

Abstract
AIM: To understand the effect of low concentration of N-methyl-N’-nitro-nitrosoguanidine (MNNG), which is a widely distributed environmental mutagen and carcinogen especially for human gastric cancer, on DNA damage and to study its possible pathway in regulating cell cycle arrest.
METHODS: The DNA damage effect was measured by Comet assay. A specific phospho-(Ser/Thr) ATM/ATR substrate antibody was used to detect the damage sensor by Western blot. p38 kinase activity was measured by direct kinase assay, and immunoprecipitation for the possible connection between ATM/ATR and p38 MAPK. Flow cytometry analysis and p38 MAPK specific inhibitor SB203580 were combined to detect the possible cell cycle arrest by p38 MAPK.
RESULTS: With the same low concentration MNNG exposure (0.2 μM 2.5 h), Comet assays indicated that strand breaks accumulated, Western blot and kinase assay showed ATM/ATR and p38 kinase activated, immunoprecipitation showed phospho-ATM/ATR substrate antibody combined with both p38 MAPK antibody and phospho-p38 MAPK antibody. p38 MAPK pathway was involved in the G1-S arrest.
CONCLUSION: Activation of ATM/ATR by MNNG induced DNA damage leads to activation of p38 MAPK, which involves in the G1 checkpoint in mammalian cells.
INTRODUCTION
Human beings are exposed to a multitude of carcinogens in their environment, and most cancers are considered to be chemically induced. Monofunctional alkylating agents like N-methyl-N’-nitro-Nitrosoguanidine (MNNG) are widely distributed environmental mutagens and carcinogens that, on activation, react with DNA and proteins and generate adducts. The main target of MNNG is believed to be chromosomal DNA damage, which in turn would provide the primary signal, triggering the DNA damage response that involves coordinate control of multiple signal transduction pathways[1-4]. MNNG is responsible for human gastric cancer, and thus MNNG-induced signal transduction should be most relevant to human gastric carcinogenesis. Since the concentration we used was close to the actual environment concentration, the results would be of much practical significance.
The complex network of DNA damage sensors, signal transmitters, and effectors (checkpoints) is evolved in all eukaryota[5]. The top level of sensors/transmitters in the signal transduction cascade that responds to DNA strand breaks is the members of the phosphatidylinositol 3-kinase family: ataxia-telangiectasia-mutated protein (ATM), ATM- and Rad3-related protein (ATR), and DNA-dependent protein kinase (DNA-PK)[6-8]. Recent findings indicate that ATM activation is not limited to the ionizing radiation-induced response and potentially plays an important role in response to DNA alkylation[9].
Some evidences have shown that ATM-dependent p53 and c-Jun N-terminal kinase (JNK) pathways are linked to UVA-induced apoptosis. On the other hand, UVC-induced apoptosis occurs through ATR-dependent p53 phosphorylation as well as the JNK pathway[10]. Recently, other data suggest a model in which activation of ATM by gamma irradiation leads to the activation of MKK6, and p38 gamma, and is essential for the proper regulation of the G (2) checkpoint in mammalian cells. But there is no report about the effect of alkylating agents on the possible ATM/ATR-p38MAPK cascade[11].
Based on the above findings, we wanted to know whether low dose of MNNG could damage DNA, what possible function of DNA damage sensor ATM/ATR was in the damage reaction, what the relationship was between ATM/ATR and p38 MAPK, and what effect was on cell cycle.
MATERIALS AND METHODS
Cell culture and MNNG treatment
African green monkey kidney Vero cells cultivated as monolayer in DMEM with 10% (v/v) heat inactivated fetal calf serum and 100 U/mL penicillin, 100 mg/mL streptomycin in a humidified 5% CO2 incubator at 37 °C. For MNNG treatment, the medium was replaced by serum-free DMEM containing 0.2 μmol/L MNNG with 0.2% solvent DMSO in the medium for indicated period of time (2.5 h). Cells treated in the same way with 0.2% (v/v) DMSO served as solvent control.
