

Abstract
Current treatment methods for advanced head and neck squamous cell carcinoma (HNSCC) include surgery, radiation therapy and chemotherapy. For recurrent and metastatic HNSCC, cisplatin is the most common treatment option, but most of patients will eventually develop cisplatin resistance. Therefore, it is imperative to define the mechanisms involved in cisplatin resistance and find novel therapeutic strategies to overcome this deadly disease. In order to determine the role of nuclear factor-kappa B (NF-κB) in contributing to acquired cisplatin resistance in HNSCC, the expression and activity of NF-κB and its upstream kinases, IKKα and IKKβ, were evaluated and compared in three pairs of cisplatin sensitive and resistant HNSCC cell lines, including a pair of patient derived HNSCC cell line. The experiments revealed that NF-κB p65 activity was elevated in cisplatin resistant HNSCC cells compared to that in their parent cells. Importantly, the phosphorylation of NF-κB p65 at serine 536 and the phosphorylation of IKKα and IKKβ at their activation loops were dramatically elevated in the resistant cell lines. Furthermore, knockdown of NF-κB or overexpression of p65-S536 alanine (p65-S536A) mutant sensitizes resistant cells to cisplatin. Additionally, the novel IKKβ inhibitor CmpdA has been shown to consistently block the phosphorylation of NF-κB at serine 536 while also dramatically improving the efficacy of cisplatin in inhibition of cell proliferation and induction of apoptosis in the cisplatin resistant cancer cells. These results indicated that IKK/NF-κB plays a pivotal role in controlling acquired cisplatin resistance and that targeting the IKK/NF-κB signaling pathway may provide a possible therapeutic method to overcome the acquired resistance to cisplatin in HNSCC.
Keywords: IKK, NF-κB, HNSCC, IKKβ inhibitors, cisplatin resistance
Introduction
Head and neck squamous cell carcinoma (HNSCC) ranks as the sixth most common cancer worldwide [1-3]. Despite advancements in basic science and improvements in clinical treatment, the overall survival rate for patients with advanced HNSCC still remains poor with a five year survival rate of less than 50% [1-3]. Currently, the standard therapies for advanced head and neck cancer include surgery, radiation therapy and chemotherapy. A combination of all three potential treatments could reduce the rate of recurrence and distant metastasis for patients with regional tumor and lymph node metastasis. For the patients with recurrent and distant metastatic cancer, however, chemotherapy is the only viable option, even though most of patients die within one year [1,4,5]. Therefore, improving the efficacy of conventional chemotherapy is desperately needed to effectively treat patients with late stage HNSCC.
Cisplatin, one of the most common chemotherapeutics for solid tumors, including HNSCC, exerts its antitumor effects through multiple mechanisms, including the generation of DNA lesions followed by the activation of the DNA damage response and the induction of apoptosis [1,4-7]. Despite many patients experiencing excellent results when they first begin taking this drug, cancers in most of patients will eventually develop resistance. Multiple signaling pathways are involved in the development of cisplatin resistance, including mutation or loss of function of tumor suppressor genes such as p53 as well as the overexpression and activation of oncogenic proteins such as HER-2, Aurora-A and members of the Bcl-2 family [1,4-7]. In order to design a mechanism-based strategy to improve the efficacy of cisplatin, it will be important to identify the more critical molecules and signaling pathways that underlie the development of cisplatin resistance.
Cumulative evidence indicates that the nuclear factor-κB (NF-κB) family of transcription factors could be important therapeutic targets in cancer treatments, including HNSCC therapy [1,5]. The NF-κB family is comprised of 5 members, including RelA (p65), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2), which are activated by both canonical and non-canonical signaling pathways. In the canonical pathway, p50 and p65 heterodimers are sequestered in the cytoplasm in an inactive state by IκBα, but, upon stimulation, IκB kinase (IKK) complex comprising IKKα, IKKβ and IKKγ is activated, leading to IκBα phosphorylation by IKKβ, which results in ubiquitination and proteasomal degradation of IκBα as well as subsequent p50 and p65 heterodimer translocation to nucleus. In the nucleus, activated NF-κB induces gene expression to regulate different cellular processes, including cell proliferation, survival, invasion, metastasis and resistance to chemotherapy [5,8-10]. Although well the concept that NF-κB is involved in chemotherapy resistance is universally accepted [11], the exact mechanism by which NF-κB contributes to acquired-cisplatin resistance is not well documented. In addition, no effective IKK and NF-κB inhibitors are used in HNSCC.
In the present study, we investigate the role of IKK/NF-κB in the regulation of acquired resistance to cisplatin in HNSCC. Our data demonstrates that the level of phosphorylated NF-κB at serine 536 in established cisplatin resistant head and neck cell lines is elevated compared to those found in their parental counterparts. Furthermore, the phosphorylation of IKKα and IKKβ, the critical kinases that phosphorylates and activates NF-κB, is also up-regulated in these cells. Moreover, knockdown of NF-κB or expression of NF-κB serine 536 alanine mutant sensitizes the resistant cells to cisplatin-induced apoptosis. As consistently shown, the specific IKKβ inhibitor, CmpdA, inhibits NF-κB phosphorylation at serine 536, while also sensitizing the resistant cells to cisplatin treatment. Combining our various results demonstrate that IKK/NF-κB could be a critical determinant of acquired resistance to cisplatin and that IKK phosphorylation of NF-κB at serine 536 may play a pivotal role in IKK/NF-κB regulation of cisplatin resistance in HNSCC.
Materials and methods
Antibodies
Antibodies were obtained from the following sources: Antibodies against IKKα (CST-2682), IKKβ (CST-8943), cleaved casepase 3 (CST-9664), phospho-IKKα/β (CST-2697), β-tubulin (CST-4466), Lamin A/C (CST-2032), p65 (CST-6956), phospho-p65 (CST-3033), GAPDH (CST-5174). HRP-labeled anti-mouse and anti-rabbit secondary antibodies were from Santa Cruz Biotechnology.
