

Abstract
AIM: To Quantitatively analyze the nitri oxide (NO) and Ca2+ in apoptosis of esophageal carcinoma cells induced by arsenic trioxide (As2O3).
METHODS: The cell line SHEEC1, a malignant esophageal epithelial cell induced by HPV in synergy with TPA in our laboratory, was cultured in a serum-free medium and treated with As2O3. Before and after administration of As2O3, NO production in cultured medium was detected quantitatively using the Griess Colorimetric method. Intracellular Ca2+ was labeled by using the fluorescent dye Fluo3-AM and detected under confocal laser scanning microscope (CLSM), which was able to acquire data in real-time enabling Ca2+ dynamics of individual cells in vitro. The apoptotic cells were examined under electron microscopy.
RESULTS: Intracellular concentration of Ca2+ increased from 1.00 units to 1.09-1.38 units of fluorescent intensity at As2O3 treatment and NO products subsequently released from As2O3-treated cells increased from 0.98-1.00 × 10-2 μmol·L-1 up to 1.48-1.52 × 10-2 μmol·L-1 and maintained in a high level continuously. Finally apoptosis of cells occurred, chromatin being agglutinated, cells shrunk, nuclei became round and mitochondria swelled.
CONCLUSION: Ca2+ and NO increased with cell damage and apoptosis in cells treated by As2O3. The Ca2+ is an initial messenger to the apoptotic pathway. To investigate Ca2+ and NO will be a new direction for studying the apoptotic signaling messenger of the esophageal carcinoma cells induced by As2O3.
INTRODUCTION
Arsenic trioxide (As2O3) has been proved to be a genotoxic and a carcinogenic agent[1-6]. Previous studies also showed that As2O3 induced cellular apoptosis in leukemia[7-15], in cancer cells of head and neck[16] and other cancer cells[17-22]. So As2O3 has antitumoral effect. We found that As2O3 induced apoptosis in esophageal squamous carcinoma cells[23]. The pathomorphological changes induced by As2O3 revealed that cells became smaller and shrank, nucleus rounded up, chromatin agglutinated and marginated, the nuclear membrane broke down followed by degenerative changes and cell mortality. All these changes indicated typical morphological changes of apoptosis[24,25]. Mitochondria, an important cellular apparatus, is related to cell breathing, oxygen metabolism, enzyme activity and energy supply. Our data demonstrated that the primary target of As2O3 inducing apoptosis of esophageal carcinoma cells might be the mitochondria[26]. It is likely that As2O3 is a mitochondriotoxic agent[27,28]. At the early stage of cellular apoptosis induced by As2O3, the mitochondria generated morphological and functional changes[29,30].
NO exerts a wide range of its biological properties via its interaction with mitochondria and NO mediated mitochondria damage[31]. In our previous data, an increase level of nitrite, a stable product of NO, was detected in the culture medium of esophageal carcinoma cells in arsenite-treated apoptosis[32]. Calcium ions (Ca2+) act as a universal second messenger in a variety of cells. Numerous functions of all types of cells are regulated by Ca2+ to a greater or lesser degree. Because of the importance of Ca2+ in biology, numerous methods of analyzing cellular Ca2+ activity have been established. Confocal laser scanning microscopy (CLSM) allows the precise spatial and temporal analysis of intracellular Ca2+ activity at the subcellular level. This optical technique has enabled scientists to document the dynamic changes of intracellular Ca2+ in vitro[33].
Arsenic may generate reactive oxygen species to exert its toxicity, which is implicated in DNA damage, signal transduction and apoptosis. What we are interested in is to see if NO and Ca2+ are involved in arsenic-induced apoptosis and to observe the changes of its target organelle—mitochondria. This study is to investigate which are the original messengers that initiate apoptosis and to detect quantitatively Ca2+ and NO in the apoptotic process of esophageal carcinoma cell line induced by As2O3.
