

Abstract
AIM: To analyze the influence of cholesterol liposome on the Ca2+ mobilization of cultured muscle cells in rabbit sphincter of Oddi’s.
METHODS: New Zealand rabbit was sacrificed and the sphincter of Oddi (SO) segement was obtained aseptically. The SO segment was cut into pieces and cultured in DMEM solution. Then the smooth muscle cells were subcultured, and the 4th-7th passage cells were used for further investigation. The intracellular Ca2+ increase was measured under confocal microscope after the addition of 20 mmol·L-1 KCl, 10-7 mol·L-1 acetylcholine and 10-7 mol·L-1 cholecystokinin, and different antagonists were added to analyze the Ca2+ mobilization pathway. After the cells were incubated with 1 g·L-1 cholesterol liposome (CL)(molar ratio was-2:1), the intracellular Ca2+ increase was measured again to determine the effect of CL on cellular Ca2+ mobilization.
RESULTS: The resting cellular calcium concentration of cultured SO cell was 108 nmol·L-1 ± 21 nmol·L-1. The intracellular Ca2+ increases induced by 20 mmol·L-1 KCl, 10-7 mol·L-1 ACh and 10-7 mol·L-1 CCK were 183% ± 56%, 161% ± 52% and 130% ± 43%, respectively. When the extracellular Ca2+ was eliminated by 2 mmol·L-1 EGTA and 5 μmol·L-1 verapamil, the intracellular Ca2+ increases induced by KCl, ACh and CCK were 20% ± 14%, 82% ± 21% and 104% ± 23%, respectively. After the preincubation with heparin, the Ca2+ increases were 62% ± 23% and 23% ± 19% induced by ACh and CCK, as for preincubation with procaine they were 72% ± 28% and 85% ± 37% induced by ACh and CCK, respectively. Pretreatment with CL for 18 h, the resting cellular Ca2+ concentration elevated to 152 nmol·L-1 ± 26 nmol·L-1, however, the cellular Ca2+ increase percentages in response to these agonists were 67% ± 32%, 56% ± 33% and 34% ± 15%.
CONCLUSION: KCl elicite the SO cellular Ca2+ increase depends on influx of extracellular Ca2+, ACh evoked the SO celllular Ca2+ increase is through the mobilization of intracellular Ca2+ pool and influx of extracellular Ca2+ as well, CCK excites the SO cells mainly through mobilization of intracellular IP3-sensitive Ca2+ store. After the incorporation with cholesterol liposome, KCl,ACh and CCK induced cellular Ca2+ increase percentages decreased.
INTRODUCTION
Biliary tract diseases are becoming more common in recent years in China[1-10]. Sphincter of Oddi (SO) is an important part locatedat the distal part of biliary tract. And it is well accepted that SO plays an important role in the regulation of biliary system hydraulic pressure and bile flow[11-13]. SO dysfunction (SOD) is one of the causes responsible for biliary tract disorders[14-16], pancreatitis[17-18]and other disorders[19-21]. The mechanism underlying the occurrence of SOD is controversial, however, several investigators have noticed the relation between hypercholesterolemia and SOD[22-24]. Szilvassy et al[23] reported that patient has an impaired SO relaxation due to high serum lipids, but normalization of serum lipids improved the sphincter of Oddi relaxation. Wei et al[24] found the abnormalities in ultrastructure of SO in rabbits with hypercholesterolemia. Therefore, hypercholesterolemia might be one of causes of SOD. Meanwhile, the effect of cholesterol on gallbladder contractility has been interpreted by many authors[25-27], who found that the cholesterol incorporated into membrane can impair the cellular signal transdution and contractility as well.
The thin optical sectioning capability of laser scanning confocal microscopy rejects light from out-of-focus planes and permits imaging of [Ca2+]i in single alive cells in optical sections less than 1ìm thick, which makes it possible to measure intracellular Ca2+ alteration instantaneously. So, this experiment was designed to analyze the effect of different agonists on Ca2+ mobilization of SO cells and cholesterol liposome on this process, in order to explore the mechanism that hypercholesterolemia affected the SO motility.
MATERIAL AND METHODS
Materials
The fluo 3/AM was obtained from Molecular Probes (USA), trypsin, HEPES, cholecystokinin-octopeptide (CCK) and bovine serum albumin (BSA) were from Sigma Chemicals (USA), and Dulbecco’s modified Eagle’s medium (DMEM) was from Gibco Laboratories(USA). The ethylene glycol-bis(β-amino ethyl ether )-N,N,N’-tetraacetic acid (EGTA), verapamil, procaine, egg phosphatidylcholine and cholesterol were obtained from Shanghai Chemical Co. (China), and the acetylcholine from Suzhou Chemical Co. The dimethyl sulfoxide (DMSO) was purchased from Beijing Chemical Co. All other chemicals were commercial products of the highest available grade of purity.
Cells preparation
The New Zealand rabbits( < 30 d) were euthanized by intravenous injection of ketamine (20 mg·kg-1), and the segment of Oddi was removed quickly and washed by PBS solution containing penicillin(500 × 103 U·L-1). The experiments were conducted in accordance with the institutional ethical guidelines. Mucosa and serosa were dissected carefully, then washed twice with culture medium. The sphincter strip was cut into 1-2 mm3 squares and placed into culture chamber, the chamber was everted and added into 3 mL DMEM medium, and pH was adjusted to 7.4 by addition of 24 mmol·L-1 NaHCO3 before the use. After 3 h incubation at 37 °C, when the tissue squares sticked to the chamber then turn over the chamber, replaced the medium every 4-5 d.