Cell culture and reagents
University of Michigan HNSCC (UM-SCC) cell lines including UM-SCC17B, UM-SCC11A and UM-SCC11B were the generous gift of T.E. Carey (University of Michigan, Ann Arbor, MI, USA). UM-SCC25 and UM-SCC25/CP were initially described by Teicher et al [12] and provided by Dr. J. Lazo, University of Pittsburgh. All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and 100 U/mL penicillin and streptomycin (Gibco). The reagents were obtained from the following sources: Protease and phosphatase inhibitor cocktails were from Roche; CHAPS was from Pierce; cisplatin was purchased from Sigma (P4394).
Small RNA interference
siRNA SMARTpool p65 (catalog # M003533) and non-targeting siRNA D-001210) were purchasedfrom Dharmacon. The cells were transfected with indicated SMARTpool siRNA or nonspecific control pool using DharmaFECT 1 reagent (Dharmacon) according to the manufacturer’s instructions. Briefly, 20 nM final concentration of siRNA was used to transfect cells at 60%-70% confluency. Cells were harvested 48-72 h after siRNA transfection.
Cell lysis and western blot analysis
Cells grown on 100-mm dishes were rinsed twice with cold PBS and then lysed on ice for 20 min in 1 mL of lysis buffer (40 mM Hepes at pH 7.5, 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 0.5 mM orthovanadate, EDTA-free protease inhibitors [Roche]) containing 1% Triton X-100. After centrifugation at 13,000 g for 10 min, samples containing 20-50 μg of protein were resolved by SDS-PAGE, and proteins were transferred to Pure Nitrocellulose Membrane (Bio-Rad), blocked in 5% nonfat milk, and blotted with the indicated antibodies.
Reporter assays
Cells were seeded in six-well plates and 200 ng of 3×κB luciferase reporter and 50 ng of pRL-SV40 (Renilla reporter control) DNA were cotransfected using Lipofectamine and Plus (Invitrogen) following the manufacturer’s instructions. Cells were harvested after 24 h of transfection, and luciferase assays were performed using the Dual Luciferase Assay System (Promega). All transfections were performed in triplicate.
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-etrazolium cellular proliferation assay
Cells were seeded in 96-well plate in triplicate at 3×103 per well and cultured in the presence or absence of cisplatin or the IKKβ inhibitors at the indicated concentrations and time course. Alternatively, cells were transiently transfected with appropriate siRNA and cultured at the indicated time points post-transfection. At the end of each time point, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) compound (Promega) was added for 1 hours at 37°C. Colorimetric readouts were read at 490 nm on a Versamax Microplate Reader (Molecular Devices). IC50 values were calculated using GraphPad Prism 5.
Clonogenic assay
UMSCC17B/CP, UMSCC25/CP, or UMSCC11B cells were plated the day before treatment at 2000 cells per well in a six-well plate. The next day, cells were pre-treated with DMSO or CmpdA for 2 hours and then treated with cisplatin for another 2 hours. After the two hour incubation with cisplatin, medium was replaced DMSO or CmpdA containing media and colonies were allowed to grow. Cells were allowed to form colonies for 10 days. The plates were then gently washed with phosphate-buffered saline and colonies stained with crystal violet. Colonies of cells containing 50 cells or larger were counted.
Luminescence-based caspase-3/7 activity assay
Cells were plated in triplicate at 2×103 per well in white-walled 96-well plates (Becton Dickinson). Cells were transiently transfected with siRNA as described above and treated with the IKK inhibitor and/or cisplatin as indicated in the figure legends. Caspase-3/7 activity was measured at 48 hours post-transfection using the Caspase-Glo 3/7 assay (Promega) according to the manufacturer’s instructions. Caspase-Glo 3/7 assay uses a caspase-3/7 tetrapeptide DEVD substrate that produces a luminescent signal on cleavage. Relative light units were measured on an Lmax Microplate Luminometer (Molecular Devices).
Statistics
Data from the in vitro experiments are expressed as mean ± SD from a minimum of 3 independent experiments. Comparison between groups were carried out by 2-way ANOVA or Student t test, and a P value of less than 0.05 was considered significant.
Results
Establishing and characterizing the cisplatin resistant head and neck cancer cell lines
In order to study cisplatin resistance in head and neck cancer, we first developed a cell line resistant to cisplatin treatment. UMSCC-17B, a human head and neck squamous cell carcinoma acquired from the University of Michigan [12], was cultured in a media with 0.5 μM of cisplatin, escalating to 7.5 μM during a 6 month span. After 6-months of culture and selection, the cells were able to grow freely in the medium with 5 μM of cisplatin. Next, we measured the sensitivity of the parental UMSCC-17B and cisplatin treated cells (UMSCC-17B/CP) to cisplatin as well as the IC50 values through MTT assay. The results showed that the UMSCC-17B cells are relatively sensitive to cisplatin treatment with an IC50 of 5.2 μM, whereas the UMSCC-17B/CP cells are more resistant to cisplatin treatment with an IC50 of 15.7 μM, which is approximately a 3-fold decrease in sensitivity compared to the parental UMSCC-17B cells (Figure 1A, upper panel). Teicher and colleagues previously reported another pair of cisplatin parental/resistant (SCC-25/SCC-25CP) lines [13], which were also used for the present study. We found that the IC50 value for cisplatin is 4.5 μM for parental SCC-25 cells and 10 μM for SCC-25CP cells in our system (Figure 1A, lower panel).
Figure 1.