MATERIALS AND METHODS
Cell line generation and cell culture
The esophageal carcinoma cell line (SHEEC1) was a malignantly transformed cell line of human embryonic esophageal epithelium induced by HPV18 E6E7 in synergy with TPA (12-O-tetradecanoyl-phorbol-13-acetate)[34]. Cells were cultured in 50 mL flasks and 24-well plate (Corning) with serum-free medium. The culture medium contained of the basal medium (MCDB151) with trace elements (M-6645 Sigma) and added transferrin, hydrocorticosone, epidermal growth factor (EGF), insulin (Sigma Chemical Co.) and extracts of bovine hypophysis (Gibco, BRL), but without calf serum, nitrite and nitrate, while containing streptomycin and penicilline (50 mg·L-1 for each).
The administration of arsenic
Arsenic trioxide (As2O3) obtained from Sigma Chemical Co. (St. Louis MO, Lot A 1010)at concertrations of 0, 1, 3 and 5 μmol·L-1 was added into the culture flasks and 24-well plates, respectively, for 0, 2, 4, 8, 12 and 24 h. The experiment was repeated once.
Transmission electron-microscopy (EM) examination
At the endpoints of As2O3 (24 h), cells of each group were digested with 0.25% trypsin, centrifuged, fixed with 2.5% glutaraldehyde, and routinely prepared for electron microscopic examination. The samples were observed under transmission electron microscope (Hitachi 300).
Cell cycle and apoptotic rate analyzed by flow cytometry (FCM)
Cells of repeated experiment were harvested to measure the ratio of apoptotic cells to survived cells. The cells were washed twice with PBS, dispersed, and filtered through 360 mesh nylon net to make a single cell suspension. It was fixed with 700 mL·L-1 precooled alcohol in ice. Before analysis cells were suspended in PBS and stained with propidium iodide. 1 × 10 9 cells·L-1 were detected by flow cytometry (FACsort, B-D Co,USA). The DNA histogram was drawn according to the fluorescent intensity value of 104 cells.
Procedure of NO detection[35]
The nitrite/nitrate colorimetric method, using the kit purchased from Boehringer Mannheim Co, was used to detect NO in culture medium. The culture medium of 0.2 mL from flask was regularly deactivated at 80 °C for 5 min and deproteinated by centrifugation in 12000 r·min-1 for 30 min, and the supernatant was determined. The procedure for NO determination was as follows: sample solution of 100 μL, 50 μL of nicotinamide adenine dinucleotide phosphate (NADPH) and 4 μL of the enzyme nitrate reductase (NR) were placed in a 3 mL test tube, mixed, incubated for 30 min at room temperature, and added 50 μL color reagent I & II, respectively, mixed,and allowed to stand in the dark at room temperature for 10 to 15 min. The NO content of the samples and the blank was estimated with Shimadzu UV/120 spectrophotometry by 450 nm and was calculated by calibration curve of standard addition method. The standards were prepared from known amounts of stock NO3 and NO2 and run in parallel with test samples in each assay.
Determination of intracellular calcium level using CLSM[33,36]
The cells were cultured on the coverslips within the glass bottom of a small cultured dish (No. 0, uncoated, and irradiated. MatTek Co., USA). At the exponential growth period, the cells were stained with 10 μmol·L-1 fluo-3/AM (Molecular Probe) for 30min at 37 °C, and washed with 135 mol·L-1 NaCl, 10 mol·L-1 HEPES, 0.4 mol·L-1 MgCl2, 1 mol·L-1 CaCl2, 1 g·L-1 D-glucose, 1 g·L-1 bovine serum albumin, pH 7.3 at least 3 times. Then the cells were placed in the culture medium 199 to maintain them in living state. Before and after administration of As2O3 the fluorescence intensity was determined by CLSM in dynamic changes for up to 900 s. Using scan-time series menu, time series was used to scan some definite cells repeatedly to monitor the dynamic changes in fluorescent intensity of intracellular Ca2+ content over time. The parameters of the CLSM (Ultima 312, Meridian Instruments Inc., USA) were as follows: the excited light 488 nm, the emission light 530 nm and pinhole 10-40 nm. The fluorescent intensity of pixel was collected and managed with the software of the instrument.