Comet assay
A base layer of 1.0% agarose was placed on microscope slides and allowed to harden. 75 μL of 1% low melting point agarose (37 °C) diluted in deionized H2O was mixed with 1.0 × 104 treated or untreated cells (5-10 μL in volume) and applied to the coated slide. A glass coverslip was then overlaid on the cell layer, and the agarose was allowed to solidify. The coverslip was then removed, and a third layer of low melting point agarose (75 μL) was added to the slide. Again, a coverslip was overlaid, and the agarose was allowed to solidify. After this, the coverslip was removed, and the slides were placed in a lysis solution (10 mM Tris, pH10.0, 2.5 M NaCl, 100 mM EDTA, 1% Triton X-100, 10% Me2SO) at 4 °C overnight.
The slides were then transferred to an electrophoresis apparatus containing an alkaline solution consisting of 300 mM NaOH and 1 mM EDTA. The slides remained in this solution for 1 h to promote DNA unwinding and were finally subjected to electric current (200 mA) for 1 h. Then the slides were removed, washed three times for 5 min in neutralizing buffer (0.4 M Tris-HCl, pH7.5) at room temperature, and stained in 50 μL 20 μg/mL dilution of EB. The stained nuclei were subsequently examined and photographed.
Western blotting
Protein bands in gels were transferred to nitrocellulose (NC) membranes for 90 min under 100 voltages. After that, all performances about the membranes including washing, primary antibody and horseradish peroxides (HRP) conjugated antibody interactions, enhanced chemiluminescence (ECL) and exposure to films were carried out according to the instruction manual provided by the manufacturer. Bands emerged on films were scanned with a scanning densitometer and quantitated with Kodak 1 D Analysis 2.0 software. Assuming the absorbance of band of DMSO control as 1, the ratio between band of MNNG treatment group and DMSO treatment group of the same kinase on the film was calculated by comparing the relative absorbances of these two bands.
Assay for p38MAPK activity
p38 kinase assay was carried out as described by the protocol of cell signaling technologies. In brief, the Vero cells were washed twice with ice-cold phosphate-buffered saline and exposed to 0.2 μmol/L MNNG for 2.5 h. Then, the cells were washed once with ice-cold phosphate-buffered saline and lysed in 500 μl of lysis buffer per sample (20 mmol/L Tris, pH7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 1 mg/mL leupeptin, 1 mmol/L PMSF). The lysates were sonicated and centrifuged and the supernatant (which contained 200 μg total protein) was incubated with an immobilized phospho-p38 kinase antibody (Thr 180/Tyr 182) with gentle shocking overnight at 4 °C. The beads were washed twice with 500 mL of lysis buffer and twice with 500 μl of kinase buffer (25 mmol/L Tris, pH7.5, 5 mmol/L β-glycerophosphate, 2 mmol/L DTT, 0.1 mmol/L Na3VO4, 10 mmol/L MgCl2). The kinase reactions were carried out in the presence of 200 μmol/L ATP, and 2 μg of ATF-2 at 30 °C for 30 min. ATF-2 phosphorylation was selectively measured by Western immunoblotting using a chemiluminescent detection system and specific antibodies against phosphorylation of ATF-2 at Thr71.
Immunoblot analysis
The cells were harvested by trypsinization, and the extracts were prepared by resuspending cell pellets in lysis buffer per sample (20 mmol/L Tris, pH7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 1 mg/mL leupeptin, 1 mmol/L PMSF). The lysates were sonicated and centrifuged and the supernatant (which contained 200 μg total protein) was incubated with a specific phospho-(Ser/Thr) ATM/ATR substrate antibody with gentle shocking overnight at 4 °C. Followed by adding 20 μL protein A agarose at 4 °C 2 h, the lysates were centrifuged at 1500 × g for 1 min at room temperature. The bands were washed five times with 500 mL of lysis buffer. The protein concentrations were determined using the Bradford assay. Prior to electrophoresis, an appropriate volume of 20 μL 3 × SDS sample buffer (150 mM Tris-HCl, pH6.8, 10% β-mercaptoethanol, 20% glycerol, 3% SDS, 0.01% bromphenol blue, 0.01% pyronin-Y) were boiled for 3 min, then it was subjected to SDS-PAGE on 10% polyacrylamide gels and electrotransferred onto nitrocellulose membranes. The membranes were probed with antibodies against total p38MAPK, phospho-p38MAPK (cell signaling technology). Quantitation of immunoblot signals was performed by densitometry of exposed X-ray films.
Flow cytometry analysis
Was carried out according to the instruction manual provided by the manufacturer.