Characterization of cisplatin resistance in head and neck cancer cell lines. A. IC50 determination of cisplatin in UMSCC-17B, UMSCC-17B/CP, UMSCC-25 and UMSCC-25/CP. Cells were treated with cisplatin (0-100 μM) for 72 hours at 37°C. Representative results from at least three experiments for each of the two treatments are shown. Error bars represent standard error of mean of three wells (*, P < 0.05). B. Cisplatin induced caspase 3/7 activity in parental but not resistant cells. Cells were treated with cisplatin (20 μM) for 48 hours. Caspase 3/7 activity was evaluated following the protocol. The results are representative of three independent experiments (**, P < 0.01). C. Cisplatin induced caspase-3 cleavage in parental but not resistant cells. The indicated cells were treated with cisplatin (0-20 μM) for 24 hours and lysed and cleaved caspase 3 was measured by western blot. The results are representative of three independent experiments.
To confirm the cell proliferation data, we evaluated the effects of cisplatin treatment on apoptosis in the two pairs of cell lines by measuring caspase 3/7 activity. As shown in Figure 1B, 48-hour treatment with 20 μM of cisplatin led to a 2.2 fold increase of caspase 3/7 activity in SCC17B cells and a 4.8 fold increase in SCC-25 cells compared to vehicle treated cells, but there was no increase of Caspase activity in either resistant cell line in response to cisplatin (Figure 1B). Next, the apoptotic response of these two pairs of cell lines to cisplatin treatment was determined by measuring caspase-3 cleavage through western blot. Following 24 hours of cisplatin treatment, both cisplatin-sensitive SCC-17B and SCC-25 cells showed a dose dependent induction of caspase-3 cleavage, but the resistant cells (both SCC-17B/CP and SCC-25/CP) showed no detectable cleaved caspase-3 (Figure 1C). These data verify that the SCC17B/CP cells and SCC-25/CP are more resistant to cisplatin compared to their parental cell lines.
IKKβ and NF-κB signaling is elevated in cisplatin resistant head and neck cancer cells
NF-κB signaling pathways play a critical role in protecting cancer cells from chemotherapy induced apoptosis [1,5,8-11]. Therefore, we determine if the NF-κB pathway is up-regulated in the SCC17B/CP and SCC-25/CP cells. Total cell lysates were prepared and the level of phosphorylation of NF-κB at serine 536, a key marker for NF-κB activity, as well as the total level of p65 was recorded. As shown in Figure 2A, the phosphorylation of p65 at serine 536 is elevated in SCC17B/CP and SCC-25/CP cells compared with those found in the parental cells, but there was no change in the total p65 level. These results demonstrate that NF-κB is activated in acquired cisplatin resistant head and neck cancer cells. The significant increase in the phosphorylation of NF-κB at serine 536 prompted us to explore whether or not the activities of IKKα and IKKβ, the upstream kinases of NF-κB, are elevated in the cisplatin resistant cells. As a result of this, we were able to detect the phosphorylation of IKKα and IKKβ in their activation loop using this specific phosphorylation antibody. The results showed that the phosphorylation of IKKα/β increased in both SCC-17B and SCC-17B/CP cells compared to those found in the parental cells, while the expression of IKKα and IKKβ experienced no change. In addition, the basal phosphorylation of IκBα, another IKK substrate, has also consistently increased, while the expression of IκBα has decreased, in comparison with the parental cells. These results demonstrated that the activities of NF-κB and its upstream kinase, IKKα and IKKβ, are upregulated in cisplatin resistant cells. The next step is determining whether or not IKKα or IKKβ phosphorylates NF-κB at serine 536. The expressions of IKKα, IKKβ and a combination of both are then reduced by siRNA transfection before the phosphorylation of NF-κB at serine 536 was measured. The results showed that knockdown of either IKKα or IKKβ decreases the phosphorylation of NF-κB at serine 536, with IKKβ knockdown appearing slightly more effective, but knockdown both IKKα and IKKβ simultaneously causes a significant reduction in phosphorylation of NF-κB serine 536 (Figure 2B). Since activated NF-κB translocate to the nucleus to regulate NF-κB target genes transcription, we next examined if there was more NF-κB in the nucleus of resistant cells compared to those in the parental cells. The proteins from the cytoplasmic and the nuclear extracts from both the parental as well as the resistant cells were separated by electrophoresis and blotted with either antibody p65, tubulin or Lamin A/C, with the latter two proteins serving as the cytoplasmic and nuclear controls, respectively. The results indicate that more NF-κB remains in the nucleus of the two resistant cells compared with those found in their parental control, while no difference in localization was observed between the parental and resistant cells with tubulin and Lamin A/C (Figure 2C). To further confirm that NF-κB signaling is upregulated in cisplatin resistant cells, we expressed NF-κB reporter in the parental and cisplatin resistant cells and then measured NF-κB luciferase activity. We found that NF-κB luciferase activity in SCC-17B/CP cells were more than twice as high as those found in parental SCC-17B cells, with similar results found in SCC-25 and SCC25/CP cells (Figure 2D). In conclusion, NF-κB/IKKβ pathway is up-regulated in cisplatin resistant HNSCC.
Figure 2.
