

Abstract
Necroptosis, a novel form of programmed cell death, was recently shown to be strongly associated with intestinal inflammation in mice and in pediatric patients with inflammatory bowel disease (IBD). Persistent inflammation of the colon is an important risk factor for colorectal cancer. Necrostatin-1 (Nec-1), known as a specific inhibitor of necroptosis, through preventing the receptor-interacting protein (RIP) 1 and RIP3 interaction. In the present study, the anti-inflammatory and antitumorigenic efficacy of necrostatin-1 was studied in mouse models of colitis and colitis-associated cancer (CAC). We found that in acute dextran sulfate sodium (DSS)-induced colitis, treatment with necrostatin-1 significantly suppressed colitis symptoms in mice, including weight loss, colon shortening, colonic mucosa damage and severity, and excessive production of interleukin-6. Necrostatin-1 administration inhibited the upregulation of RIP1 and RIP3 and enhanced the expression of caspase-8 in DSS-induced colitis. In addition, the anti-inflammatory effect of necrostatin-1 was confirmed by in vitro analyses. Necrostatin-1 treatment reduced the production of proinflammatory cytokine and extracellular HMGB1 release in HT-29 cells in active necroptosis. Furthermore, In a mouse model of colitis-associated tumorigenesis, necrostatin-1 administration significantly suppressed tumor growth and development through inhibiting JNK/c-Jun signaling. Taken together, these findings suggest that necrostatin-1 might be a promising therapeutic option for the treatment of colitis-associated colorectal cancer in patients with IBD.
Keywords: Inflammatory bowel diseases, necroptosis, necrostatin-1, murine colitis models, colon cancer
Introduction
Chronic inflammation is considered a risk factor for many common malignancies including cancers of the breast [1], lung [2], and colon [3]. Colorectal cancer is the third most common type of cancer and the third leading cause of cancer-associated mortality in both males and females in the United States [4]. Inflammatory bowel diseases are a salient example of the link between chronic inflammation and cancer [5], and one of the most serious consequences of persistent inflammation of the colon is an increased risk for developing colorectal cancer [6]. Animal models that reproduce many aspects of the human disease have provided significant clues regarding the crucial roles of inflammatory mediators and related molecular events in promoting the development of CAC [7].
Traditionally, apoptosis and necrosis have been considered as the two main forms of cell death involved in the regulation of intestinal homeostasis in the intestinal epithelium [8]. Recent evidence has suggested that necroptosis, a novel form of cell death that exhibits features of both apoptosis and necrosis, is associated with intestinal inflammation [9-12]. Mouse model experiments have shown that epithelial cell necroptosis induces intestinal inflammation, indicating that necroptosis could also contribute to the pathogenesis of IBD in humans [13,14]. A more recent study demonstrated that in the inflamed tissues of pediatric IBD patients, the expression of RIP3 and mixed lineage kinase domain-like protein (MLKL) is increased, whereas caspase-8 expression is reduced, indicating that necroptosis is strongly associated with intestinal inflammation and contributes to strengthen the inflammatory process [15]. Therefore, inhibition of necroptosis has been suggested as a novel therapeutic approach for reducing inflammation.
Necroptosis can be induced by various stimuli, such as tumor necrosis factor-α (TNF-α) [16], which has been extensively studied, as well as toll-like receptors, intracellular RNA and DNA sensors, and other mediators [12]. Execution of necroptosis requires the assembly of a RIP1 and RIP3 containing necrosome [17]. Upon activation of the TNF receptor, mutual direct and indirect phosphorylation of RIP1 and RIP3 in the necrosome activates necrotic signaling, whereas caspase-8, a cysteine protease that is critically involved in regulating cellular apoptosis, is inhibited during this process [18,19].