RESULTS
Accumulation of strand breaks indicated by Comet assays
After treatment with 0.2 μM MNNG 2.5 h, Comet assay (single cell gel electrophoresis, SCGE) showed significant Comet tail formation, similar to the positive control group (4NQO 2.5 mM 30 min). While the control group, both the DMSO treatment group and PBS treatment group, showed no Comet tail formation (Figure 1). Evidently even low concentration of MNNG (0.2 μM) could cause DNA strand broken.
Figure 1.
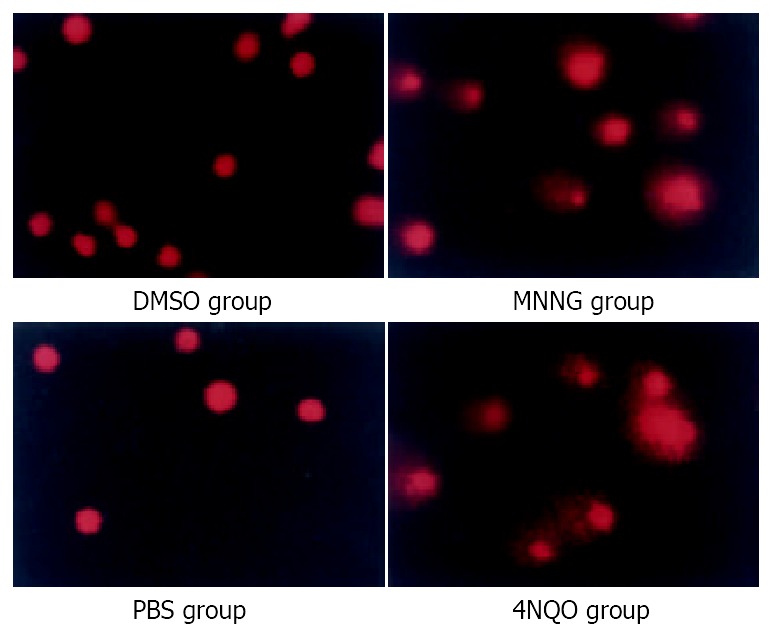
Comet assays indicated accumulation of strand breaks. With low concentration MNNG 0.2 μM 2.5 h treatment, SCGE showed significant comet tail formation, just like the positive control group (4NQO 2.5 mM 30 min). While the control group, both the DMSO treatment group and PBS treatment group, showed no comet tail formation.
ATM/ATR activated by low concentration MNNG exposure
To test if ATM/ATR was activated in Vero cells following 0.2 μM MNNG exposure, we selected specific phospho-(Ser/Thr) ATM/ATR substrate antibody. We observed increased phosphorylation of ATM/ATR substrate after 2.5 h treatment. The molecular weight of one activated substrate was about 38 kDa. Pretreatment with 100 μM wortmannin abrogated the upregucation. Positive control 4-NQO 2.5 μM for 30 minutes showed similar activated band as the MNNG treatment group (Figure 2). ATM/ATR up-regulation/phosphorylation suggested that DNA strand breaks arising during the repair process activate ATM. These findings indicated that ATM activation was not limited to the ionizing radiation-induced response and potentially played an important role in response to DNA alkylation.
Figure 2.
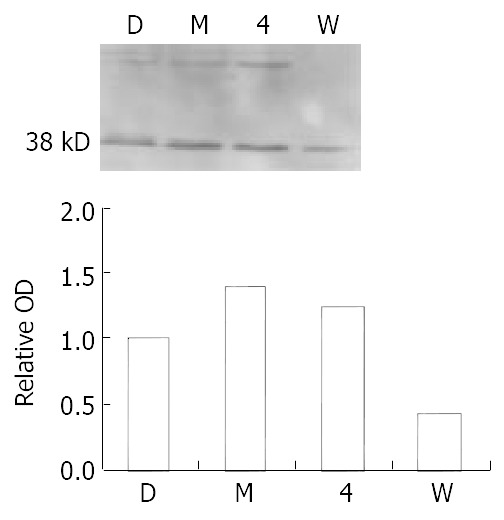
MNNG exposure activates ATM/ATR in Vero cells. Increased phosphorylation of ATM/ATR was observed after 0.2 μM MNNG 2.5 h exposure. One activated substrate mo-lecular weight is about 38 kDa. Pretreatment with 100 μM wortmannin abrogated the upregulation. Positive control 4-NQO 2.5 mM 30 minutes.
p38 kinase activated by MNNG treatment
Results showed the exposure to 0.2 μmol/L MNNG for 2.5 h, p38 kinase in Vero cells was activated by about 1.47 fold (Figure 3).