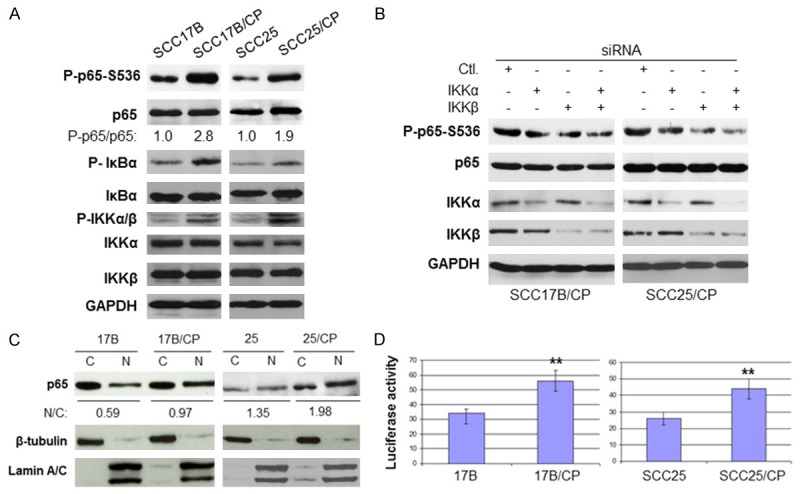
NF-κB/IKK signaling is upregulated in cisplatin resistant HNSCC.A. IKK/NF-κB activity is elevated in the cisplatin resistant cell lines. The levels of phosphorylation of IKKα/β and p65, IKKα, IKKβ and GAPDH in the cells were determined by western blot. Densitometric analysis of 3 independent experiments shows a statistically-significant increase in levels of phosphorylated p65 in both cell lines. B. Cells were transfected with siRNA control, IKKα, IKKβ or IKKα plus IKKβ and blotted with the indicated antibodies. C. NF-κB p65 translocate to nucleus in cisplatin resistant cell lines. The levels of p65, β-tublin and Lamin A/C of the cytoplasmic and nuclear extracts of the cells were determined by immunoblotting with the indicated antibodies. Densitometry analyses for p65 were performed employing ImageJ software (NIH) and presented as ratio of nuclear p65 to cytoplasmic p65 band signal intensity. D. NF-κB reporter activity increased in cisplatin resistant cells. Cells were harvested 24 hours after transfection with luciferase reporter and Renilla reporter control and luciferase assays were performed. The experiments were repeated three times (**, P < 0.01).
Knockdown of NF-κB p65 sensitizes cisplatin resistant cells to cisplatin treatment
Next, we determined precisely how NF-κB is involved in cisplatin resistance in the established cisplatin resistant cells. If NF-κB is vital in protecting against cisplatin-induced cell death, one would expect that NF-κB depletion would re-sensitize the resistant cells to cisplatin-induced cell death. To begin, siRNA to NF-κB p65 was utilized in parallel with a control siRNA. Western blot showed that 48 hours of siRNA p65 treatment led to a dramatic reduction of p65 expression in both SCC-17B/CP and SCC-25/CP cells (Figure 3A). Next, we tested for the effects of knocking down p65 protein expression on apoptosis in response to cisplatin treatment by measuring caspase 3/7 activity. SCC17B/CP cells were treated with either siRNA control or siRNA p65 before (48 hours post transfection) being exposed to either 20 μM cisplatin or vehicle control for 24 hours, and caspase activity was measured. As shown in Figure 3B, after either vehicle or 20 μM cisplatin treatment, the induction of caspase 3/7 activity was similar in siRNA control treated cells (SCC-17B/CP and SCC25/CP), whereas the caspase 3/7 activity was significantly higher in siRNA p65 treated cells, with 1.5 fold and 3 fold increases in p65 knockdown17-B/CP and SCC-25/CP cells respectively (Figure 3B). In a parallel experiment, with the same cisplatin treatment, Caspase-3 cleavage was measured by western blot in either siRNA control or siRNA p65 treated cells. We found that siRNA against p65 or cisplatin treatment alone does not induce caspase-3 cleavage in both SCC-17B/CP and SCC-25/CP cells, whereas the combination of siRNA p65 and cisplatin treatment causes obvious caspase-3 cleavage (Figure 3C). These results suggested that NF-κB is involved in acquired cisplatin resistance.
Figure 3.
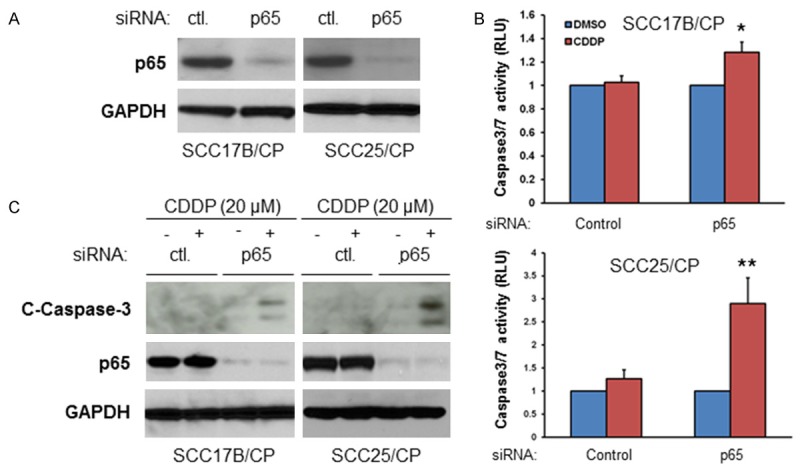
Depletion of NF-κB sensitizes cisplatin resistant cells to cisplatin treatment. A. The siRNA against p65 is effective in reducing p65 expression in the cisplatin resistant cells. Cells were transfected with siRNA control or siRNA NF-κB p65 for 48 hours and lysed and the levels of the levels of p65 and GAPDH were determined by immunoblotting. The experiments were carried out on three separate occasions. B. Cells were transfected with siRNA control or siRNA NF-κB p65 for 48 hours and then treated (DMSO) or Cisplatin (20 μM) for additional 24 hours. Caspase 3/7 activity was evaluated following the protocol. The results are representative of three independent experiments (*, P < 0.05, **, P < 0.01). C. Cells were transfected with siRNA control or siRNA NF-κB p65 for 48 hours and then left untreated (DMSO) or treated with 20 μM Cisplatin for additional 48 hours. The levels of cleaved caspase 3, p65 and GAPDH were determined by immunoblotting. The experiments were repeated three times.
