometric analysis
Cells were incubated in medium containing 4 mmol/L 5-FU, 10 mmol/L CDDP alone or both (4 μmol/L 5-FU + 10 μmol/L CDDP) continuously, and then were fixed in ice-cold 70% ethanol at 0, 12, 24, 48, 72 and 96 h after initial treatment. Then approximately 10000 cells each specimen stained by 10 μg/mL PI were analyzed by Flow Cytometry (FACS®), as described by Bunz et al[4].
Statistical analysis
Data of MTT assay were mean values of at least three different experiments and expressed as mean ± SD, analyzed by two-tailed Student's t-test and General Linear Model, P value of less than 0.05 was considered as statistically significant.
RESULTS
Response to CDDP in LoVo cells and SW480 cells
We compared the sensitivity and responsiveness of LoVo cells and SW480 cells to CDDP and the combination of CDDP + 5-FU in cytotoxicity assays. As shown by the dose-effect curve, both cell lines are sensitive to CDDP, but more for SW480 cells (Figure 1A). Neither of the cell lines examined showed significantly synergetic response when the two drugs were combined simultaneously, when compared with single-therapy of 5-FU, indicating that CDDP, to some degree, may block the effects of 5-FU (Figure 1B).
Figure 1.
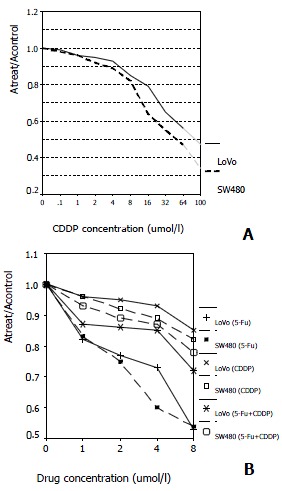
Comparison of sensitivity to 5-FU, CDDP or combination therapy in LoVo cells and SW480 cells. (A) SW480 cells show increased sensitivity to CDDP than LoVo cells at the concentrations above 16 μmol/L (Ratio of absorption is 0.64 and 0.79 at 16 μmol/L, respectively; P < 0.01). (B) The dose-effect curve tells that the cytotoxicity diminishes in combination therapy of 5-FU and CDDP in both cell lines when compared with 5-FU single-therapy (Ratio of absorption in LoVo + 5-FU, SW480 + 5-FU, LoVo + 5-FU + CDDP and SW480 + 5-FU + CDDP is 0.53, 0.54, 0.72, 0.78, respectively; P < 0.01).
Distribution of cell cycle by FCM analysis
LoVo cells demonstrated an apparent peak of cells with hypodiploid DNA 48 h after 5-FU exposure, 48 h earlier than SW480 cells, which indicated that LoVo cells are more sensitive to 5-FU (Figure 2, Figure 3A). When treated with CDDP for 48 h, SW480 cells showed more cells with hypodiploid DNA than LoVo cells, about 2-fold increase at 96 h (55.1% and 28.5%, respectively) (Figure 2, Figure 3B). However, when the two lines were exposed to 5-FU in combination with CDDP, this cytotoxicity significantly diminished. SW480 cells showed an 18% cells with hypodiploid DNA, but not in LoVo cells in 96 h (Figure 2, Figure 3C). Both cell lines exposed to 5-FU showed an accumulation in G1-phase and a significantly decreased proportion of S-phase. On the contrary, CDDP arrests both lines mainly in S-phase instead of G1-phase. Percentage of cells in G1-phase and S-phase dominates alternatively in both lines treated with combination of 5-FU and CDDP (Figure 4).
Figure 2.
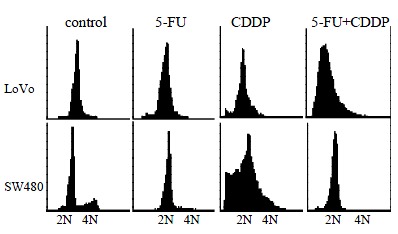
DNA content distribution of LoVo and SW480 cells after 48 h exposure to 5-FU and/or CDDP.5-FU causes a more apparent increase of the proportion of hypo diploid DNA cells in LoVo than in SW480. CDDP renders an apparent increase of the hypodiploid DNA cells in both cell lines, but SW480 has more than LoVo cells. Few hypodiploid DNA cells are observed in combination of 5-FU and CDDP treatment.
Figure 3.