Figure 3.
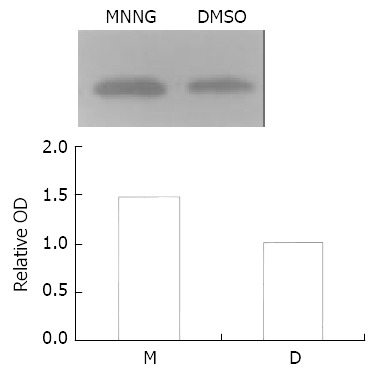
MNNG treatment activated p38 kinase. p38 kinase in Vero cells was activated by about 1.47 fold after the exposure to 0.2 μmol/L MNNG for 2.5 h.
ATM/ATR-P38MAPK pathway activated by MNNG
With same treatment (0.2 μM MNNG 2.5 h), immunoprecipitation (IP) showed phospho-ATM/ATR substrate antibody combined with both p38 MAPK antibody and phospho-p38MAPK antibody. The band of MNNG group was 1.78 (p38MAPK) and 2.37 fold (phospho-p38MAPK) stronger than that of DMSO group (Figure 4). Combined with the result of P38MAPK activated by the same treatment, we concluded that ATM/ATR-P38MAPK pathway was activated by MNNG.
Figure 4.
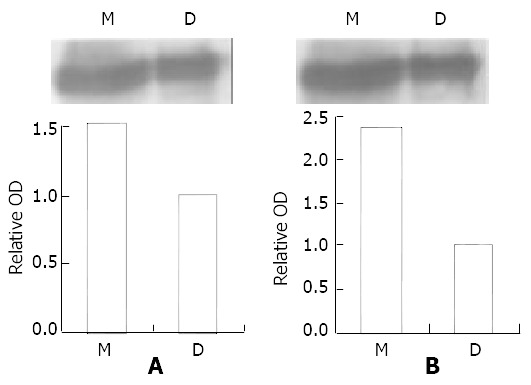
ATM/ATR-P38MAPK pathway is activated by MNNG. Immunoprecipitation showed phospho-ATM/ATR substrate antibody connected with both the p38 MAPK antibody and the phospho-p38MAPK antibody. The band of MNNG group is 1.78 fold (p38MAPK) and 2.37 fold (phospho-p38MAPK) stron-ger than DMSO group. Combined with the result that P38MAPK was activated by the same treatment, we conclude that ATM/ATR-P38MAPK pathway is activated by MNNG.
P38MAPK involved in G1-S arrest
In the flow cytometry analysis (Figure 5), it was found that the population of S phase of cell cycle in MNNG treatment group was decreased as compared with the controls (DMSO group). Pretreatment with p38MAPK specific inhibitor SB203580 for 1 h, the G1-S arrest disappeared after the same MNNG treatment (Table 1). It implied that P38MAPK was involved in the G1-S arrest.
Figure 5.
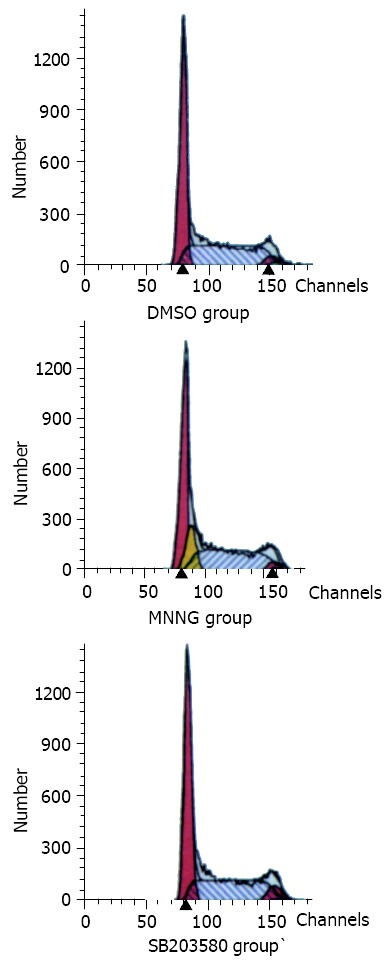
P38MAPK joined G1-S arrest. In the flow cytometry analysis work, it was found that the population of S phase of cell cycle in MNNG treatment group was decreased as com-pared with the controls (DMSO group). Pretreatment with p38MAPK specific inhibitor SB203580 for 1 h, the G1-S arrest disappeared after the same MNNG treatment.