Expression of NF-κB p65 serine 535 alanine mutant sensitizes cisplatin resistant cells to cisplatin treatment
Next, we determined the role of NF-κB phosphorylation at serine 536 in the regulation cisplatin resistance. SCC17B/CP and SCC25/CP cells were transfected with either the vector control or flag tagged p65 with serine 536 mutation to alanine (flag-p65-S536A). The cells were either left untreated or treated with cisplatin for 48 hours before caspase activity was measured. The results showed that significant induction of caspase activity was detected in p65-S536A transfected cells, but not in vector control transfected cells in response to cisplatin treatment (Figure 4A). In a parallel experiment, caspase 3 cleavage was examined, with the results suggesting that cisplatin induced caspase 3 cleavage in p65-S536A transfected cells, but not in vector transfected cells (Figure 4B). These data suggested that NF-κB phosphorylation at serine 536 plays an important role in acquired cisplatin resistance in HNSCC.
Figure 4.

Overexpression of NF-κB serine 536 alanine mutant sensitizes cisplatin resistant cells to cisplatin. A. Cells were transfected with vector or flag-p65 S536A and left untreated or treated with cisplatin for 48 hours and caspase cleavage was measured by western blot. B. Cells were transfected with vector or flag-p65 S536A and left untreated or treated with cisplatin for 48 hours and caspase activity was measured (*, P<0.05).
Inhibition of IKKβ synergize with cisplatin to induce apoptosis in cisplatin resistant cells
Our data suggests that both IKK and NF-κB were up-regulated in cisplatin resistant cells and that the depletion of NF-κB expression increases the sensitivity of the resistant cells to cisplatin. If IKKβ/NF-κB cascade is vital to protecting cells from cisplatin-induced apoptosis, we would expect that the IKK inhibitors would sensitize cells to cisplatin-induced cell death. To test this hypothesis, a recently identified IKKβ inhibitor, Bay-65-1942 [14], also known as CmpdA, was employed. Treating both SCC-17B/CP and SCC-25/CP with 5 μM of CmpdA for 48 hours resulted in a dramatic decrease of phosphorylation of p65 at serine 536 without affecting the total p65 level (Figure 5A). Next, In order to evaluate the effect of the IKK inhibitor (CmpdA) on cisplatin induced apoptosis, we first measured caspase 3/7 activity. As shown in 5B (left panel), treatment of SCC-17B/CP cells with CmpdA or cisplatin alone for 48 hours showed no induction of caspase-3/7 activity, whereas the combination of cmpdA and cisplatin resulted in a 40% increase in caspase 3/7 activity. Similar results were observed in SCC-25/CP cells (Figure 5B, right panel), with the combination of cmpdA and cisplatin causing a 50% increase in induction of caspase 3/7. Next, we employed western blot to assess caspase 3 cleavage in these cells. The results indicate that cisplatin treatment alone for 48 hours showed no induction of caspase-3 cleavage, while CmpdA only slightly induced the cleavage of caspase-3, but the combination led to dramatic induction of caspase-3 cleavage in both SCC-17B/CP and SCC-25/CP cell lines (Figure 5C). To further determine the survival and proliferation effects of combined IKK inhibitor and cisplatin, we performed a clonogenic assay with either CmpdA or cisplatin alone, as well as one with a combination of both CmpdA and cisplatin in both resistant cell lines. As shown in Figure 5D, the combination of CmpdA and cisplatin demonstrated a significantly reduced number of colonies compared to either agent alone. These data indicate that compA sensitizes SCC-17B/CP and SCC-25/CP cells to cisplatin induced apoptosis.
Figure 5.
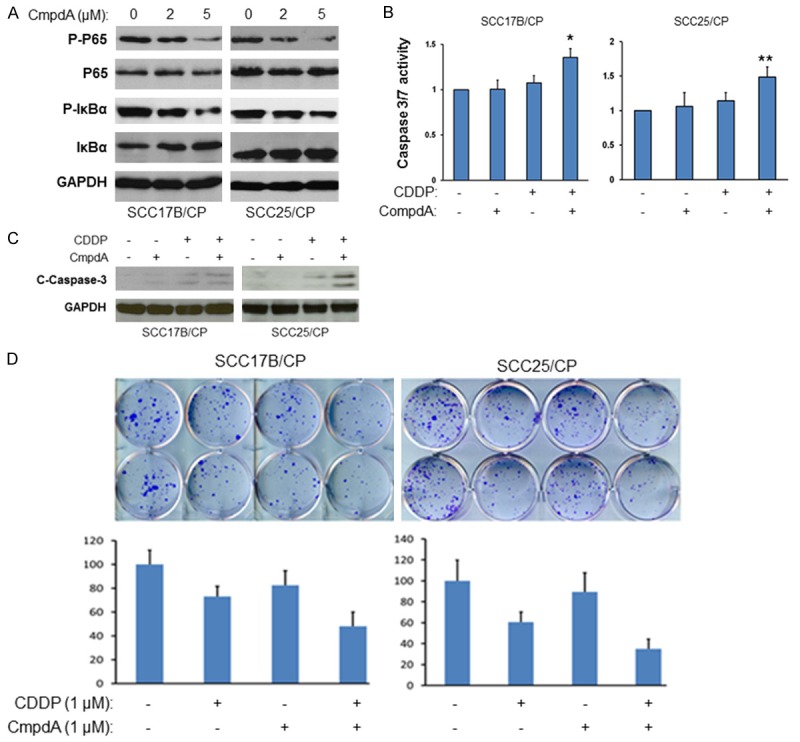
Inhibition of NF-κB/IKKβ signaling by the IKKβ inhibitor, CompA sensitizes cisplatin resistant cells to cisplatin. A. Cells were treated with different doses of CompA for 2 hours, lysed and analyzed by western blots. B. Cells were treated with CompA alone, cisplatin alone, or CompA and cisplatin together for 48 hours and Caspase 3/7 activity was evaluated (*, P < 0.05; **, P < 0.01). C. Cells were treated with CompA alone, cisplatin alone, or CompA and cisplatin together for 48 hours and cleaved caspase 3 and GAPDH were determined. The experiments were repeated three times. D. Cells were pre-treated with CompA, cisplatin, or CompA plus cisplatin and colony formation was observed and the numbers of colony were counted. The results are representative of three independent experiments.