Subculture: Cells migrated from the explants 10-15 d, as the chamber was confluenced with cells, then subcultured by addition with 2.5 g·L-1 trypsin at 37 °C for 10 min. The trypsin was inactived by bovine serum, and the cell suspension was centrifuged at 1000 r·min-1 for 7 min. The supernant was removed and cell pellet resuspended in fresh medium to a concentration of 5-7 × 105·mL-1 and repassaged into several chambers.
Differentiation: Three glass cover slips (18 mm × 18 mm) placed into a 6 cm diameter chamber, then cell suspension was added and incubated for 48 h. The slips covered with cells and fixed with 950 mL·L-1 ethanol for 30 min, washed with PBS for 5 min and desiccated naturally. The β-actin staining was performed with immunohistological kits. The positive stain was localized in long, straight, noninterrupted fibrils scattered densely along the longitudinal axis. Under phase-contrast microscope, the cultured cells showed a characteristic “hill- and - valley” growth pattern, and the 4th-7th passage cells were used in this experiments.
Preparation of cholesterol liposome
Liposomes were prepared as described previously[28,29]. Cholesterol (200 mg) and phosphatidylcholine (100 mg) were added into 5 mL chloroform and soluted completely. After the organic solution was evaporated, the container was placed in the desiccator overnight at 4 °C. Then PBS (pH 7.2) was added into the container, the finalconcentration of CL was 10 g·L-1. After the sonication, the mixture was centrifuged at 21000 × g for 30 min to sediment the undispersed lipid, then the supernatant was collected. The cholesterol-to-phospholipid molar ratio (FC/ PL) of the liposome was -2:1. Control liposome (Cholesterol 100 mg and phosphatidylcholine 200 mg) was prepared in the same way, FC/PL molar ratio was -0.5:1. Both liposomes were sterilized by filtration through a 0.45 µm filter and mixed with sterile DMEM (Dulbecco’s modified Eagle’s medium) at a concentration of 1 g·L-1, which is in accordance with the serum concentration of rabbit model with hypercholesterolemia[57], pH was adjusted to 7.4, control and cholesterol-rich media were determined to be isomolar before experimentation.
Calcium measurement
A 10 mm hole was made in a plastic chamber ( Made in Denmark), then a 22 mm glass coverslip was used to seal the hole tightly from the bottom. Then it was cleaned thoroughly and sterilized by ultra-violet lamp for 2 h. The cell suspension was added into the chambers and incubated for 48h, they were incubated with or without CL overnight.
Preparation of fluo-3/AM: The concentration of free cytosolic Ca2+ in SMCs was determined using the fluorescent Ca2+ indicator fluo-3/ AM. 50 µg fluo-3 was dissolved in 50 µL dimethyl sulfoxide (DMSO) (about 885 µmol·L-1), and mixed thoroughly. Then it was subdivided into ten vials and stored at -20 °C. Five µL of vial of stock solution was diluted by D-Hanks with a proporation of 1:200(V/V). The final concentration of fluo-3 was about 4.5 µmol·L-1. This loading solution should be used in 3 h, to maximize loading efficiency. Ca2+ measurement: The SO cells culture media was removed and washed for 10 min with D-Hanks solution. Remove the final wash solution, add the loading solution, incubate about 50 min at 37 °C. Then remove the loading solution, wash the cells with D-Hanks, measure the fluorescence soon after loading. The measurement was performed under Bio-Rad MRC 1024 laser scanning confocal microscope, select cellular Ca2+ indicator from the method menu, then select the button for fluo-3, numerical aperture being 1.3, and pixel 512. The data was treated at Compaq Pentium 90. The peak excitation was about 506 nm and the peak emission was about 526 nm.
Solution
HEPES buffered solution (in mmol·L-1): 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 Glucose, 0.49 EDTA; D-Hanks: 138 NaCl, 5.4 KCl, 0.37 Na2HPO4·H2O, 0.44 KH2PO4, 4.17 NaHCO3. PBS: 138 NaCl, 2.7 KCl, 10 Na2HPO4·H2O, 1.6 KH2PO4; High K+ solution contained the following (in mmol·L-1): 120 NaCl, 20 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 Glucose. Heparin was diluted by D-Hanks with 7 × 105 U·L-1, procaine concentration was 10 mmol·L-1.
Statistical analyses
Results were expressed as -x ± S-x. Student’s test or an analysis of variance (ANOVA) was performed to test the statistical significance as necessary, P < 0.05 was regarded as significant.
RESULTS
Cultured SO cells showed a typical smooth muscle cell characteristics which presented as “hill-and -valley” and α-SM actin positive staining, filament was demonstrated along the longitudinal axis of the cells(Figure 1).Loading with fluo-3/ AM for 50 min, the fluorescence distributed inhomogenously under LSCM, which may represent the Ca 2+ pool, is inhomogenous. All experiments were conducted at room temperature(21 °C-23 °C). The value of Ca2+ concentration was calculated and based on the following equation:[Ca2+]i = Kd × (F-Fmin) / (Fmax-F), where Kd is the dissociation constant for Ca2+ (316 nmol·L-1), Fmax is fluorescence maxima which was obtained by saturating intracellular signal values; Fmin is fluorescence minima which represented zero-Ca2+ signal. The resting cellular Ca2+ concentration was 108 ± 21 nmol·L-1.
Figure 1.

Cultured SO cells characterized with á-SM actin positive staining, filament were demonstrated along the longitudinal axis of the cells.
Distinctive agonists induced alteration of intracellular fluorescence
At the presence of extracellular Ca2+, intracellular Ca2+ concentration changed under the LSCM after the addition of agonists, and it showed spatially heterogeneous alteration of fluorescent intensity in SO cells (Figure 2).
Figure 2.