Table 1.
Cell cycle analysis by flow cytometer
| Cell |
Cell cycle phase fraction (%) |
||
| G0-G1 | S | G2-M | |
| DMSO group | 50.22 | 42.24 | 4.54 |
| MNNG group | 90.36 | 0.00 | 9.64 |
| SB203580 group | 51.78 | 41.79 | 6.43 |
DISCUSSION
Genotoxic events activate a number of signaling pathways that serve, for example, to activate DNA repair mechanisms, halt cell cycle progression and/or trigger advancement into apoptosis. Although all genotoxins produce such general responses, the mechanisms governing response to divergent forms of DNA damage are potentially diverse themselves. γ-irradiation produces double and single strand breaks as well as numerous oxidative changes to bases and deoxyribose moieties, whereas MNNG alkylates (methylates) several nucleophilic centers in bases but does not directly induce strand breaks. MNNG exposure produces mutagenic and cytotoxic O6-methylguanine (O6MeG) adducts that can force O6MeG-T mispairing following replication. In addition to O6MeG, MNNG causes base alkylation at numerous nucleophilic centers within DNA, such as the N3 position of adenine. The presence of such adducts triggers DNA glycosylases to generate apurinic/apyrimidinic sites during repair. Apurinic/apyrimidinic sites, in turn, activate an apurinic/apyrimidinic-specific endonuclease resulting in cleavage of DNA.
MNNG can activate JNK/SAPK in human 293 cells at concentrations as high as 70 μmol/L[12]. By our experience, MNNG at concentration of 20 μmol/L is enough to kill over 80% Vero cells while at concentration of 0.2 μmol/L the highest nontargeted mutation frequency is induced without remarkable mortality[13]. Although it has been verified that with ultraviolet and high concentration of chemical DNA damaging agents, signal transduction is activated not only by damaged but also by undamaged DNA pathways. Insufficient knowledge has been obtained on details of cellular response to low concentration of chemicals especially MNNG. Comet assay (Single cell gel electrophoresis assay, SCGE) is a new and sensitive test for DNA damage studies[14-19]. In our study, with low concentration of MNNG 0.2 μM 2.5 h treatment, Comet assay showed significant Comet tail formation, indicating that strand breaks are accumulated. So even with very low concentration of 0.2 μM of MNNG, DNA strand can also be broken.
It is widely believed that it is the presence of broken DNA strands that activates the catalytic activity of ATM. The ATM gene is mutated in ataxia-telangiectasia, a pleiotropic autosomal recessive disorder characterized by progressive cerebellar degeneration, oculocutaneous telangiectasia, immunodeficiency, cancer predisposition, and an extreme sensitivity to ionizing radiation (IR). Cells derived from ataxia-telangiectasia patients exhibit chromosomal instability and a profound defect in all cellular responses to DNA double strand breaks (DSBs). ATM is a serine/threonine protein kinase that is located mainly in the cell nucleus. Upon infliction of DSBs, ATM mediates the rapid induction of numerous cellular responses that lead to damage repair, and activation of cell cycle checkpoints and other survival pathways[20,21]. To test if ATM/ATR was activated in Vero cells following 0.2 μM MNNG exposure, we selected specific phospho-(Ser/Thr) ATM/ATR substrate antibody. We observed increased phosphorylation of ATM/ATR substrate after 2.5 h treatment. The molecular weight of one activated substrate was about 38 kDa. Pretreatment with 100 μM wortmannin abrogated the upregulation while in positive control it showed similar activated bands as in the MNNG treatment group. ATM/ATR up-regulation/phosphorylation suggested that DNA strand breaks activated ATM/ATR. These findings also indicate that ATM activation is not limited to the ionizing radiation-induced response and potentially plays an important role in response to DNA alkylation.