Necrostatin-1, 5-(1Hindol-3-ylmethyl)-3-methyl-2-sulfanylideneimidazolidin-4-one, is a selective and potent allosteric inhibitor of RIP1 kinase that has been confirmed to be a special inhibitor of necroptosis [10]. Nec-1 has been shown to be a cellular protectant in animal models of retinal detachment [20], intracerebral hemorrhage [21] and spinal cord injury [22]. However, the effects of Nec-1 in mouse models of colitis and CAC are still unknown.
In this study, we investigated for the first time the anti-inflammatory potency of Nec-1 and the effect of Nec-1 on the expression of RIP1, RIP3, MLKL and caspase-8 in mouse models of experimental colitis. To further establish an additional tumor-preventing capacity of Nec-1, colitis-associated carcinogenesis was studied in mouse models induced by azoxymethane (AOM) and DSS. Pathways possibly involved were assessed at the cellular level in vitro and ex vivo.
Materials and methods
Mice
Six-to-eight week old female C57BL/6 mice were purchased from the Fourth Military Medical University, Laboratory Animal Co. Ltd. Animal studies were approved by the Animal Experiment Administration Committee of the University.
Models of acute colitis
Acute colitis was achieved by feeding C57BL/6 mice with 4% DSS (MP Biomedicals, molecular weight 35,000-50,000 Da) dissolved in sterile, distilled water ad libitum from day 1 to 5, followed by 5 days of regular drinking water.
CAC protocol
CAC was induced as described [23]. Briefly, mice were injected intraperitoneally with 12.5 mg/kg AOM (Sigma-Aldrich) and after 5 days, received drinking water containing 3% DSS for 5 days. Mice were then given regular drinking water for 14 days, followed by two additional DSS treatment cycles (Figure 5A). Colons were removed on day 120 and flushed with PBS, and tumors were counted. Macroscopic tumors were measured with calipers and software for microscopic tumors. Portions of the distal colons were either frozen in liquid nitrogen or fixed with formaldehyde (4%) and paraffin embedded for histological analyses.
Figure 5.
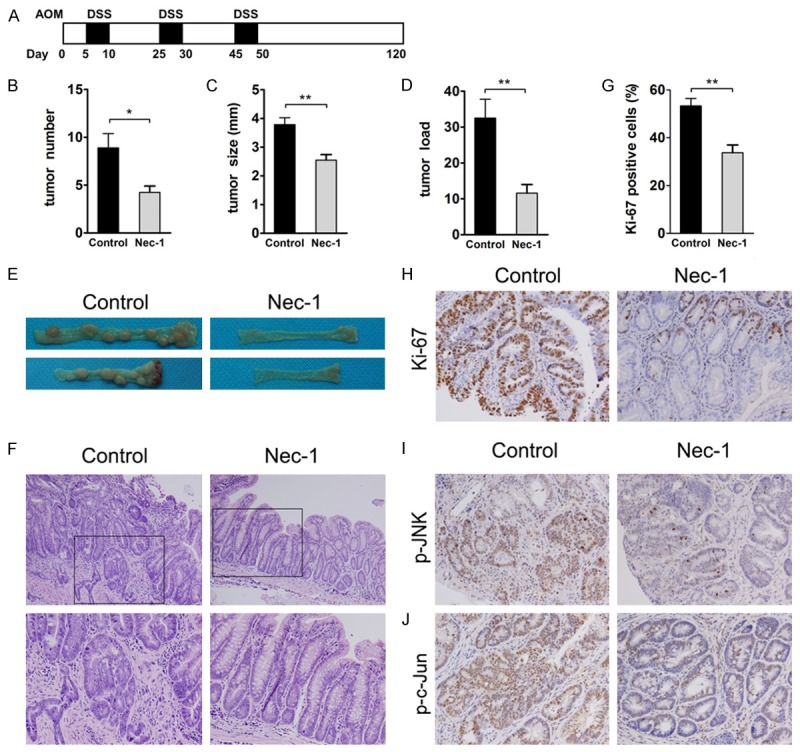
Nec-1 prevents colitis-associated tumor development. (A) Schematic overview of Nec-1 administration during CAC induction. Mice were injected with AOM followed by three cycles of 3% DSS in drinking water. Vehicle or Nec-1 (4.5 mg/kg) was administered intraperitoneally daily from day 1 to day 120. Intestinal tumors were analyzed on day 120. (B) Tumor number, (C) Tumor size, and (D) Tumor load, the sum of the diameters of all tumors in a given mouse, were determined. (E) Macroscopic evaluation of the tumors. The colons were removed from mice on day 120 treated with Nec-1 or vehicle. Representative results from 10 independent animals are shown here. (F) The colons were processed for H&E staining, and representative histological sections from each group are shown here. Original magnification, ×200 (upper rows), ×400 (lower rows). (H) Proliferation was determined through Ki-67 staining and (G) Percentage of Ki-67 positive cells within colonic crypts was calculated. Original magnification, ×400. Immunohistochemical analysis of (I) Phospho-JNK and (J) Phospho-c-Jun in CAC tumors treated with vehicle or Nec-1. Original magnification, ×400. Data represent the means (SEM), n = 7-8 per group; *P < 0.05, **P < 0.01.