RESULTS
Cell apoptosis
Ultrastructural morphological changes of mitochondria in As2O3 treated cells were described in the previous reports[25,26]. Cells treated with As2O3 at different concentrations for 24 h displayed an apoptotic appearance. Under electron microscope, condensed and marginated chromatins in most of the nuclei appeared accompanying swelling mitochondria (Figure 1). By flow cytometry, time course study on As2O3 induced apoptosis revealed that apoptotic peak can be observed as early as 12 h after the incubation of arsenic trioxide in 3 μmol·L-1. The apoptotic cells accounted for 5.0% of total cell population at 12 h and 28.3% at 24 h (Figure 2).
Figure 1.
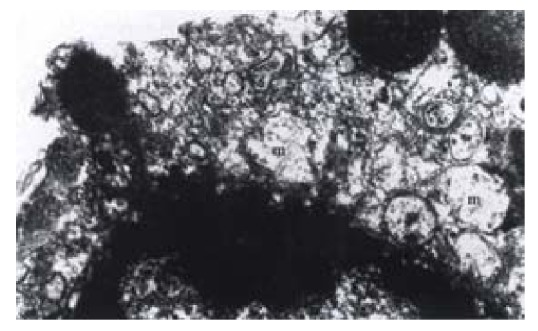
Ultrastructure of SHEEC1 cell treated with 3 μmol·L-1 As2O3. Apoptotic appearance displayed with swelling of mitochondria and nuclear chroma-tin coagulating and merginating. m, mitochondria. n, nucleus. EM, × 7000
Figure 2.
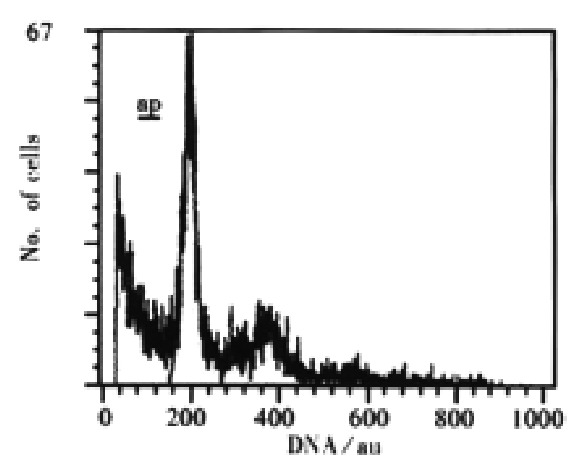
DNA histogram of SHEEC1 cell in 24 h after 3 μmol·L-1 of As2O3 adding. ap, apoptotic peak.
NO determination
When As2O3 acted on the SHEEC1 for 2-24 h, in 0, 1, 3 and 5 μmol·L-1 As2O3, NO in cultured medium was increased at the time points. The amount of NO released from SHEEC1 was increased from the basal condition (0.98-1.00 × 10-2 μmol·L-1) up to the high level (1.48-1.52 × 10-2 μmol·L-1) (8h) and maintained for 24 h (Figure 3). The concentration of NO in different groupsvaried, high concentration of NO in 5 μmol·L-1 of As2O3 and low concentration of NO in 1 μmol·L-1 of As2O3.
Figure 3.
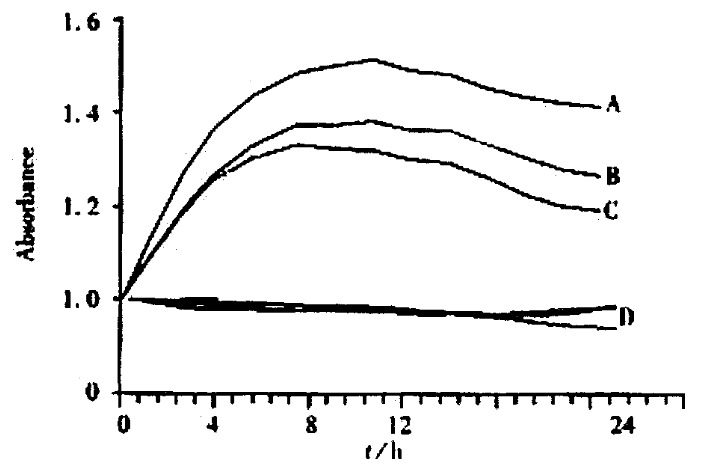
NO determination of SHEEC1 treated with different concentra-tion of As2O3. NO increased markedly in 5 μmol·L-1 of As2O3 group (A), intermediately in 3 μmol·L-1 of As2O3 group (B) and lowly in 1 μmol·L-1 of As2O3 group (C). The control group, 0 μmol·L-1 of As2O3, were remained on the basal lines (D).