Downstream targets of ATM/ATR kinase activity that partially delineate DNA damage-activated cell cycle checkpoint signaling pathways have been recently described. The best characterized checkpoint pathways involve p53, Chk1, Chk2, c-Abl, and BRCA1[7,22-27], but the participating signaling pathways are less clear[28,29]. The complex phenotype of AT cells suggests that it must have other cellular substrates as well[30,31]. Our Western blot result showed that the molecular weight of one activated phosphorylated ATM/ATR substrate was about 38 kDa. To identify the possible substrates for ATM and the related kinases ATR, we selected immunoprecipitation test and found that phospho-ATM/ATR substrate antibody was connected with both p38 MAPK antibody and phospho-p38MAPK antibody. By in vitro kinase assays, with the same MNNG treatment, p38 MAPK was activated. Taken together, we conclude that ATM/ATR-P38MAPK pathway is activated by MNNG.
MAPKs (mitogen-activated protein kinase) are evolutionarily conserved enzymes connecting cell surface receptors to critical regulatory targets with in cells. MAPKs respond to chemical and physical stresses, thereby controlling cell survial and adaptation. It is becoming clear that MAPKs regulate almost all cellular processes, from gene expression to cell death[32-35]. Mammals express at least four distinctly regulated groups of MAPKs, extracellular signal related kinase (ERK)-1/2, C-Jun N-terminal kinase (JNK1/2/3), p38 MAPK and ERK5. Among MAPKs, both JNK/SAPK and p38MAPK play important roles in cellular stress signal transduction. The involvement of MAP kinases in cell cycle arrest has been studied in a number of organisms. Perhaps due to differences in substrate specificity and regulation among the MAP kinases, their roles in cell cycle regulation appear to be different. Activation of p42MAPK is required for the G2/M transition in the maturation of Xenopus oocytes[36]. BMK1 (ERK5) was reported to be required for epidermal growth factor-induced progression through S phase[37]. p38α has been reported to be involved in Cdc42-induced G1 arrest as well as the spindle assembly checkpoint[38,39]. In our experiment, treatment of Vero cells with SB203580, a selective inhibitor of the p38 MAPK pathway, effectively inhibited the G1-S arrest which could be induced by MNNG. These data support an important interplay between the p38 pathway and G1 cell cycle checkpoint control.
In conclusion, low concentration of MNNG can damage DNA, activate ATM/ATR-p38MAPK cascade and induce G1-S arrest. To our knowledge, this is the first report about the involvement of the ATM/ATR-p38MAPK cascade in the G1-S arrest induced by monofunctional alkylating agent MNNG.
Footnotes
Supported by Natural Science Foundation of Zhejiang Province, No. 29801
Edited by Zhu LH and Wang XL
References
- 1.Schär P. Spontaneous DNA damage, genome instability, and cancer--when DNA replication escapes control. Cell. 2001;104:329–332. doi: 10.1016/s0092-8674(01)00220-3. [DOI] [PubMed] [Google Scholar]
- 2.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 3.Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 4.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 5.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 6.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 7.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 8.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 9.Adamson AW, Kim WJ, Shangary S, Baskaran R, Brown KD. ATM is activated in response to N-methyl-N'-nitro-N-nitrosoguanidine-induced DNA alkylation. J Biol Chem. 2002;277:38222–38229. doi: 10.1074/jbc.M204409200. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Ma WY, Kaji A, Bode AM, Dong Z. Requirement of ATM in UVA-induced signaling and apoptosis. J Biol Chem. 2002;277:3124–3131. doi: 10.1074/jbc.M110245200. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S, Han J. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20:4543–4552. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilhelm D, Bender K, Knebel A, Angel P. The level of intracellular glutathione is a key regulator for the induction of stress-activated signal transduction pathways including Jun N-terminal protein kinases and p38 kinase by alkylating agents. Mol Cell Biol. 1997;17:4792–4800. doi: 10.1128/mcb.17.8.4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Yu Y, Chen X. Evidence for nontargeted mutagenesis in a monkey kidney cell line and analysis of its sequence specificity using a shuttle-vector plasmid. Mutat Res. 1994;323:105–112. doi: 10.1016/0165-7992(94)90083-3. [DOI] [PubMed] [Google Scholar]