NF-κB/IKKβ signaling is up-regulated in human tumor derived cisplatin resistant HNSCC cells
Next, we investigated if NF-κB/IKKβ pathways are upregulated in HNSCC cells derived from a patient whose tumor had acquired cisplatin resistance with treatment. The UM-SCC-11A cell originated from a pretreatment biopsy, while the UM-SCC-11B was derived from the same patient after a session of chemotherapy undertaken during surgery [12]. We wanted to determine if NF-κB/IKKβ pathway was activated in UM-SCC-11B cells compared to that found in UM-SCC-11A cells. The MTT assay showed that UMSCC-1A cells were relatively sensitive to cisplatin with an IC50 of 6.7 μM, whereas UMSCC-11B cells are relatively resistant to cisplatin with an IC50 of 10 μM, which is an approximately 50% increase over UMSCC-11A cells (Figure 6A). In addition, the western blot experiment showed that the phosphorylation of NF-κB p65 at serine 536 in UMSCC-11B is markedly increased compared with those found in UMSCC-11A cells (Figure 6B). These data indicate that NF-κB/IKKβ is elevated in UMSCC-11B compared to UMSCC-11A, which corroborates the SSC17-B/SCC17-B/CP and SCC-25/SCC25/CP data.
Figure 6.
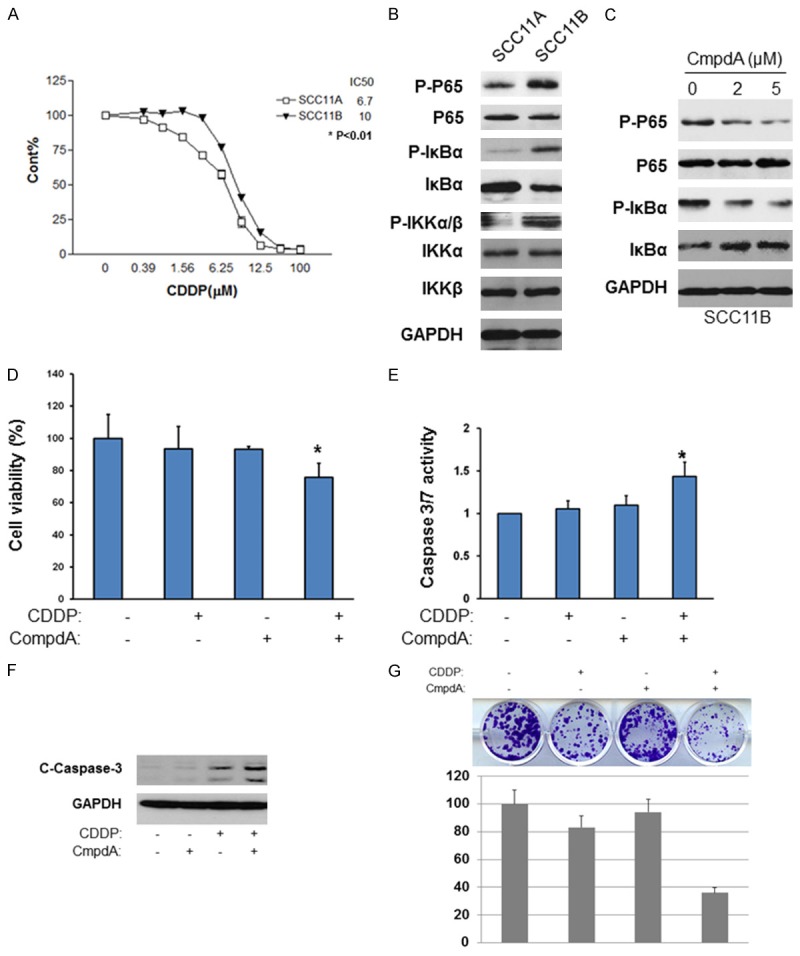
NF-κB is activated in patient-derived HNSCC cell lines and cisplatin resistance is reversed with IKKβ inhibition. A. IC50 values of UMSCC-11A and UMSCC-11B cells were determined. Cells were treated with Cisplatin (0-100 μM) for 72 hours. Cell proliferation was measured through MTT Assay. IC50 values were determined. B. Cell lysates from UMSCC-11A and UMSCC-11B cells were analyzed by western blots. C. Cells were treated with different doses of CompA for 2 hours, lysed and analyzed by western blots. D. UMSCC-11B cells were treated with CompA alone, Cisplatin alone, or CompA and Cisplatin as indicated for 48 hours and cell proliferation was measured by MTT assay. The results are representative of three independent experiments (*, P < 0.05). E. Cells were treated same with (C) for 48 hours and Caspase 3/7 activity was measured. The results are representative of three independent experiments (*, P < 0.05). F. Cells were treated same with (C) and the levels of cleaved caspase 3 and GAPDH. The results are representative of three independent experiments. G. Cells were treated with CompA, Cisplatin, or CompA and Cisplatin as indicated and colony formation was observed and the numbers of colony were counted. Each experiment was repeated three times.
Afterward, we treated UMSCC-11B cells with either cisplatin, CompA or a combination of both to test their effects on cell proliferation and apoptosis. Our data showed that CmpdA effectively blocks phosphorylation of NF-κB at serine 536 dose-dependently (Figure 6C). In addition, as shown in Figure 6D, both 2 μM CmpdA and 20 μM cisplatin alone did not inhibit cell proliferation, whereas the combination of CmpdA and cisplatin caused a 30% decrease in cell proliferation within SCC11B cells. We also measured the caspase activity and caspase-3 cleavage in the cells following these treatments. In Figure 6E, UMSCC-11B cells treated with either CmpdA or cisplatin alone did not induce caspase-3/7 activity, whereas the combination treatment led to a dramatic activation of caspase 3/7. In accordance with the caspase 3/7 activity results, western blot showed that there is no detectable cleaved caspase-3 in either CompA or cisplatin treatments, but showed a dramatic caspase-3 cleavage in the combination treatment (Figure 6F). Finally, in Figure 6G, the combination of CmpdA and cisplatin demonstrated a significant reduction in the numbers of colonies compared to either CmpdA or cisplatin treatment alone. These data demonstrate that NF-κB activity is upregulated in a human tumor derived cisplatin resistant cell, and the inhibition of IKK/NF-κB enhances the ability of cisplatin to increase apoptosis as well as decrease colony formation in the human derived cisplatin resistant cells.