Dynamic change calcium of intracellular calcium
To show the time course of changes in Ca2+ in individual cells, the changes in fluorescence intensity (arbitrary unit, au) at different representative cells were measured. Upon the initiation of stimulation by As2O3, all the cells responded with a rapid rise in [Ca2+] from 1.00 au. to 1.09-1.38 au of fluorescent intensity. The peak levels of Ca2+ in all cells were consistently reached at about 900 s after stimulation (Figure 4A). In the control group, without being treated with As2O3, the fluorescent intensity of cell, were remained on the basal line (Figure 4B).
Figure 4.
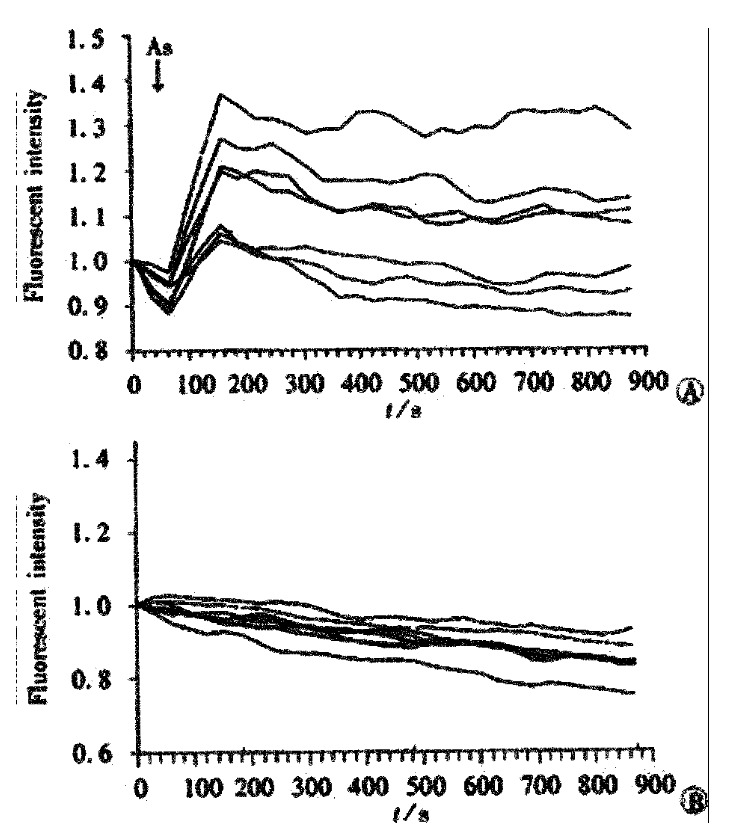
Dynamic changes of intracellular calcium in 7 cells of SHEEC1 treated with As2O3. A, SHEEC1 cells treated with As2O3 in 3 μmol·L-1. B, Control group without adding As2O3.
DISCUSSION
In general, the process of cell apoptosis involved three phases: the initiation phase, the effector phase and the degradation phase[37]. The initiation (or signal transduction) phase is the stage in which specific or non-specificpro-apoptotic signal transduction pathways are activated. The effecter (or central control) phase mainly occurs in the mitochondria[38] where mitochondria membranes are unstable as a result of the action of the permeability alternation. Some genes such as p53 and bcl-2, activate in this phase[39-41]. The degradation (or morphological and biochemical changes) phase manifest the postmitochondrial features of apoptosis, in which soluble intermembrane proteins released from mitochondria played an active role in the activation of proteolytic destruction. In our previous reports, we investigated the early changes of the apoptotic cells induced by As2O3 and defined the phase in which As2O3 was involved[26,27]. Our results demonstrated that As2O3 acted directly on mitochondria for the early stage of apoptosis. The alteration of mitochondria in arsenic trioxide treated tumor cells could be observed as early as 2 h after the treatment[27,30]. In this study we investigated signal messengers of apoptosis, by first selecting both messengers of NO and Ca2+ in the apoptotic pathway.