Treatment with necrostatin-1
In all experiments, Nec-1 (Sigma-Aldrich) was given simultaneously with the start of colitis and CAC induction. During the colitis regimen, 10% DMSO (Sigma-Aldrich) or Nec-1 (4.5 mg/kg) was intraperitoneally administered twice daily. Nec-1 (4.5 mg/kg) was intraperitoneally administered once daily during CAC induction.
Clinical assessment of colitis
Body weight, stool consistency and rectal bleeding were assessed daily. Values assessed prior to DSS exposure served as baseline. Weight changes were calculated in relation to the weight at baseline (100%). Stool consistency was scored as follows: 0, well-formed pellets; 2, pasty and semi-formed stools; and 4, liquid stools that adhered to the anus. Rectal bleeding was scored as follows: 0, no blood using hemoccult (Beckman Coulter); 2, positive hemoccult; and 4, gross bleeding. Mice were sacrificed by cervical dislocation. The entire colon was removed, and the length was measured.
Histological scoring
Entire colons were excised postmortem. In DSS induced colitis, 4-6 rings from each colon were fixed with formaldehyde. From AOM-DSS-treated mice, complete colons were fixed. Paraffin sections were stained with H&E. Histological signs of inflammation were evaluated as a combined score of inflammatory cell infiltration (0-3) and tissue damage (0-3), resulting in a score ranging from 0 to 6 as described previously [24,25].
Cell culture
The colonic adenocarcinoma cell line HT-29 was cultured in Dulbecco’s Modified Eagle’s Medium containing 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C under 5% CO2. In all, 4.4×106 cells per well were seeded in six multi-well plates for protein analysis. TNF-α (100 ng/ml; R&D Systems), either alone or in combination with z-VAD-fmk (20 μM; Sigma), Smac (1 μM; Selleckchem) (without serum), and necrostatin-1 (100 μM) was added to culture medium to induce inflammation and cell death and to inhibit necroptosis.
Real-time quantitative PCR
Expression of IL-8 and IL-1β was detected by real-time PCR. Total RNA was extracted with TRIzol reagent (OMEGA Bio-Tek) and reverse-transcribed into cDNA by a PrimeScript RT reagent Kit (TaKaRa Biotechnology). The following primers were used: IL-8 forward primer, 5’-ATGACTTCCAAGCTGGCCGTGGCT-3’; IL8 reverse primer, 5’-TCTCAGCCCTCTTCAAAAACTTCTC-3’; IL-1β forward primer, 5’-GCACGATGCACCTGTACGAT-3’; IL-1β reverse primer, 5’-AGACATCACCAAGCTTTTTGCT-3’; GAPDH forward primer, 5’-GCTCCTCCTGTTCGACAGTCA-3’; and GAPDH reverse primer, 5’-ACCTTCCATGGTGTCTGA-3’. GAPDH mRNA was used as an internal control. Real-time PCR was performed using the SYBR Premix Ex Taq II Kit (TaKaRa Biotechnology).