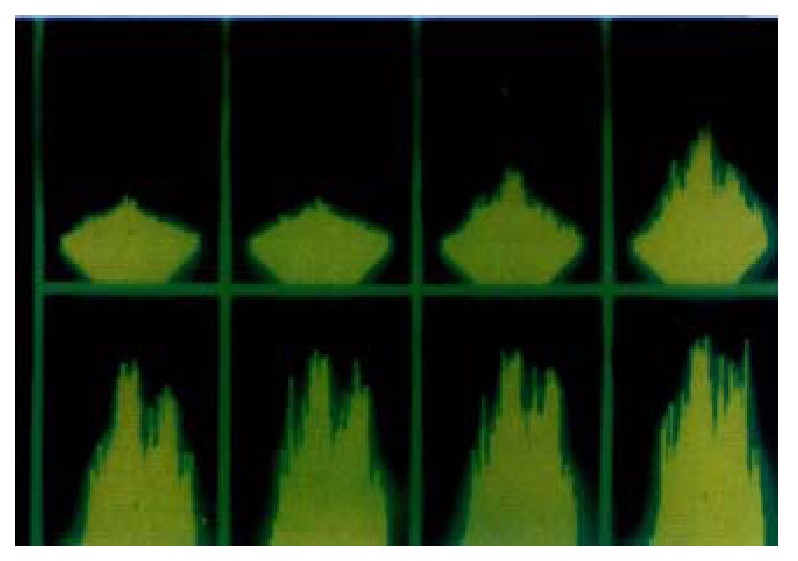
At the presence of extracellular Ca2+, intracellular Ca2+ concentra-tion changed under the LSCM(1 image·s-1) after the addition of agonists, and it shows spatially heterogeneous alteration of fluorescent intensity in SO cells.
After the addition of 20 mmol·L-1 KCl, the increase of intracellular fluorescence was 183% ± 56% ( n = 4), 10-7 mol·L-1 ACh caused fluorescence increase was 161% ± 52% ( n = 4), 10-7 mol·L-1 CCK agonized an increase of 130% ± 43% ( n = 4), the maximum Ca2+ concentration were 297 ± 66 nmol·L-1, 275 ± 58 nmol·L-1 and 251 ± 45 nmol·L -1, respectively.(n = 4, Figure 3).
Figure 3.
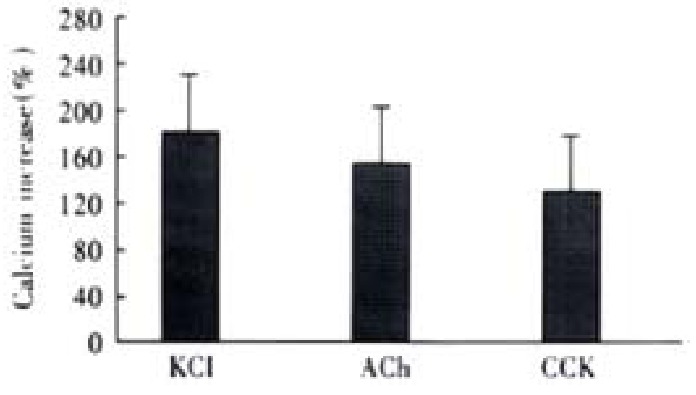
Intracellular fluorescence increases induced by different agonists. Ca2+ Concentration increased from resting level of 108 ± 21 nmol·L-1 to 297 ± 66 nmol·L-1, 275 ± 58 nmol·L-1 and 251 ± 45 nmol·L-1, respectively. ANOVA was performed for the analysis, F = 0.9184, no significant difference be-tween three groups (n = 4).
The elimination of extracellular Ca2+ was performed by incubation of 2 mmol·L-1 EGTA and 10 μmol·L-1 verapamil for 10 min. The extracellular Ca2+ was chelated by EGTA (a highly specific chelator for free Ca2+ ions) and inward Ca2+ current was inhibited by verapamil (inhibitor of L-type Ca2+ channels). Compared with the presence of extracellular Ca2+, under this circumstance, the cellular fluorescence decreased from the peak rapidly.
At absence of extracellular Ca2+ treated by EGTA and verapamil, 20 mmol·L-1 KCl induced an Ca2+ increase of 20% ± 14% (P < 0.01, vs control, t = 4.882), with maximal Ca2+ concentration of 131 ± 17 nmol·L-1, which indicated the induction of KCl depends on the presence of extracellular Ca2+. Under the same condition, ACh increased the cellular Ca2+ by 82% ± 21%, with a maximal Ca2+ concentration of 192 nmol·L-1 ± 22 nmol·L-1. After pretreatment of heparin ( inhibitor of IP3-sensitive Ca2+ release channel ) and procaine(inhibitor of IP3-insensitive Ca2+ release channel), the cellular influoscence increase by 62% ± 23% and 72% ± 28%, respectively, the maximal cellular calcium was 175 nmol·L-1 ± 26 nmol·L -1 and 186 nmol·L-1 ± 30 nmol·L-1, and there was no significant difference from that of untreated cells (Figure 4)
Figure 4.
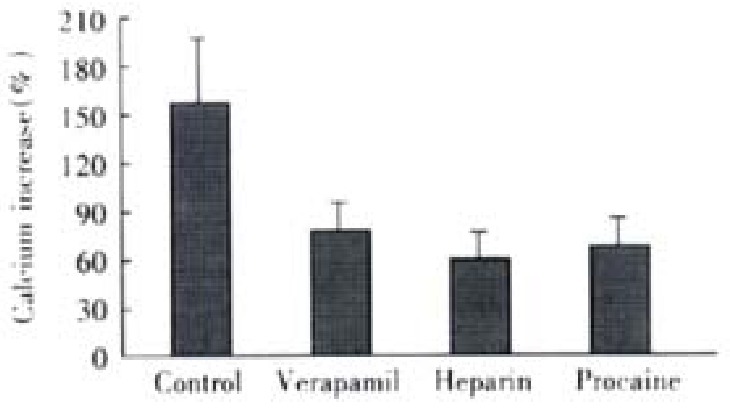
Effect of different antagonists on ACh induced cellular Ca2+ increase (-x ± S-x, n = 4). Maximal Ca2+ concentration increased to 192 ± 22 nmol·L-1,175 nmol·L-1 ± 26 nmol·L-1 and 186 ± 30 nmol·L-1, respectively. ANOVA was performed for the analysis, F = 1.324, with no significant difference be-tween the three experimental groups (n = 4 ).