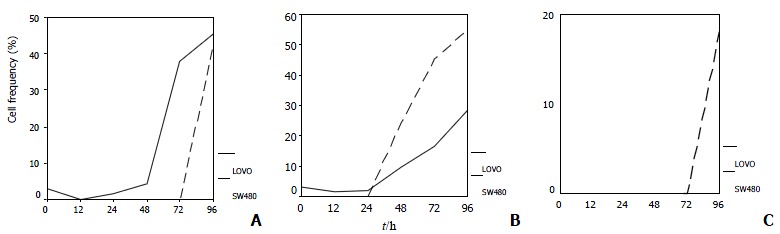
Hypodiploid DNA induced by 5-FU, CDDP or a combination therapy in LoVo cells and SW480 cells. LoVo cells treated with 5-FU show an earlier presence and higher percentage of cells with hypodiploid DNA. B When exposed to CDDP, SW480 cells showed a more dramatic increase in presence of hypodiploid DNA than LoVo cells after 48 h. C When the two cell lines were exposed to 5-FU in combination with CDDP, SW480 cells have an 18% hypodiploid DNA, but not in LoVo cells in 96 h after treatment.
Figure 4.
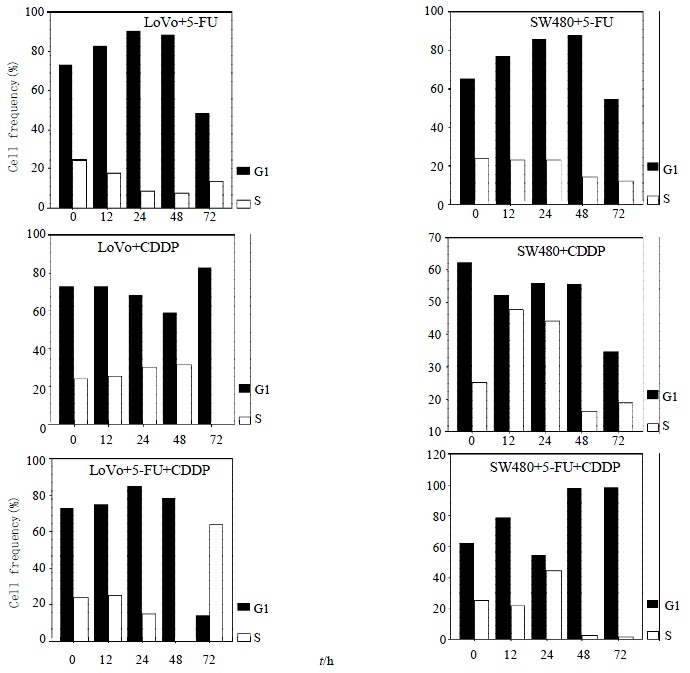
Effects on cell cycle distribution of LoVo cells & SW480 cells treated with5-FU, CDDP or combination therapy. Both cell lines show an accumulation of G1-phase exposed to5-FU. On the contrary, CDDP mainly renders an accumulation of S-phase. Percentage of G1-and S-phase dominates alternatively in both lines treated with combination of 5-FU and CDDP.
P53 expression by immunocytochemical staining
P53 staining scattered nestedly in LoVo cells, predominantly in nuclei, and the proportion of positive cells only accounts for less than 1%, which was thus considered as wtP53. However, SW480 cells showed extensively and strongly P53 expression, and more than 98% cells were labeled in nuclei, so it suggested a mutated P53 in this cell line (Figure 5).
Figure 5.