Experiments on the effects of various modulators (dose and time lag) of arsenic in the level of Ca2+ and NO were carried out. Nitric oxide (NO) is a free radical generated in cells by nitric oxide synthases (NOS)[42]. It is a gaseous inter- and intra-cellular messenger that plays as a signaling molecular in many physiological and pathological processes and it is also a cytotoxic agent involved in many diseases, whichhas been elaborated extensively during the last decade. Various intra- or extra-cellular factors act on mitochondria to produce NO. NO binds to cytochrome oxidase[43], blocks respiratory chain and induces apoptosis[44,45].
Cells themselves control intracellular Ca2+ concentration ([Ca2+]) strictly with several Ca2+ regulatory mechanisms, such as Ca2+ channels, Ca2+ pumps, and Ca2+ exchangers. The role of calcium is as the important intracellular signal element in regulating cell death[46]. As revealed by previous reports, it seems that calcium changes in apoptosis vary with stimuli and cell lines[47]. This data suggested that an early, gradual and sustained increase in intracellular Ca2+ is necessary for the appearance of apoptotic characteristics. In the examination of CLSM with Fuo-3 AM as a calcium indicator, we found that a rise in intracellular calcium was elicited at once after application of As2O3. The mechanism of how arsenic increases intracellular calcium levels was not clear at this moment. Arsenic has been shown to disrupt mitochondria and may elevate intracellular calcium via a signal transduction pathway. Arsenite has also been reported to activate protein kinase C and mitogen-activated protein kinase[48]. These kinases are known to be involved in the calcium signal transduction pathway.
According to the previous reports, the relationship between NO, Ca2+ and mitochondria in apoptosis is as follows:various extracellular factors can induce the increase of intracellular Ca2+ levels ([Ca2+]i), modulating cellular signaling and gene expression, and the increased ([Ca2+]i) effect on NO production through the iNOS pathway[49,50]; mitochondria are a source of NO[51], the production of which may affect energy metabolism, O2 consumption and O2 free radical formation[52]; mitochondrial Ca2+ uptake in combination with NO production triggers the collapse of mitochondrial membrane potential, affecting mitochondrial respiration and culminating in delayed cell death[53].
In conclusion, our data proved that increased calcium ions and nitric oxide triggered by As2O3 may play an important role in arsenite-induced apoptosis in esophageal carcinoma cells. The demonstration of the involvement of Ca2+ and NO in arsenite-induced apoptosis suggests a new direction for studying the apoptotic pathway.
Footnotes
Supported by the National Natural Science Foundation of China. No. 39830380
Edited by Ma JY
References
- 1.Matsui M, Nishigori C, Toyokuni S, Takada J, Akaboshi M, Ishikawa M, Imamura S, Miyachi Y. The role of oxidative DNA damage in human arsenic carcinogenesis: detection of 8-hydroxy-2'-deoxyguanosine in arsenic-related Bowen's disease. J Invest Dermatol. 1999;113:26–31. doi: 10.1046/j.1523-1747.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 2.Goering PL, Aposhian HV, Mass MJ, Cebrián M, Beck BD, Waalkes MP. The enigma of arsenic carcinogenesis: role of metabolism. Toxicol Sci. 1999;49:5–14. doi: 10.1093/toxsci/49.1.5. [DOI] [PubMed] [Google Scholar]