CCK induced an cellular Ca2+ increase of 104% ± 23% after the elimination of extracellular Ca2+. After incubation of heparin and procaine, CCK induced calcium increase were 23% ± 19% and 85% ± 37%, which was different from that of acetylcholine(Figure 5).
Figure 5.
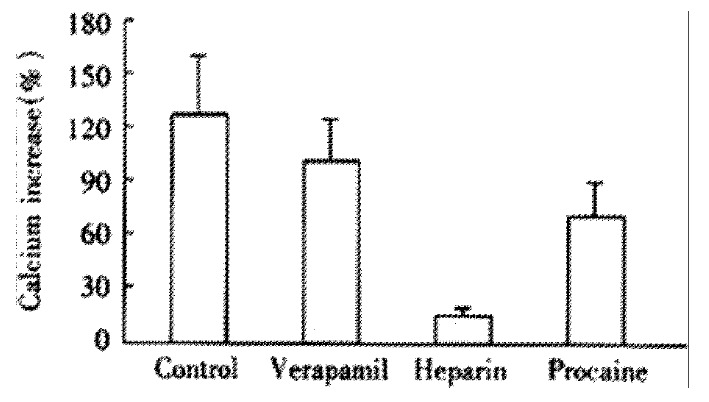
Effect of different antagonists on CCK induced cellular Ca2+ increase (-x ± S-x, n = 4). Maximal Ca2+concentration increased to 220 ± 26 nmol·L-1, 133 ± 21 nmol·L-1 and 201 ± 40 nmol·L-1, respectively. SNK-q test was performed, there was a statistical difference between heparin treated group and other two groups, P < 0.01.
Intracellular calcium increase after the incubation of cholesterol liposome
After the incubation of 1 g·L-1 CL for 18 h, many CL incorporated into the membrane, the cells changed slightly as mentioned by others[45]. The intracellular calcium concentration of control liposome(FC/PL molar ratio with-0.5:1) treated cells were 117 ± 19 nmol·L-1, there was no significant difference as compared with untreated cells. However, the resting cellular Ca2+ concentration was elevated obviously by incubation of CL(FC/PL molar ratio with -2:1 ), which was 152 ± 26 nmol·L-1. But, after addition of agonists, the Ca2+ concentrations increased to 257 ± 54 nmol·L-1, 238 ± 57 nmol·L-1 and 204 nmol·L-1 ± 26 nmol·L-1, respectively, the calcium increase percentages were significantly decreased, the celllular influorescence increases induced by KCl, ACh and CCK were 67% ± 32%( t = 3.597), 56% ± 33% ( t = 3.410) and 34% ± 15%( t = 4.216), respectively,(P < 0.05,vs control) (Figure 6).
Figure 6.
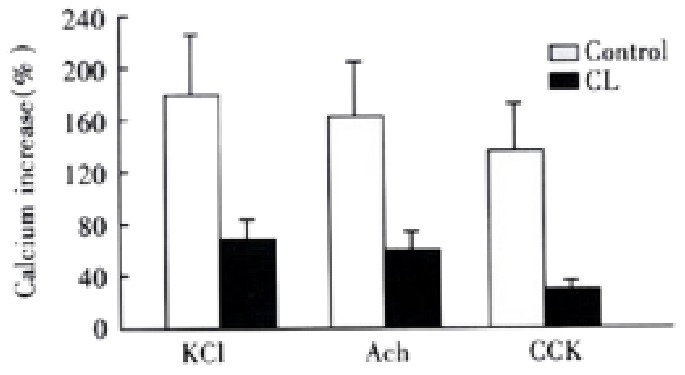
Effect of cholesterol liposome on cellular Ca2+ mobilization (-x ± S-x, n = 4). Agonists induced cellular influorescence increase percentages were markedly less than that of control group.
DISCUSSION
The present study shows that Ca2+ mobilization of SO cells evoked by potassium, acetylcholine and cholecystokinin were impaired after the cells incubated with cholesterol liposome (CL). And it also indicated that CL affects the different pathways of Ca2+ mobilization, for our results demonstrated that KCl induced intracellular Ca2+ increase depe nds on the extracellular Ca2+ influx, acetylcholine may agonize the Ca2+ increase through both intra- and extracellular pathway, while CCK elicited SO cells via mobilizing intracellular Ca2+ store. These observations agree with other authors’ in gallbladder muscle cells[30-34].
Based on literatures and our results, potassium excited the smooth muscle cells depend on Ca2+ influx through L-type voltage-dependent Ca2+ channel which could be inhibited by verapamil. As for acetylcholine and cholecystokinin, 2 mmol·L-1 EGTA and 10ìm verapamil could not inhibit the cellular Ca2+ increase completely, demonstrating that both Ach and CCK elicit SO cells do not depend on influx of extracellular calcium. However, heparin could inhibit the cellular Ca2+ increase induced by CCK, which indicate a CCK evoked Ca2+ increase of SO cells through IP3-sensetive pathway.