Western blot analysis
Tissue samples were lysed in RIPA lysis buffer (Beyotime, Shanghai, China) containing 1 mM PMSF and a cocktail of protease and phosphatase inhibitors. The protein quantification was determined by BCA Protein Assay Kit (Thermo Scientific), and equal amounts of proteins were subjected to SDS/PAGE (10% gels) under reducing conditions. After electrophoresis, proteins were transferred onto PVDF membranes (Millipore) in running buffer with 10% methanol. Non-specific sites were blocked with 5% (w/v) non-fat dried skimmed milk powder in PBST (10 mM phosphate buffer, pH 7.4; 0.15 M NaCl; and 0.05% Tween-20) for 30 min at room temperature (25°C). The membranes were then incubated overnight at 4°C with the following antibodies, which were diluted in PBST containing 5% (w/v) non-fat dried skimmed milk powder: anti-RIP3 (dilution of 1:800; Abcam); anti-RIP1 (dilution of 1:200; Biorbyt); anti-MLKL (dilution of 1:1000; Biorbyt); anti-caspase-8 (dilution of 1:1000; Novus); anti-HMGB1 (dilution of 1:2000; Antibody Revolution); anti-JNK (dilution of 1:500; Proteintech); anti-c-Jun, anti-p-JNK and anti-p-c-Jun (dilution of 1:1000; Cell Signaling); and anti-α-tubulin (dilution of 1:1000; Santa Cruz). After three washes with PBST, the membranes were incubated with horseradish-peroxidase conjugated secondary antibodies (Pierce) for 1 h in PBST containing 5% (w/v) non-fat dried skimmed milk powder (dilution of 1:5000). Protein bands were visualized using enhanced chemiluminescence (ECL) Plus Western blotting detection kit (Amersham Biosciences) according to the manufacturer’s instructions. A PageRuler Prestained protein Ladder Plus (Fermentas Life Sciences) was used for sizing the proteins.
Immunohistochemistry analysis
Formaldehyde-fixed and paraffin-embedded sections of colon tissue were deparaffinized using xylene and alcohol and then subjected to antigen retrieval with citrate buffer (pH 6.0). The sections were subsequently incubated with 0.3% H2O2 and normal goat serum for blocking. After washing with PBS, the sections were incubated with primary antibodies at 4°C overnight in a moist chamber. Following incubation, immunoperoxidase staining was completed using a Streptavidin-Peroxidase Kit (Zhongshan Jinqiao Co., Beijing, China), and 3,3’-diaminobenzidine (Zhongshan Jinqiao Co., Beijing, China) was employed to detect the target proteins. The primary antibodies used in these experiments were anti-RIP1 (1:200; Biorbyt), anti-RIP3 (1:200; Biorbyt), anti-caspase-8 (1:100; Novus), anti-Ki-67 (1:100; Abcam), anti-p-JNK (1:60; Abcam), and anti-p-c-Jun (1:60; Abcam).
Enzyme-linked immunosorbent assay (ELISA)
IL-6 in serum and the cell medium was measured via ELISA. Serum IL-6 levels were assessed using a specific Mouse IL-6 precoated ELISA Kit (Dakewe Biotech Co., Shenzhen, China) according to the manufacturer’s protocol. IL-6 levels in the cell medium were measured with a human IL-6 precoated ELISA Kit (Dakewe Biotech Co., Shenzhen, China). The concentration in each sample well was determined through interpolation from a standard curve. Each sample was tested in duplicate.
Statistical analysis
The data were expressed as the mean value ± SEM. Statistical differences were assessed via analysis of variance (ANOVA) followed by appropriate post hoc tests including Student’s t-test and multiple comparison tests. All statistical analyses were performed using GraphPad Prism V5.0 software. Differences were considered statistically significant when P < 0.05.