- 3.Schaumlöffel N, Gebel T. Heterogeneity of the DNA damage provoked by antimony and arsenic. Mutagenesis. 1998;13:281–286. doi: 10.1093/mutage/13.3.281. [DOI] [PubMed] [Google Scholar]
- 4.Ho IC, Yih LH, Kao CY, Lee TC. Tin-protoporphyrin potentiates arsenite-induced DNA strand breaks, chromatid breaks and kinetochore-negative micronuclei in human fibroblasts. Mutat Res. 2000;452:41–50. doi: 10.1016/s0027-5107(00)00035-x. [DOI] [PubMed] [Google Scholar]
- 5.Gebel T. Suppression of arsenic-induced chromosome mutagenicity by antimony. Mutat Res. 1998;412:213–218. doi: 10.1016/s1383-5718(97)00181-2. [DOI] [PubMed] [Google Scholar]
- 6.Gebel T, Birkenkamp P, Luthin S, Dunkelberg H. Arsenic(III), but not antimony(III), induces DNA-protein crosslinks. Anticancer Res. 1998;18:4253–4257. [PubMed] [Google Scholar]
- 7.Zhu XH, Shen YL, Jing YK, Cai X, Jia PM, Huang Y, Tang W, Shi GY, Sun YP, Dai J, et al. Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. J Natl Cancer Inst. 1999;91:772–778. doi: 10.1093/jnci/91.9.772. [DOI] [PubMed] [Google Scholar]
- 8.Look AT. Arsenic and apoptosis in the treatment of acute promyelocytic leukemia. J Natl Cancer Inst. 1998;90:86–88. doi: 10.1093/jnci/90.2.86. [DOI] [PubMed] [Google Scholar]
- 9.Shao W, Fanelli M, Ferrara FF, Riccioni R, Rosenauer A, Davison K, Lamph WW, Waxman S, Pelicci PG, Lo Coco F, et al. Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer Inst. 1998;90:124–133. doi: 10.1093/jnci/90.2.124. [DOI] [PubMed] [Google Scholar]
- 10.Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- 11.Tamm I, Paternostro G, Zapata JM. Treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1999;340:1043; author reply 1044–1045. doi: 10.1056/NEJM199904013401313. [DOI] [PubMed] [Google Scholar]
- 12.Jing Y, Dai J, Chalmers-Redman RM, Tatton WG, Waxman S. Arsenic trioxide selectively induces acute promyelocytic leukemia cell apoptosis via a hydrogen peroxide-dependent pathway. Blood. 1999;94:2102–2111. [PubMed] [Google Scholar]
- 13.Huang XJ, Wiernik PH, Klein RS, Gallagher RE. Arsenic trioxide induces apoptosis of myeloid leukemia cells by activation of caspases. Med Oncol. 1999;16:58–64. doi: 10.1007/BF02787360. [DOI] [PubMed] [Google Scholar]
- 14.Lallemand-Breitenbach V, Guillemin MC, Janin A, Daniel MT, Degos L, Kogan SC, Bishop JM, de Thé H. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J Exp Med. 1999;189:1043–1052. doi: 10.1084/jem.189.7.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li YM, Broome JD. Arsenic targets tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res. 1999;59:776–780. [PubMed] [Google Scholar]
- 16.Seol JG, Park WH, Kim ES, Jung CW, Hyun JM, Lee YY, Kim BK. Potential role of caspase-3 and -9 in arsenic trioxide-mediated apoptosis in PCI-1 head and neck cancer cells. Int J Oncol. 2001;18:249–255. [PubMed] [Google Scholar]
- 17.Chen HY, Liu WH, Qin SK. Induction of arsenic trioxide on apoptosis of hepatocarcinoma cell lines. Shijie Huaren Xiaohua Zazhi. 2000;8:532–535. [Google Scholar]
- 18.Gu QL, Li NL, Zhu ZG, Yin HR, Lin YZ. A study on arsenic trioxide inducing in vitro apoptosis of gastric cancer cell lines. World J Gastroenterol. 2000;6:435–437. doi: 10.3748/wjg.v6.i3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tu SP, Jiang SH, Tan JH, Jiang XH, Qiao MM, Zhang YP, Wu YL, Wu YX. Proliferation inhibition and apoptosis induction by arsenic triox-ide on gastric cancer cell SGC 7901. Shijie Huaren Xiaohua Zazhi. 1999;7:18–21. [Google Scholar]