The normal function of SO is the precondition of biliary tract homeostasis, and the motility was under the coordination of hormone and innervation[35-38]. SOD is responsible for many disorders including gallbladder stasis,stone formation and unexplainable upper abdominal ache post-cholecystectomy[13]. Additionnally, it was proved that the occurrence of SOD was correlated with intestinal dysmotility[20,39,40] and it was also correlated with recurrent and chronic pancreatitis[41,42]. On the other hand, most researches on SO were concentrated on the SO manometry, in such a situation, the results and conclusions were always full of discrepancy due to the complex effect of hormone and nerve or differences in species[12,13]. Therefore, isolated and cultured SO cells were used to study its characteristics in order to rule out complex influences in vivo[43-46].
It was well known that motility regulation of smooth muscle cells depends on several mechanisms, including: influx of extracellular Ca2+ , intracellular Ca2+ mobilization and sensitivity modulation of intracellular Ca2+. If this process was inhibited, the contractility will be impaired. In our previous experiments, high molar ratio CL (molar ratio-2:1) impaired the SO muscle cell contractility[43], whereas, low molar ratio CL(0.5:1) had no effect on cells contractility. According to previous literatures,cholesterol enrichment of the SMC membrane occurs rapidly, and the aortic smooth muscle cells in hypercholesterolemic rabbit has impaired relaxation[47-49], and reduced contraction as well[50]. Cholesterol incorporation into membrane could also result in an alteration of membrane conduction of ions[51,63]. Because the cholesterol liposome was readily incorporated into cells membrane, that the duration, CL and cells incubation needed, was not a routine one. In another previous experiment, we used 2h for incubation[43]. Broderick et al used 3h in the study of CL effect on arterial smooth muscle[50].
Our aim was mainly to observe the Ca2+ mobilization alteration affected by CL. There are many evidence in vivo and in vitro showing an impaired contractility in human and animal gallbladders with cholesterol stones[28,52]. Li et al[53] measured the actin and myosin isoform in gallbladders smooth muscle following feeding in prairie dogs and found that cholesterol feeding induced a shift in actin isoforms, but whether it is really responsible for the decreased contractility is uncertain. It was proved that cholesterol could alter smooth muscle membrane and cell function by changing the physical state of the membrane phospholipid bilayer[54], and therefore affect the function of integral membrane proteins, such as Ca2+ and potassium channels, as well as transmembrane receptors[26]. Membrane fluidity decreased with excessive cholesterol incorporation and subsequently restricted optimal function of membrane proteins, such as receptor binding of ligands, receptor coupling with G proteins and activation of enzymes[55,56]. Thus, it could explain the impaired activation of signal transduction pathways responsible for contractile responses to receptor-dependent agonists, such as CCK and ACh. It has been shown that muscle cell contraction, membrane fluidity, membrane cholesterol and phospholipid content are reversible after the membrane cholestrol was leached out by incubation of cholesterol-free liposomes for several hours[27,28]. Whether the effects of cholesterol liposome observed in this study were due to cytotoxicity Other authors reported that high concentration of CL might have influence to cultured muscle cells[46]. The similar procedures had been done by many reseachers[27,29,64], and the cholesterol was within the serum concentration range of hypercholesterolemia rabbit model[57]. So, the cytotoxicity of liposomes to cells in this study, if any, might be unconsiderable.
The effect of cholesterol on SO has not been elucidated yet. SO manometry of hypercholesterolemic rabbit shows that abnormal SO motility had been observed before the formation of gallstone[57], basal pressure rose and amplitude of phasic contraction decreased that represented an impaired relaxation and decreased contraction, too. Szilvassy et al[58] observed that the SO of hypercholesterolemic rabbits restored the nitrergic transmitter mediated relaxation by farnesol treatment. The SO segments of rabbits, prairie dogs and guinea pigs belong to extraduodenal type[59-61] and were able to pump fluid from the bile duct to duodenum. Therefore, it could be concluded that cholesterol affected the SO contractility which was responsible for a reduced peristaltic function to pump bile into duodenum. CCK receptors located in SO neurons and muscle cells were G-protein coupled receptors[62] which could be affected by excessive cholesterol incorporation resulting in decreased release of neural mediators or an impaired contraction. Therefore, we can conclude that hypercholesterolemia could not only impair relaxation but also contraction of rabbits sphincter of Oddi, which could lead to the occurance of SO dysfunction.
In summary, the present study shows that potassium induced rabbits SO cells Ca2+ increase depends on Ca2+ influx through L-type channel; acetylcholine induces SO cells Ca2+ increase from both intra- and extra-cellular Ca2+ release; cholecystokinin evokes the SO cells by mobilizing IP3-sensitive Ca2+ stores; and cholesterol liposome could affect the intracellular Ca2+ increase induced by different agonists.