Discussion
Cisplatin is the most common anticancer drug used to treat head and neck cancer. Currently, the standard of care for cisplatin-based chemotherapy regimen as treatment of recurrent and metastatic head and neck cancer is a combination of cisplatin, fluorouracil and cetuximab, but its efficacy is limited due to toxicity and the development of cisplatin resistance [1,4,6,15-17]. Therefore, a better understanding of the mechanisms involved in developing cisplatin resistance, especially specifically acquired resistance, is needed to improve the efficacy of treatment for head and neck cancer. Identifying the critical signaling pathways that underlie cisplatin resistance would offer an opportunity for patients to maintain or even regain sensitivity to drugs used through combination therapy.
The transcription factor NF-κB is involved in many cellular functions, including the regulation of apoptosis and chemotherapy resistance [10,11]. Earlier studies have demonstrated that NF-κB is related to intrinsic cisplatin resistance and that targeting NF-κB signaling can increase the sensitivity of cancer cells to cisplatin treatment, but the exact role and mechanisms by which NF-κB regulates this acquired resistance to cisplatin are unclear [18-22]. Several studies published by different groups have already shown that the phosphorylation of NF-κB at serine 536 plays pivotal roles in NF-κB regulation of tumorigenesis [23-25]. Our data first demonstrate that the phosphorylation of NF-κB p65 at serine 536 is dramatically elevated in several head-neck cancer cell lines that were subjected to long term exposure of cisplatin and in a patient derived head-neck cancer cell line from a patient treated with cisplatin, which serve to indicate the key role of NF-κB phosphorylation at serine 536 in regulation of cisplatin resistance. In addition, phosphorylation of IKK (both IKKα and IKKβ), the NF-κB upstream kinase that phosphorylate NF-κB at serine 536, is also dramatically elevated in these resistant cell lines. Furthermore, NF-κB p65 knockdown or overexpression of NF-κB p65-Serine 536A mutant sensitizes the resistant cells to cisplatin. As has been consistently demonstrated, the IKKβ inhibitor CmpdA blocks NF-κB p65 serine 536 phosphorylation and significantly sensitizes the resistant cells to cisplatin treatment. These data demonstrated that NF-κB/IKK is a critical factor that contributes to the acquired cisplatin resistance in HNSCC and the blocking of IKK/NF-κB activity by the IKK kinase inhibitor could be a strategy to improve the efficacy of cisplatin treatment in human trials. Our data are consistent with the observation by Duarte and colleagues showing that Curcumin, the major component of the spice turmeric derived from the rhizome of the East Indian plant, enhances the effect of cisplatin in suppressing head and neck squamous cell carcinoma through the inhibition of IKKβ/NF-κB pathway [22].
Toxicity has proven to be another challenge when treating head and neck cancer patients with cisplatin. Our data indicate that lower doses of cisplatin, in combination with the IKK inhibitor, caused marked induction of apoptosis, whereas cisplatin alone was less effective even at high doses in the cisplatin resistant cells. An IKK inhibitor, in combination with cisplatin, could allow a lower dosage of cisplatin for patients, which would improve its toxicity profile and provide patients with better outcomes.
The mechanisms associated with cisplatin resistance involve many different cellular processes [1,6]. NF-κB mediates apoptosis through transcriptional regulation of downstream target genes, including Bcl-2, BCL-xl, XIAP, c-IAP2 and Survivin [26-29]. Specimens derived from patients, both before and after chemotherapy, demonstrated that Survivin expression levels were elevated during or after chemotherapy and the up-regulation of Survivin was associated with both resistance to chemotherapy and poor prognosis [26-28]. It would be interesting to determine which NF-κB target genes are critical for conferring cisplatin resistance in HNSCC.
In summary, the data presented here indicates that IKK/NF-κB is involved in acquired resistance to cisplatin in HNSCC cells. Currently, there are no effective IKK inhibitors approved for use to treat cancer patients, including those with head and neck cancer.
The next steps should include the testing of CmpdA on in-vivo efficacy models of cisplatin sensitive and resistant HNSCC.
Acknowledgements
We thank Dr. Thomas E. Carey for the cells and Albert Baldwin for providing the IKKβ inhibitor. We also thank Drs. Amy Fulton and Antonino Passaniti for critical reading of this manuscript. This work was supported in part by National Institutes of Health Grants R00CA149178 to H.C.D and startup funds from Marlene and Stewart Greenebaum Cancer Center, University of Maryland School of Medicine to H.C.D.
Disclosure of conflict of interest
None.