- 20.Xu HY, Yang YL, Gao YY, Wu QL, Gao GQ. Effect of arsenic trioxide on human hepatoma cell line BEL-7402 cultured in vitro. World J Gastroenterol. 2000;6:681–687. doi: 10.3748/wjg.v6.i5.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Qin SK, Chen BA, Chen HY. Experimental study on antitumor effect of arsenic trioxide in combination with cisplatin or doxorubicin on hepatocellular carcinoma. World J Gastroenterol. 2001;7:702–705. doi: 10.3748/wjg.v7.i5.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu HY, Gao YY, Wu QL, Gao GQ, Yang YL, Chen SX, Liu TF. Proliferation inhibition and apoptosis induction by arsenic trioxide on human hepatoma cell line in vitro. Shijie Huaren Xiaohua Zazhi. 2000;8:1233–1237. [Google Scholar]
- 23.Shen ZY, Tan LJ, Cai WJ, Shen J, Chen C, Tang XM, Zheng MH. Arsenic trioxide induces apoptosis of oesophageal carcinoma in vitro. Int J Mol Med. 1999;4:33–37. doi: 10.3892/ijmm.4.1.33. [DOI] [PubMed] [Google Scholar]
- 24.Shen ZY, Tan LJ, Cai WJ, Shen J, Chen CY, Tang XM. Morphologic study on apoptosis of esophageal carcinoma cell line induced by arsenic trioxide. Shijie Huaren Xiaohua Zhahi. 1998;6:226–229. [Google Scholar]
- 25.Shen ZY, Chen CY, Shen J, Cai WJ. Ultrastructural study of apoptosis and necrosis in the esophageal carcinoma cell line induced by arsenic trioxide. Zhongguo Yixue Wulixue Zazhi. 1999;16:91–94. [Google Scholar]
- 26.Shen ZY, Shen J, Chen MH, Li QS, Hong CQ. Morphological changes of mitochondria in apoptosis of esophageal carcinoma cells induced by As2O3. Zhonghua Binlixue Zazhi. 2000;29:200–203. [PubMed] [Google Scholar]
- 27.Shen ZY, Shen J, Cai WJ, Hong C, Zheng MH. The alteration of mitochondria is an early event of arsenic trioxide induced apoptosis in esophageal carcinoma cells. Int J Mol Med. 2000;5:155–158. doi: 10.3892/ijmm.5.2.155. [DOI] [PubMed] [Google Scholar]
- 28.Kroemer G, de Thé H. Arsenic trioxide, a novel mitochondriotoxic anticancer agent. J Natl Cancer Inst. 1999;91:743–745. doi: 10.1093/jnci/91.9.743. [DOI] [PubMed] [Google Scholar]
- 29.Shen ZY, Chen MH, Li QS, Shen J. An ultrastructural study on the programmed cell death of human amniotic epithelium. Dianzi Xianwei Xuebao. 2000;19:259–260. [Google Scholar]
- 30.Shen ZY, Shen J, Li QS, Chen CY, Chen JY, Yi Z. Morphological and functional changes of mitochondria in apoptotic esophageal carcinoma cells induced by arsenic trioxide. World J Gastroenterol. 2002;8:31–35. doi: 10.3748/wjg.v8.i1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rachmilewitz D. Role of nitric oxide in gastrointestinal tract. World J Gastroenterol. 1998;4:28–29. [Google Scholar]
- 32.Shen ZY, Shen WY, Chen MH, Hong CQ, Shen J. Alterations of nitric oxide in apoptosis of esophageal carcinoma cells induced by arsenite. Shijie Huaren Xiaohua Zhahi. 2000;8:1101–1104. [Google Scholar]
- 33.Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol Rev. 1999;79:1089–1125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- 34.Shen Z, Cen S, Shen J, Cai W, Xu J, Teng Z, Hu Z, Zeng Y. Study of immortalization and malignant transformation of human embryonic esophageal epithelial cells induced by HPV18 E6E7. J Cancer Res Clin Oncol. 2000;126:589–594. doi: 10.1007/pl00008469. [DOI] [PubMed] [Google Scholar]
- 35.Shen WY, Chen MH, Shen ZY, Zhang LM. Microspectrophotometric determination of nitric oxide. J Shantou Univ Med College. 2000;13:10–11. [Google Scholar]
- 36.Satoh Y, Nishimura T, Kimura K, Mori S, Saino T. Application of real-time confocal microscopy for observation of living cells in tissue specimens. Hum Cell. 1998;11:191–198. [PubMed] [Google Scholar]