Footnotes
Edited by Ma JY
References
- 1.Fang CH, Yang JZ, Kang HG. A PCR study on Hp DNA of bile, mucosa and stone in gallstones patients and its relation to stone nuclear formation. Shijie Huaren Xiaohua Zazhi. 1999;7:233–235. [Google Scholar]
- 2.Gu SW, Luo KX, Zhang L, Wu AH, He HT, Weng JY. Relationship between ductule prol iferation and liver fibrosis of chronic liver disease. Shijie Huaren Xiaohua Zazhi. 1999;7:845–847. [Google Scholar]
- 3.Zhou LS, Shi JS, Wang ZR, Wang L. Tumor necrosis factor α in gallbladder and gallstone. Shijie Huaren Xiaohua Zazhi. 2000;8:426–428. [Google Scholar]
- 4.Fang CH, Yang J. A study on DNA of aerobic and anaerobic bacte-ria in bile, mucosa and stone in gallstone patients. Shijie Huaren Xiaohua Zazhi. 2000;8:66–68. [Google Scholar]
- 5.Li XP, Mao XZ. Effect of estrogen, cholic acid loading and bile draining on hepatobiliary functions in rats. Shijie Huaren Xiaohua Zazhi. 2000;8:1009–1012. [Google Scholar]
- 6.He XS, Huang JF, Liang LJ, Lu MD, Cao XH. Surgical resection for hepatoportal bile duct cancer. World J Gastroenterol. 1999;5:128–131. doi: 10.3748/wjg.v5.i2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhi QH. New development of biliary surgery in China. World J Gastroenterol. 2000;6:187–192. doi: 10.3748/wjg.v6.i2.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li LB, Cai XJ, Li JD, Mu YP, Wang YD, Yuan XM, Wang XF, Bryner B, Finley RK Jr. Will intraoperative cholangiography prevent bil-iary duct injury in laparoscopic cholecystectomy. World J Gastroenterol. 2000;6:21. [Google Scholar]
- 9.Yang YK, Qiu KX, Zhan YZ, Zhan EY, Yang HM, Zheng P. Deter-mination of cholesterol in human biliary calculus by TLC scanning. World J Gastroenterol. 2000;6:62. [Google Scholar]
- 10.Chen XM, LaRusso NF. Human intestinal and biliary cryptosporidiosis. World J Gastroenterol. 1999;5:424–429. doi: 10.3748/wjg.v5.i5.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coelho JC, Moody FG. Certain aspects of normal and abnormal motility of sphincter of Oddi. Dig Dis Sci. 1987;32:86–94. doi: 10.1007/BF01296692. [DOI] [PubMed] [Google Scholar]
- 12.Toouli J. What is sphincter of Oddi dysfunction. Gut. 1989;30:753–761. doi: 10.1136/gut.30.6.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dodds WJ. Biliary tract motility and its relationship to clinical disorders. AJR Am J Roentgenol. 1990;155:247–258. doi: 10.2214/ajr.155.2.2115247. [DOI] [PubMed] [Google Scholar]
- 14.Herman F, Delforge M, Bastens B, Masy V, Lilet H, Brassine A. [Dysfunction of the sphincter of Oddi in cholecystectomy patients] Rev Med Liege. 1998;53:193–198. [PubMed] [Google Scholar]
- 15.Toouli J. Biliary motility disorders. Baillieres Clin Gastroenterol. 1997;11:725–740. doi: 10.1016/s0950-3528(97)90018-x. [DOI] [PubMed] [Google Scholar]
- 16.Eversman D, Fogel EL, Rusche M, Sherman S, Lehman GA. Frequency of abnormal pancreatic and biliary sphincter manometry compared with clinical suspicion of sphincter of Oddi dysfunction. Gastrointest Endosc. 1999;50:637–641. doi: 10.1016/s0016-5107(99)80011-x. [DOI] [PubMed] [Google Scholar]
- 17.Guelrud M, Morera C, Rodriguez M, Jaen D, Pierre R. Sphincter of Oddi dysfunction in children with recurrent pancreatitis and anomalous pancreaticobiliary union: an etiologic concept. Gastrointest Endosc. 1999;50:194–199. doi: 10.1016/s0016-5107(99)70224-5. [DOI] [PubMed] [Google Scholar]
- 18.Tomás A, Vida F, Ponce J. [Acute recurrent pancreatitis in a patient with Oddi's sphincter dysfunction] Gastroenterol Hepatol. 1999;22:279–281. [PubMed] [Google Scholar]
- 19.Chen SZ, Sha JP, Chen XC, Hou CC, Fu WH, Liu W. Dysrelaxation of sphincter of Oddi in patients with bile reflux gastritis: study on effect of nifedipine of gallbladder emptying. Shijie Huaren Xiaohua Zazhi. 1999;7:1020–1023. [Google Scholar]
- 20.Chen SZ, Bu X, Hou CC, Li S, Chen XC. Mechanism of nifedipine for improving abnormal gallbladder emptying function in patients with irritable bowel syndrome. Shijie Huaren Xiaohua Zazhi. 1998;6:423–426. [Google Scholar]
- 21.Chen SZ, Zhao H, Wu CY, Fu WJ, Chen XC. Gallbladder emptying function in patients with bile reflux gastritis. Shijie Huaren Xiaohua Zazhi. 1998;6:427–429. [Google Scholar]
- 22.Szilvassy Z, Nagy I, Szilvassy J, Jakab I, Csati S, Lonovics J. Impaired nitrergic relaxation of the sphincter of Oddi of hyperlipidaemic rabbits. Eur J Pharmacol. 1996;301:R17–R18. doi: 10.1016/0014-2999(96)00159-8. [DOI] [PubMed] [Google Scholar]
- 23.Szilvássy Z, Nagy I, Madácsy L, Hajnal F, Velösy B, Takács T, Lonovics J. Beneficial effect of lovastatin on sphincter of Oddi dyskinesia in hypercholesterolemia and hypertriglyceridemia. Am J Gastroenterol. 1997;92:900–902. [PubMed] [Google Scholar]