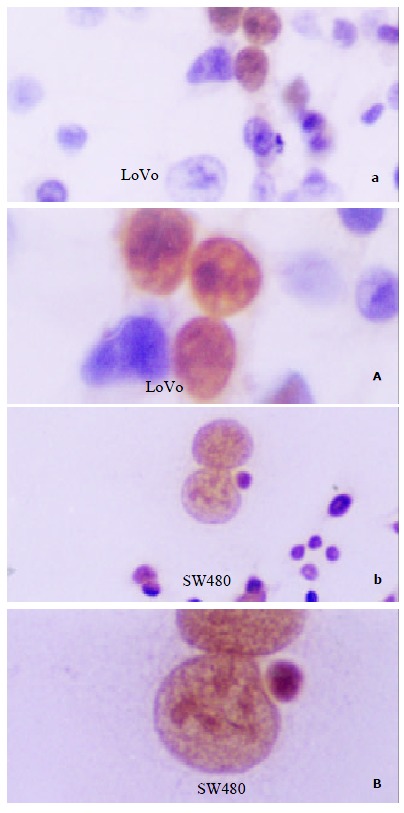
P53 staining with the anti-P53 mAb Do-7 in LoVo and SW480 cells. (a, b × 100; A, B × 200)
DISCUSSION
Our previous studies showed increased sensitivity to 5-FU in colon cancer cell lines with microsatellite instability, and considered that mutations of TGF-R II, IGF IIR and RIZ gene maybe enhance the potentials of cell growth and proliferation, which increases the sensitivity to 5-FU. This assumption is supported greatly by findings that the generation time of LoVo cells is significantly shorter than that of SW480 cells[4], and by our results here, we found that both cell lines demonstrated an accumulation of G1-phase after 12 h 5-FU exposure, but apoptosis occurred earlier in LoVo cells.
Many researches have been done to reveal biochemical factors associated with 5-FU response, meanwhile, efforts are made to improve the efficacy of chemotherapy by combining 5-FU with other second-line drugs, such as paclitaxel, oxaliplatin, mitomycin, calcium folinate, INF-α, irinotecan, leucovorin, suramin, tegafur, and so on[5-10]. Considering mechanistic differences exist between 5-FU and CDDP, we supposed the possibility of their synergism and analyzed the efficacy of combination therapy of these two drugs.
However, our data didn't agree with this supposition. We know that after administration 5-FU is rapidly taken up by cells and metabolized by enzymes by several pathways to produce two active metabolites, i.e.5-FUTP, which may be incorporated directly into RNA, and 5-FdUMP. 5-FdUMP in the presence of reduced folates inhibits thymidylate synthase (TS) activity and depletes dTTP, a necessary precursor of DNA synthesis. Alternatively, it may be phosphorylated to the triphosphate and 5-FdUTP incorporated directly into DNA, inhibiting chain elongation and altering DNA stability, resulting in the production of single-strand breaks and DNA fragmentation[11-13]. Thus, 5-FU belongs to the phase-specific anticancer drug that means improved cytotoxicity to cells in S-phase. However, CDDP acts differently, it binds to DNA base pairs, creating adducts, crosslinks, and strand breaks that inhibit DNA replication.
As pointed above, the two cell lines both arrest in G1-phase, and LoVo cells precede SW480 cells in presence of hypodiploid nuclei with treatment of 5-FU. But CDDP rendered an apparent peak of cells with hypodiploid DNA after 48 hour, and LoVo cells showed less percentage of hypodiploid DNA cells, which suggested that, from view of population, SW480 cells are more sensitive to CDDP than LoVo cells. This result is consistent with most reports, i.e. colorectal cancer cells with microsatellite instability are more resistant to CDDP, and several assumptions have been made to explain this phenomenon: firstly, an assumption so-called "Recognition, Excision, Futility of repairing"[14,15]. The DNA-CDDP adducts are recognized and then excised by the mismatch repair system (MMR), the underlying molecular mechanism responsible for correction of mismatch base pairs or some sorts of DNA damages, but incapable to be repaired because of certain reasons. This failure may then lead to permanent single- or double-strand breaks which are now considered to be the initiation of cell death or apoptosis. Secondly, "Protection mechanism"[16]. DNA-CDDP adducts are recognized by MMR system or other nuclear factors, then the following binding functions a shelter which protects the damage from repairing by other mechanisms independent of MMR system, which renders cells to death or apoptosis. Thirdly, "Cell cycle pathway". It is supposed that response of cancer cells to CDDP depends on the ability of G2/M arresting. Some workers considered that P53 is responsible for the shift of G1/S phases, whereas MMR system can inactivate CDK1-CylinB complex by phosphorylation of two amino acid residues, Thr14 and Thr15, of CDK1, and blocks cells in G2/M phase for repair, unrepairable DNA damage often results in activation of the apoptotic pathway (Hawn et al, 1995). This assumption is supported by many data[17-19]. Here, the first explanation disclaims itself because of the homogenous loss of hMSH2, one of most important members responsible for DNA-CDDP adducts recognition in MMR system. We also failed to detect the G2/M phase arrest which is emphasized in the third assumption. As for the second supposition, further evidences are required to confirm it.