- 37.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 38.Brenner C, Kroemer G. Apoptosis. Mitochondria--the death signal integrators. Science. 2000;289:1150–1151. doi: 10.1126/science.289.5482.1150. [DOI] [PubMed] [Google Scholar]
- 39.Zhang CS, Wang WL, Peng WD, Hu PZ, Chai YB, Ma FC. Promotion of apoptosis of SMMC7721cells by bcl-2 ribozyme. Shijie Huaren Xiaohua Zazhi. 2000;8:417–419. [Google Scholar]
- 40.Yuan RW, Ding Q, Jiang HY, Qin XF, Zou SQ, Xia SS. Bcl-2, p53 protein expression and apoptosis in pancreatic cancer. Shijie Huaren Xiaohua Zazhi. 1999;7:851–854. [Google Scholar]
- 41.Wang LD, Zhou Q, Wei JP, Yang WC, Zhao X, Wang LX, Zou JX, Gao SS, Li YX, Yang C. Apoptosis and its relationship with cell proliferation, p53, Waf1p21, bcl-2 and c-myc in esophageal carcinogenesis studied with a high-risk population in northern China. World J Gastroenterol. 1998;4:287–293. doi: 10.3748/wjg.v4.i4.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuai XL, Ge ZJ, Meng XY, Ni RZ. Expression of nitric oxide synthase in human gastric carcinoma. Shijie Huaren Xianohua Zazhi. 2000;8:22–24. [Google Scholar]
- 43.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, Lin B, Chen G, Lu J, Lin F, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 44.Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 45.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504:46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 46.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529 Pt 1:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang M, Zhang H, Xue S. Role of calcium in apoptosis of HL-60 cells induced by harringtonine. Sci China C Life Sci. 1998;41:600–607. doi: 10.1007/BF02882901. [DOI] [PubMed] [Google Scholar]
- 48.Jun CD, Oh CD, Kwak HJ, Pae HO, Yoo JC, Choi BM, Chun JS, Park RK, Chung HT. Overexpression of protein kinase C isoforms protects RAW 264.7 macrophages from nitric oxide-induced apoptosis: involvement of c-Jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and CPP-32 protease pathways. J Immunol. 1999;162:3395–3401. [PubMed] [Google Scholar]
- 49.Korhonen R, Kankaanranta H, Lahti A, Lähde M, Knowles RG, Moilanen E. Bi-directional effects of the elevation of intracellular calcium on the expression of inducible nitric oxide synthase in J774 macrophages exposed to low and to high concentrations of endotoxin. Biochem J. 2001;354:351–358. doi: 10.1042/0264-6021:3540351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurr JR, Liu F, Lynn S, Jan KY. Calcium-dependent nitric oxide production is involved in arsenite-induced micronuclei. Mutat Res. 1998;416:137–148. doi: 10.1016/s1383-5718(98)00076-x. [DOI] [PubMed] [Google Scholar]
- 51.Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J Biol Chem. 1998;273:11038–11043. doi: 10.1074/jbc.273.18.11038. [DOI] [PubMed] [Google Scholar]
- 52.Nishikawa M, Takeda K, Sato EF, Kuroki T, Inoue M. Nitric oxide regulates energy metabolism and Bcl-2 expression in intestinal epithelial cells. Am J Physiol. 1998;274:G797–G801. doi: 10.1152/ajpgi.1998.274.5.G797. [DOI] [PubMed] [Google Scholar]
- 53.Umansky V, Ushmorov A, Ratter F, Chlichlia K, Bucur M, Lichtenauer A, Rocha M. Nitric oxide-mediated apoptosis in human breast cancer cells requires changes in mitochondrial functions and is independent of CD95 (APO-1/Fas) Int J Oncol. 2000;16:109–117. doi: 10.3892/ijo.16.1.109. [DOI] [PubMed] [Google Scholar]