- 24.Wei JG, Wang YC, Du F, Yu HJ. Dynamic and ultrastructural study of sphincter of Oddi in early-stage cholelithiasis in rabbits with hypercholesterolemia. World J Gastroenterol. 2000;6:102–106. doi: 10.3748/wjg.v6.i1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Q, Amaral J, Oh S, Biancani P, Behar J. Gallbladder relaxation in patients with pigment and cholesterol stones. Gastroenterology. 1997;113:930–937. doi: 10.1016/s0016-5085(97)70189-6. [DOI] [PubMed] [Google Scholar]
- 26.Chen Q, De Petris G, Yu P, Amaral J, Biancani P, Behar J. Different pathways mediate cholecystokinin actions in cholelithiasis. Am J Physiol. 1997;272:G838–G844. doi: 10.1152/ajpgi.1997.272.4.G838. [DOI] [PubMed] [Google Scholar]
- 27.Chen Q, Amaral J, Biancani P, Behar J. Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology. 1999;116:678–685. doi: 10.1016/s0016-5085(99)70190-3. [DOI] [PubMed] [Google Scholar]
- 28.Yu P, Chen Q, Biancani P, Behar J. Membrane cholesterol alters gallbladder muscle contractility in prairie dogs. Am J Physiol. 1996;271:G56–G61. doi: 10.1152/ajpgi.1996.271.1.G56. [DOI] [PubMed] [Google Scholar]
- 29.Bialecki RA, Tulenko TN. Excess membrane cholesterol alters calcium channels in arterial smooth muscle. Am J Physiol. 1989;257:C306–C314. doi: 10.1152/ajpcell.1989.257.2.C306. [DOI] [PubMed] [Google Scholar]
- 30.Xu QW, Shaffer EA. The potential site of impaired gallbladder contractility in an animal model of cholesterol gallstone disease. Gastroenterology. 1996;110:251–257. doi: 10.1053/gast.1996.v110.pm8536864. [DOI] [PubMed] [Google Scholar]
- 31.Mansour A, Dawoud I, Gad-El-Hak N. The potential site of disordered gallbladder contractility during the early stage of cholesterol gallstone formation. Hepatogastroenterology. 1998;45:1404–1409. [PubMed] [Google Scholar]
- 32.Yu P, Chen Q, Harnett KM, Amaral J, Biancani P, Behar J. Direct G protein activation reverses impaired CCK signaling in human gallbladders with cholesterol stones. Am J Physiol. 1995;269:G659–G665. doi: 10.1152/ajpgi.1995.269.5.G659. [DOI] [PubMed] [Google Scholar]
- 33.Behar J, Rhim BY, Thompson W, Biancani P. Inositol trisphosphate restores impaired human gallbladder motility associated with cholesterol stones. Gastroenterology. 1993;104:563–568. doi: 10.1016/0016-5085(93)90427-e. [DOI] [PubMed] [Google Scholar]
- 34.Yu P, De Petris G, Biancani P, Amaral J, Behar J. Cholecystokinin-coupled intracellular signaling in human gallbladder muscle. Gastroenterology. 1994;106:763–770. doi: 10.1016/0016-5085(94)90713-7. [DOI] [PubMed] [Google Scholar]
- 35.Hogan WJ, Geenen JE. Biliary dyskinesia. Endoscopy. 1988;20 Suppl 1:179–183. doi: 10.1055/s-2007-1018172. [DOI] [PubMed] [Google Scholar]
- 36.Luman W, Williams AJ, Pryde A, Smith GD, Nixon SJ, Heading RC, Palmer KR. Influence of cholecystectomy on sphincter of Oddi motility. Gut. 1997;41:371–374. doi: 10.1136/gut.41.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mawe GM, Kennedy AL. Duodenal neurons provide nicotinic fast synaptic input to sphincter of Oddi neurons in guinea pig. Am J Physiol. 1999;277:G226–G234. doi: 10.1152/ajpgi.1999.277.1.G226. [DOI] [PubMed] [Google Scholar]
- 38.Middelfart HV, Matzen P, Funch-Jensen P. Sphincter of Oddi manometry before and after laparoscopic cholecystectomy. Endoscopy. 1999;31:146–151. doi: 10.1055/s-1999-13663. [DOI] [PubMed] [Google Scholar]
- 39.Soffer EE, Johlin FC. Intestinal dysmotility in patients with sphincter of Oddi dysfunction. A reason for failed response to sphincterotomy. Dig Dis Sci. 1994;39:1942–1946. doi: 10.1007/BF02088129. [DOI] [PubMed] [Google Scholar]
- 40.Evans PR, Dowsett JF, Bak YT, Chan YK, Kellow JE. Abnormal sphincter of Oddi response to cholecystokinin in postcholecystectomy syndrome patients with irritable bowel syndrome. The irritable sphincter. Dig Dis Sci. 1995;40:1149–1156. doi: 10.1007/BF02064214. [DOI] [PubMed] [Google Scholar]
- 41.Tarnasky PR, Hoffman B, Aabakken L, Knapple WL, Coyle W, Pineau B, Cunningham JT, Cotton PB, Hawes RH. Sphincter of Oddi dysfunction is associated with chronic pancreatitis. Am J Gastroenterol. 1997;92:1125–1129. [PubMed] [Google Scholar]
- 42.Di Francesco V, Brunori MP, Rigo L, Toouli J, Angelini G, Frulloni L, Bovo P, Filippini M, Vaona B, Talamini G, et al. Comparison of ultrasound-secretin test and sphincter of Oddi manometry in patients with recurrent acute pancreatitis. Dig Dis Sci. 1999;44:336–340. doi: 10.1023/a:1026658618605. [DOI] [PubMed] [Google Scholar]
- 43.Wang XJ, Wei JG, Wang YC, Xu JK, Wu QZ, Wu DC, Yang XX. Effect of cholesterol liposome on contractility of rabbit Oddi's sphincter smooth muscle cells. Shijie Huaren Xiaohua Zazhi. 2000;8:633–637. [Google Scholar]