Combination therapy of 5-FU and CDDP showed less efficacy than single-therapy of 5-FU. Here we found that there was less cells with hypodiploid DNA in both cell lines treated with a combination of 5-FU and CDDP, which suggested that the 5-FU-induced cytotoxicity may, at least partially, diminish by the concomitant presence of CDDP. There are at least two mechanisms may explain the observed dominance of CDDP over 5-FU. One may simply involve a CDDP-induced cell cycle blockade, we called it "Cell cycle disturbance", analogous to that recently described by Judson et al[20] in paclitaxel. By arresting cells in S-phase of cell cycle, CDDP inhibited both cell lines undergoing apoptosis after exposure to 5-FU. Both cell lines firstly showed a G1-phase increase, a "5-FU-like response" and then followed by S-phase increase, a "CDDP-like response" In SW480 cells, particularly in SW480 cells, the percentage of G1-phase and S-phase dominates alternatively. These results demonstrate clearly that disturbance of cell cycle arrest and apoptosis occurred in the combination therapy. An alternative mechanism by which CDDP may exert dominance over 5-FU centers on the ability of each drug to modulate level of many biochemical molecules, we called it "molecular antagonism". CDDP intercalates into DNA, forming adducts, and has been shown to both activate and block a variety of biochemical molecules, including transcription factors, such as c-myc, AP-1/AP-2, Oct-1, E2F1, P53 and P73; or molecules involved in cell signal transduction, such as Ras, PKA, EGF4, PKC-α/-ε/-theta;; or factors associated with proliferation, DNA replication and cell cycle regulation, such as PCNA, TS, DNA pol-α/β, Topo I, Cyclin E/D, P16, P21, P27; or Bax and Bcl-2, and so on. We have recently shown that there is a direct correlation between cytotoxicity and 5-FU induced transcriptional activation, i.e. some of these factors are also downstream elements induced by 5-FU and, in turn, affects sensitivity to 5-FU[21-24], leading us to postulate that at least part of the mechanism involves the antagonism of factors induced by each drug. In fact, these two possibilities are compatible with each other, the latter might just be the underlying biochemical explanation of the former.
Many evidences have shown that sensitivity of cancer cells to 5-FU is associated with a variety of mechanisms, including the key enzyme required for its activation and catabolism, folate substrate and the TS activity, and so on. The concept that P53 is involved in chemotherapy-induced cell cycle arrest and apoptosis is accepted by most scientists[5,12,25-32]. Yoshikawa et al[1] found there was no relationship between the sensitivity to 5-FU and P53 in colorectal cancer chemotherapy according to evidences from clinical trials, combining with these findings, they proposed that that 5-FU might act via two different pathways, depending on dose: (a) G1/S-phase cell cycle arrest and apoptosis at 1000 ng/mL, and (b) G2/M-phase cell cycle arrest and mitotic catastrophe at 100 ng/mL in SW480. Our results accord with the higher concentration group they reported, i.e. cells undergo G1-phase or S-phase arrest and apoptosis. Controversies exist in the role of P53 in CDDP chemosensitivity, evidences from malignancies of lung, esophagus, cervix and bladder showed that wtP53 is a favorable prognostic predictor in chemotherapy, and that mutation of P53 will lead cells to chemoresistance[33-36]. However, in agreement with the findings of Pestell et al[37] in ovarian cancer, colon cancer cell line with mutant P53 exhibited more sensitivity to CDDP. We think this discrepancy may result from the different type of tissue. Up-regulation of P53 in response to 5-FU/CDDP-induced DNA damage may activates P21 and wee1/mik1, which inhibits the CDK activity, and consequentially, E2F1 failed to release itself from E2F1:RB complex due to down-regulation of RB phosphorylation, as a result, cells arrest in G1/S-phase. Alternatively, it is recently reported that P53-induced increase of P21 activity may also be mediated by the PI3K-AKT1/AKT2 signal transduction pathway[38]. Lin et al[39] found that activation of ATM induced by DNA damage can directly phosphorylate specific residues at the NH2-terminal of E2F1 and can increase P53 expression. Nagashima et al[40] described that P53 can also be acetylated and activated by DNA damage-induced P33ING2 in CDDP and paclitaxel exposure. All these evidences proved that P53 plays an important role in chemotherapy-induced cell cycle arrest and apoptosis. We've known that P21 and P53 are both mutant in SW480 cells, So blockage of cell cycle in G1/S-phase in this cell line may be P53-independent, compared with LoVo cells, apoptosis of SW480 cells treated with 5-FU or CDDP delayed, which may imply that apoptosis induced by P53 pathway is more effective than others. Huang once reported that a few or even one double-strand break of DNA would be enough to increase expression of P53, and led cells to cycle arrest for repairing, if failed, undergoing apoptosis[21,41,42]. So P53, cell cycle status, damage repair system and apoptotic pathway together determine cells to survive or not.