References
- 1.Pendleton KP, Grandis JR. Cisplatin-Based Chemotherapy Options for Recurrent and/or Metastatic Squamous Cell Cancer of the Head and Neck. Clin Med Insights Ther. 2013;5:103–116. doi: 10.4137/CMT.S10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Pignon JP, le Maitre A, Maillard E, Bourhis J MACH-NC Collaborative Group. Meta-analysis of chemo-therapy in head and neck cancer (MACH-NC): an update on 93 randomised trials and 17,346 patients. Radiother Oncol. 2009;92:4–14. doi: 10.1016/j.radonc.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Molin Y, Fayette J. Current chemotherapies for recurrent/metastatic head and neckcancer. Anticancer Drugs. 2011;22:621–625. doi: 10.1097/CAD.0b013e3283421f7c. [DOI] [PubMed] [Google Scholar]
- 5.Wang F, Arun P, Friedman J, Chen Z, Van Waes C. Current and potential inflammation targeted therapies in head and neck cancer. Curr Opin Pharmacol. 2009;9:389–395. doi: 10.1016/j.coph.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. doi: 10.1038/cddis.2013.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto Y, Gaynor RB. IkappaB kinases. Key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 10.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 11.Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999;5:412–417. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 12.Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, Bradford CR, Carey TE. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–426. doi: 10.1002/hed.21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teicher BA, Holden SA, Kelley MJ, Shea TC, Cucchi CA, Rosowsky A, Henner WD, Frei E 3rd. Characterization of a human squamous carcinoma cell line resistant to cis-diamminedichloroplatinum (II) Cancer Res. 1987;47:388–393. [PubMed] [Google Scholar]
- 14.Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K, Niki T, Sakai K, Inbe H, Takeshita K, Ishimori M, Komura H, Murata T, Lowinger T, Bacon KB. A selective novel low-molecularweight inhibitor of IkappaB kinase-beta (IKKbeta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br J Pharmacol. 2005;145:178–192. doi: 10.1038/sj.bjp.0706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, De Raucourt D, Bokemeyer C, Schueler A, Amellal N, Hitt R. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 16.Vermorken JB, Remenar E, van Herpen C, Gorlia T, Mesia R, Degardin M, Stewart JS, Jelic S, Betka J, Preiss JH, van den Weyngaert D, Awada A, Cupissol D, Kienzer HR, Rey A, Desaunois I, Bernier J, Lefebvre JL EORTC /TAX Study Group. Cisplatin, fluorouracil, and docetaxel in unresectable head and neck cancer. N Engl J Med. 2007;357:1695–1704. doi: 10.1056/NEJMoa071028. [DOI] [PubMed] [Google Scholar]
- 17.Burtness B. Cetuximab and cisplatin for chemotherapy-refractory squamous cell cancer of the head and neck. J. Clin. Oncol. 2005;23:5440–5442. doi: 10.1200/JCO.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Ruan HY, Masuda M, Ito A, Umezawa K, Nakashima T, Yasumatsu R, Kuratomi Y, Yamamoto T, Weinstein IB, Komune S. Effects of a novel NF-kappaB inhibitor, dehydroxymethylepoxyquinomicin (DHMEQ), on growth, apoptosis, gene expression, and chemosensitivity in head and neck squamous cell carcinoma cell lines. Head Neck. 2006;28:158–165. doi: 10.1002/hed.20304. [DOI] [PubMed] [Google Scholar]
- 19.Lee TK, Poon RT, Wo JY, Ma S, Guan XY, Myers JN, Altevogt P, Yuen AP. Lupeol suppresses cisplatin-induced nuclear factor-kappaB activation in head and neck squamous cell carcinoma and inhibits local invasion and nodal metastasis in an orthotopic nude mouse model. Cancer Res. 2007;67:8800–8809. doi: 10.1158/0008-5472.CAN-07-0801. [DOI] [PubMed] [Google Scholar]
- 20.Li B, Li YY, Tsao SW, Cheung AL. Targeting NFkappaB signaling pathway suppresses tumor growth, angiogenesis, and metastasis of human esophageal cancer. Mol Cancer Ther. 2009;8:2635–2644. doi: 10.1158/1535-7163.MCT-09-0162. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee A, Chang X, Sen T, Ravi R, Bedi A, Sidransky D. Regulation of p53 family member isoform DeltaNp63alpha by the nuclear factorkappaB targeting kinase IkappaB kinase beta. Cancer Res. 2010;70:1419–1429. doi: 10.1158/0008-5472.CAN-09-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duarte VM, Han E, Veena MS, Salvado A, Suh JD, Liang LJ, Faull KF, Srivatsan ES, Wang MB. Curcumin enhances the effect of cisplatin in suppression of head and neck squamous cell carcinoma via inhibition of IKKβ protein of the NFκB pathway. Mol Cancer Ther. 2010;9:2665–2675. doi: 10.1158/1535-7163.MCT-10-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Shao L, Creighton CJ, Zhang Y, Xin L, Ittmann M, Wang J. Function of phosphorylation of NF-kB p65 ser536 in prostate cancer oncogenesis. Oncotarget. 2015;6:6281–6294. doi: 10.18632/oncotarget.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. J Biol Chem. 2005;280:34538–34547. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 25.Hu J, Nakano H, Sakurai H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25:1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- 26.Kato J, Kuwabara Y, Mitani M, Shinoda N, Sato A, Toyama T, Mitsui A, Nishiwaki T, Moriyama S, Kudo J, Fujii Y. Expression of survivin in esophageal cancer: correlation with the prognosis and response to chemotherapy. Int J Cancer. 2001;95:92–95. doi: 10.1002/1097-0215(20010320)95:2<92::aid-ijc1016>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 27.Kumar B, Yadav A, Lang JC, Cipolla MJ, Schmitt AC, Arradaza N, Teknos TN, Kumar P. YM155 reverses cisplatin resistance in head and neck cancer by decreasing cytoplasmic survivin levels. Mol Cancer Ther. 2012;11:1988–1998. doi: 10.1158/1535-7163.MCT-12-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan Z, Tiwari RP, Khan N, Prasad GB, Bisen PS. Induction of apoptosis and sensitization of head and neck squamous carcinoma cells to cisplatin by targeting survivin gene expression. Curr Gene Ther. 2012;12:444–453. doi: 10.2174/156652312803519805. [DOI] [PubMed] [Google Scholar]
- 29.Dutta J, Fan Y, Gupta N, Fan G, Gélinas C. Current insights into the regulation of programmed cell death by NF-κB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]