- 44.Zhang JS, Wei JG, Zhang ML, Wang D, Shi YH, Ji ZL. Effects of cholesterol liposome on cytoskeleton of rabbit sphincter of Oddi cells in culture. J Fourth Milit Med Univ. 1997;18:528–531. [Google Scholar]
- 45.Zhang JS, Wei JG, Wu JZ, Chen JY. Culture and morphologic observation of rabbit Oddi's sphincter cells. Shijie Huaren Xiaohua Zazhi. 1999;7:316–319. [Google Scholar]
- 46.Zhang JS, Wei JG, Wu JZ, Zhang YH, Wang H. Experimental study of the effects of cholesterol liposome on rabbit sphincter of Oddi cells proliferation in culture. J Fourth Milit Univ. 1998;19:494–297. [Google Scholar]
- 47.Tulenko TN, Laury-Kleintop L, Walter MF, Mason RP. Cholesterol, calcium and atherosclerosis: is there a role for calcium channel blockers in atheroprotection. Int J Cardiol. 1997;62 Suppl 2:S55–S66. doi: 10.1016/s0167-5273(97)00242-8. [DOI] [PubMed] [Google Scholar]
- 48.Weisbrod RM, Griswold MC, Du Y, Bolotina VM, Cohen RA. Reduced responsiveness of hypercholesterolemic rabbit aortic smooth muscle cells to nitric oxide. Arterioscler Thromb Vasc Biol. 1997;17:394–402. doi: 10.1161/01.atv.17.2.394. [DOI] [PubMed] [Google Scholar]
- 49.Kitagawa S, Yamaguchi Y, Shinozuka K, Kwon YM, Kunitomo M. Dietary cholesterol enhances impaired endothelium-dependent relaxations in aortas of salt-induced hypertensive Dahl rats. Eur J Pharmacol. 1996;297:71–76. doi: 10.1016/0014-2999(95)00729-6. [DOI] [PubMed] [Google Scholar]
- 50.Van Diest MJ, Herman AG, Verbeuren TJ. Influence of hypercholesterolaemia on the reactivity of isolated rabbit arteries to 15-lipoxygenase metabolites of arachidonic acid: comparison with platelet-derived agents and vasodilators. Prostaglandins Leukot Essent Fatty Acids. 1996;54:135–145. doi: 10.1016/s0952-3278(96)90071-x. [DOI] [PubMed] [Google Scholar]
- 51.Cox RH, Tulenko TN. Altered contractile and ion channel function in rabbit portal vein with dietary atherosclerosis. Am J Physiol. 1995;268:H2522–H2530. doi: 10.1152/ajpheart.1995.268.6.H2522. [DOI] [PubMed] [Google Scholar]
- 52.Behar J, Lee KY, Thompson WR, Biancani P. Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology. 1989;97:1479–1484. doi: 10.1016/0016-5085(89)90392-2. [DOI] [PubMed] [Google Scholar]
- 53.Li YF, Bowers RL, Haley-Russell D, Moody FG, Weisbrodt NW. Actin and myosin isoforms in gallbladder smooth muscle following cholesterol feeding in prairie dogs. Gastroenterology. 1990;99:1460–1466. doi: 10.1016/0016-5085(90)91176-7. [DOI] [PubMed] [Google Scholar]
- 54.Gleason MM, Medow MS, Tulenko TN. Excess membrane cholesterol alters calcium movements, cytosolic calcium levels, and membrane fluidity in arterial smooth muscle cells. Circ Res. 1991;69:216–227. doi: 10.1161/01.res.69.1.216. [DOI] [PubMed] [Google Scholar]
- 55.Squier TC, Bigelow DJ, Thomas DD. Lipid fluidity directly modulates the overall protein rotational mobility of the Ca-ATPase in sarcoplasmic reticulum. J Biol Chem. 1988;263:9178–9186. [PubMed] [Google Scholar]
- 56.Fong TM, McNamee MG. Stabilization of acetylcholine receptor secondary structure by cholesterol and negatively charged phospholipids in membranes. Biochemistry. 1987;26:3871–3880. doi: 10.1021/bi00387a020. [DOI] [PubMed] [Google Scholar]
- 57.Du F, Wei JG. Function of Oddi's sphincter disorders in formation of cholelithiasis (abstract). Zhonghua Waike Zazhi. 1999;37:383. [Google Scholar]
- 58.Szilvassy Z, Sari R, Nemeth J, Nagy I, Csati S, Lonovics J. Improvement of nitrergic relaxation by farnesol of the sphincter of Oddi from hypercholesterolaemic rabbits. Eur J Pharmacol. 1998;353:75–78. doi: 10.1016/s0014-2999(98)00449-x. [DOI] [PubMed] [Google Scholar]
- 59.Calabuig R, Ulrich-Baker MG, Moody FG, Weems WA. The propulsive behavior of the opossum sphincter of Oddi. Am J Physiol. 1990;258:G138–G142. doi: 10.1152/ajpgi.1990.258.1.G138. [DOI] [PubMed] [Google Scholar]
- 60.Calabuig R, Weems WA, Moody FG. Choledochoduodenal flow: effect of the sphincter of Oddi in opossums and cats. Gastroenterology. 1990;99:1641–1646. doi: 10.1016/0016-5085(90)90469-h. [DOI] [PubMed] [Google Scholar]
- 61.Toouli J. Sphincter of Oddi motility. Br J Surg. 1984;71:251–256. doi: 10.1002/bjs.1800710402. [DOI] [PubMed] [Google Scholar]
- 62.Wank SA. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: an exemplary family. Am J Physiol. 1998;274:G607–G613. doi: 10.1152/ajpgi.1998.274.4.g607. [DOI] [PubMed] [Google Scholar]
- 63.Zhou Q, Jimi S, Smith TL, Kummerow FA. The effect of cholesterol on the accumulation of intracellular calcium. Biochim Biophys Acta. 1991;1085:1–6. doi: 10.1016/0005-2760(91)90224-6. [DOI] [PubMed] [Google Scholar]
- 64.Broderick R, Bialecki R, Tulenko TN. Cholesterol-induced changes in rabbit arterial smooth muscle sensitivity to adrenergic stimulation. Am J Physiol. 1989;257:H170–H178. doi: 10.1152/ajpheart.1989.257.1.H170. [DOI] [PubMed] [Google Scholar]