In conclusion, we have demonstrated that colon cancer cell lines with microsatellite instability are more sensitive to 5-FU, but CDDP goes conversely. Combination therapy of 5-FU and CDDP can lead cells to cycle arrest, but it shows less cytotoxicity than single-therapy of 5-FU. P53 may be involved in cell cycle shift of G1-phase to S-phase, but inessentially.
ACKNOWLEDGMENTS
We sincerely thank Mrs. Mi-Wei Li, from the Infective Disease Institute of No.1 Affiliated Hospital of Medical School of Zhe Jiang University, for assistance with Flow Cytometry.
Footnotes
Edited by Zhao M
References
- 1.Yoshikawa R, Kusunoki M, Yanagi H, Noda M, Furuyama JI, Yamamura T, Hashimoto-Tamaoki T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: a novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res. 2001;61:1029–1037. [PubMed] [Google Scholar]
- 2.Bleiberg H. Colorectal cancer: the challenge. Eur J Cancer. 1996;32A Suppl 5:S2–S6. doi: 10.1016/s0959-8049(96)00319-x. [DOI] [PubMed] [Google Scholar]
- 3.Lu JG, Lin C, Huang ZQ, Wu JS, Fu M, Zhang XY, Liang X, Yao X, Wu M. Inhibitory effects of human cholangiocarcinoma cell line by recombinant adenoviruses p16 with CDDP. Shijie Huaren Xiaohua Zazhi. 2000;8:641–645. [Google Scholar]
- 4.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 5.Trainer DL, Kline T, McCabe FL, Faucette LF, Feild J, Chaikin M, Anzano M, Rieman D, Hoffstein S, Li DJ. Biological characterization and oncogene expression in human colorectal carcinoma cell lines. Int J Cancer. 1988;41:287–296. doi: 10.1002/ijc.2910410221. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy AS, Harrison GH, Mansfield CM, Zhou XJ, Xu JF, Balcer-Kubiczek EK. Survival of colorectal cancer cell lines treated with paclitaxel, radiation, and 5-FU: effect of TP53 or hMLH1 deficiency. Int J Cancer. 2000;90:175–185. [PubMed] [Google Scholar]
- 7.Chester JD, Dent JT, Wilson G, Ride E, Seymour MT. Protracted infusional 5-fluorouracil (5-FU) with bolus mitomycin in 5-FU-resistant colorectal cancer. Ann Oncol. 2000;11:235–237. doi: 10.1023/a:1008356017611. [DOI] [PubMed] [Google Scholar]
- 8.Smith R, Wickerham DL, Wieand HS, Colangelo L, Mamounas EP. UFT plus calcium folinate vs 5-FU plus calcium folinate in colon cancer. Oncology (Williston Park) 1999;13:44–47. [PubMed] [Google Scholar]
- 9.Sabaawy HE, Farley T, Ahmed T, Feldman E, Abraham NG. Synergetic effects of retrovirus IFN-alpha gene transfer and 5-FU on apoptosis of colon cancer cells. Acta Haematol. 1999;101:82–88. doi: 10.1159/000040929. [DOI] [PubMed] [Google Scholar]
- 10.Van Cutsem E, Cunningham D, Ten Bokkel Huinink WW, Punt CJ, Alexopoulos CG, Dirix L, Symann M, Blijham GH, Cholet P, Fillet G, et al. Clinical activity and benefit of irinotecan (CPT-11) in patients with colorectal cancer truly resistant to 5-fluorouracil (5-FU) Eur J Cancer. 1999;35:54–59. doi: 10.1016/s0959-8049(98)00353-0. [DOI] [PubMed] [Google Scholar]
- 11.Falcone A, Pfanner E, Brunetti I, Allegrini G, Lencioni M, Galli C, Masi G, Danesi R, Antonuzzo A, Del Tacca M, et al. Suramin in combination with 5-fluorouracil (5-FU) and leucovorin (LV) in metastatic colorectal cancer patients resistant to 5-FU+LV-based chemotherapy. Tumori. 1998;84:666–668. doi: 10.1177/030089169808400610. [DOI] [PubMed] [Google Scholar]