- 14.Kassie F, Parzefall W, Knasmüller S. Single cell gel electrophoresis assay: a new technique for human biomonitoring studies. Mutat Res. 2000;463:13–31. doi: 10.1016/s1383-5742(00)00041-7. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber V, Amé JC, Dollé P, Schultz I, Rinaldi B, Fraulob V, Ménissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 16.Héron-Milhavet L, Karas M, Goldsmith CM, Baum BJ, LeRoith D. Insulin-like growth factor-I (IGF-I) receptor activation rescues UV-damaged cells through a p38 signaling pathway. Potential role of the IGF-I receptor in DNA repair. J Biol Chem. 2001;276:18185–18192. doi: 10.1074/jbc.M011490200. [DOI] [PubMed] [Google Scholar]
- 17.Klaude M, Eriksson S, Nygren J, Ahnström G. The comet assay: mechanisms and technical considerations. Mutat Res. 1996;363:89–96. doi: 10.1016/0921-8777(95)00063-1. [DOI] [PubMed] [Google Scholar]
- 18.Fairbairn DW, Olive PL, O'Neill KL. The comet assay: a comprehensive review. Mutat Res. 1995;339:37–59. doi: 10.1016/0165-1110(94)00013-3. [DOI] [PubMed] [Google Scholar]
- 19.McKelvey-Martin VJ, Green MH, Schmezer P, Pool-Zobel BL, De Méo MP, Collins A. The single cell gel electrophoresis assay (comet assay): a European review. Mutat Res. 1993;288:47–63. doi: 10.1016/0027-5107(93)90207-v. [DOI] [PubMed] [Google Scholar]
- 20.Pandita TK, Lieberman HB, Lim DS, Dhar S, Zheng W, Taya Y, Kastan MB. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene. 2000;19:1386–1391. doi: 10.1038/sj.onc.1203444. [DOI] [PubMed] [Google Scholar]
- 21.Khanna KK. Cancer risk and the ATM gene: a continuing debate. J Natl Cancer Inst. 2000;92:795–802. doi: 10.1093/jnci/92.10.795. [DOI] [PubMed] [Google Scholar]
- 22.Caspari T. How to activate p53. Curr Biol. 2000;10:R315–R317. doi: 10.1016/s0960-9822(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 23.Turenne GA, Paul P, Laflair L, Price BD. Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal serine residues of p53. Oncogene. 2001;20:5100–5110. doi: 10.1038/sj.onc.1204665. [DOI] [PubMed] [Google Scholar]
- 24.Saito S, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E, Anderson CW. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem. 2002;277:12491–12494. doi: 10.1074/jbc.C200093200. [DOI] [PubMed] [Google Scholar]
- 25.Ye R, Bodero A, Zhou BB, Khanna KK, Lavin MF, Lees-Miller SP. The plant isoflavenoid genistein activates p53 and Chk2 in an ATM-dependent manner. J Biol Chem. 2001;276:4828–4833. doi: 10.1074/jbc.M004894200. [DOI] [PubMed] [Google Scholar]
- 26.Kharbanda S, Yuan ZM, Weichselbaum R, Kufe D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene. 1998;17:3309–3318. doi: 10.1038/sj.onc.1202571. [DOI] [PubMed] [Google Scholar]
- 27.Gatei M, Scott SP, Filippovitch I, Soronika N, Lavin MF, Weber B, Khanna KK. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000;60:3299–3304. [PubMed] [Google Scholar]
- 28.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme. Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 29.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 30.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 31.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–37543. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 32.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 33.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 35.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 36.Palmer A, Gavin AC, Nebreda AR. A link between MAP kinase and p34(cdc2)/cyclin B during oocyte maturation: p90(rsk) phosphorylates and inactivates the p34(cdc2) inhibitory kinase Myt1. EMBO J. 1998;17:5037–5047. doi: 10.1093/emboj/17.17.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- 38.Molnár A, Theodoras AM, Zon LI, Kyriakis JM. Cdc42Hs, but not Rac1, inhibits serum-stimulated cell cycle progression at G1/S through a mechanism requiring p38/RK. J Biol Chem. 1997;272:13229–13235. doi: 10.1074/jbc.272.20.13229. [DOI] [PubMed] [Google Scholar]
- 39.Takenaka K, Moriguchi T, Nishida E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science. 1998;280:599–602. doi: 10.1126/science.280.5363.599. [DOI] [PubMed] [Google Scholar]