- 12.Otake Y, Tanaka F, Yanagihara K, Hitomi S, Okabe H, Fukushima M, Wada H. Expression of thymidylate synthase in human non-small cell lung cancer. Jpn J Cancer Res. 1999;90:1248–1253. doi: 10.1111/j.1349-7006.1999.tb00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinsella AR, Smith D, Pickard M. Resistance to chemotherapeutic antimetabolites: a function of salvage pathway involvement and cellular response to DNA damage. Br J Cancer. 1997;75:935–945. doi: 10.1038/bjc.1997.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cory JG, Breland JC, Carter GL. Effect of 5-fluorouracil on RNA metabolism in Novikoff hepatoma cells. Cancer Res. 1979;39:4905–4913. [PubMed] [Google Scholar]
- 15.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehmé A, Christen RD, Howell SB. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 16.Swann PF, Waters TR, Moulton DC, Xu YZ, Zheng Q, Edwards M, Mace R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273:1109–1111. doi: 10.1126/science.273.5278.1109. [DOI] [PubMed] [Google Scholar]
- 17.Crul M, Schellens JH, Beijnen JH, Maliepaard M. Cisplatin resistance and DNA repair. Cancer Treat Rev. 1997;23:341–366. doi: 10.1016/s0305-7372(97)90032-3. [DOI] [PubMed] [Google Scholar]
- 18.Brown R, Hirst GL, Gallagher WM, McIlwrath AJ, Margison GP, van der Zee AG, Anthoney DA. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene. 1997;15:45–52. doi: 10.1038/sj.onc.1201167. [DOI] [PubMed] [Google Scholar]
- 19.Thiebaut F, Enns R, Howell SB. Cisplatin sensitivity correlates with its ability to cause cell cycle arrest via a wee1 kinase-dependent pathway in Schizosaccharomyces pombe. J Cell Physiol. 1994;159:506–514. doi: 10.1002/jcp.1041590315. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda M, Orimo H, Moriyama H, Nakajima E, Matsubara N, Mibu R, Tanaka N, Shimada T, Kimura A, Shimizu K. Close correlation between mutations of E2F4 and hMSH3 genes in colorectal cancers with microsatellite instability. Cancer Res. 1998;58:594–598. [PubMed] [Google Scholar]
- 21.Judson PL, Watson JM, Gehrig PA, Fowler WC, Haskill JS. Cisplatin inhibits paclitaxel-induced apoptosis in cisplatin-resistant ovarian cancer cell lines: possible explanation for failure of combination therapy. Cancer Res. 1999;59:2425–2432. [PubMed] [Google Scholar]
- 22.Dempke W, Voigt W, Grothey A, Hill BT, Schmoll HJ. Cisplatin resistance and oncogenes--a review. Anticancer Drugs. 2000;11:225–236. doi: 10.1097/00001813-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 2000;57:1229–1235. doi: 10.1007/PL00000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iqbal S, Lenz HJ. Determinants of prognosis and response to therapy in colorectal cancer. Curr Oncol Rep. 2001;3:102–108. doi: 10.1007/s11912-001-0008-5. [DOI] [PubMed] [Google Scholar]
- 25.D'Amico TA, Harpole DH. Molecular biology of esophageal cancer. Chest Surg Clin N Am. 2000;10:451–469. [PubMed] [Google Scholar]
- 26.Buyse M, Piedbois Y, Piedbois P, Gray R. Tumour site, sex, and survival in colorectal cancer. Lancet. 2000;356:858. doi: 10.1016/S0140-6736(05)73441-3. [DOI] [PubMed] [Google Scholar]
- 27.Rosty C, Chazal M, Etienne MC, Letoublon C, Bourgeon A, Delpero JR, Pezet D, Beaune P, Laurent-Puig P, Milano G. Determination of microsatellite instability, p53 and K-RAS mutations in hepatic metastases from patients with colorectal cancer: relationship with response to 5-fluorouracil and survival. Int J Cancer. 2001;95:162–167. doi: 10.1002/1097-0215(20010520)95:3<162::aid-ijc1028>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 28.Bras-Gonçalves RA, Pocard M, Formento JL, Poirson-Bichat F, De Pinieux G, Pandrea I, Arvelo F, Ronco G, Villa P, Coquelle A, et al. Synergistic efficacy of 3n-butyrate and 5-fluorouracil in human colorectal cancer xenografts via modulation of DNA synthesis. Gastroenterology. 2001;120:874–888. doi: 10.1053/gast.2001.22440. [DOI] [PubMed] [Google Scholar]
- 29.Elsaleh H, Powell B, Soontrapornchai P, Joseph D, Goria F, Spry N, Iacopetta B. p53 gene mutation, microsatellite instability and adjuvant chemotherapy: impact on survival of 388 patients with Dukes' C colon carcinoma. Oncology. 2000;58:52–59. doi: 10.1159/000012079. [DOI] [PubMed] [Google Scholar]
- 30.Arango D, Corner GA, Wadler S, Catalano PJ, Augenlicht LH. c-myc/p53 interaction determines sensitivity of human colon carcinoma cells to 5-fluorouracil in vitro and in vivo. Cancer Res. 2001;61:4910–4915. [PubMed] [Google Scholar]
- 31.Luo F, Kan B, Lei S, Yan LN, Mao YQ, Zou LQ, Yang YX, Wei YQ. Study on P53 protein and C-erbB2 protein expression in primary hepatic cancer colorectal cancer by flow cytometry. World J Gastroenterol. 1998;4(Suppl 2):87. [Google Scholar]
- 32.Xu QW, Li YS, Zhu HG. Relationship between expression p53 protein, PCNA and CEA in colorectal cancer and lymph node metastasis. World J Gastroenterol. 1998;4:218. [Google Scholar]
- 33.Choi JH, Ahn KS, Kim J, Hong YS. Enhanced induction of Bax gene expression in H460 and H1299 cells with the combined treatment of cisplatin and adenovirus mediated wt-p53 gene transfer. Exp Mol Med. 2000;32:23–28. doi: 10.1038/emm.2000.5. [DOI] [PubMed] [Google Scholar]
- 34.Huang TG, Ip SM, Yeung WS, Ngan HY. Mitomycin C and cisplatin enhanced the antitumor activity of p53-expressing adenovirus in cervical cancer cells. Cancer Invest. 2001;19:360–368. doi: 10.1081/cnv-100103131. [DOI] [PubMed] [Google Scholar]
- 35.Nakashima S, Natsugoe S, Matsumoto M, Kijima F, Takebayashi Y, Okumura H, Shimada M, Nakano S, Kusano C, Baba M, et al. Expression of p53 and p21 is useful for the prediction of preoperative chemotherapeutic effects in esophageal carcinoma. Anticancer Res. 2000;20:1933–1937. [PubMed] [Google Scholar]
- 36.Miyake H, Hara I, Hara S, Arakawa S, Kamidono S. Synergistic chemosensitization and inhibition of tumor growth and metastasis by adenovirus-mediated P53 gene transfer in human bladder cancer model. Urology. 2000;56:332–336. doi: 10.1016/s0090-4295(00)00567-7. [DOI] [PubMed] [Google Scholar]
- 37.Pestell KE, Hobbs SM, Titley JC, Kelland LR, Walton MI. Effect of p53 status on sensitivity to platinum complexes in a human ovarian cancer cell line. Mol Pharmacol. 2000;57:503–511. doi: 10.1124/mol.57.3.503. [DOI] [PubMed] [Google Scholar]
- 38.Mitsuuchi Y, Johnson SW, Selvakumaran M, Williams SJ, Hamilton TC, Testa JR. The phosphatidylinositol 3-kinase/AKT signal transduction pathway plays a critical role in the expression of p21WAF1/CIP1/SDI1 induced by cisplatin and paclitaxel. Cancer Res. 2000;60:5390–5394. [PubMed] [Google Scholar]
- 39.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 40.Nagashima M, Shiseki M, Miura K, Hagiwara K, Linke SP, Pedeux R, Wang XW, Yokota J, Riabowol K, Harris CC. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc Natl Acad Sci USA. 2001;98:9671–9676. doi: 10.1073/pnas.161151798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makin G, Hickman JA. Apoptosis and cancer chemotherapy. Cell Tissue Res. 2000;301:143–152. doi: 10.1007/s004419900160. [DOI] [PubMed] [Google Scholar]