Results
Treatment with the necroptosis inhibitor Nec-1 reduces inflammation in acute DSS-induced colitis
The anti-inflammatory potency of Nec-1 was evaluated in a mouse model of DSS-induced colitis. Mice were subjected to a single 5-day course of DSS to induce acute colitis. The results showed that daily treatment with Nec-1 significantly suppressed colitis symptoms in mice. Within 10 days, the weight curves of Nec-1-treated mice were superimposed on those of DSS-fed mice that received vehicle only. Starting at day 5, Nec-1-treated mice lost markedly less weight than the DSS-exposed control mice (Figure 1A), and the colons of Nec-1-treated mice were significantly longer than the colons in the DSS control group (Figure 1B). Moreover, the Nec-1 treatment group exhibited decreased bleeding and diarrhea scores compared with their control littermates (Figure 1C and 1D).
Figure 1.
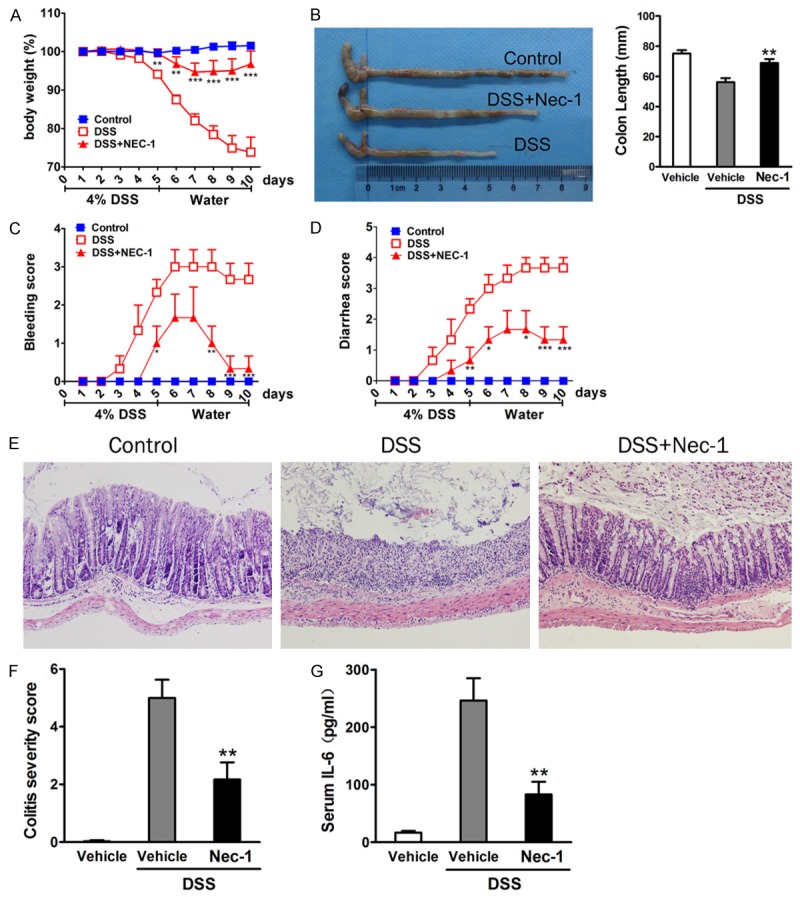
Nec-1 treatment suppresses inflammation in acute DSS-induced colitis. Mice exposed to DSS intraperitoneally received Nec-1 or vehicle (10% DMSO) throughout the entire experimental period. A. Body weight was determined daily. B. The length of the excised colon was measured. C. Bleeding and D. Diarrhoea scores were determined at the end of the experiment. E. Mucosal histology was examined via H&E staining. Original magnification, ×200. F. Colitis severity scores were determined in a double-blind manner. G. IL-6 levels in serum were measured via ELISA. Data represent the mean (SEM), n = 6 per group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. DSS vehicle.
Accompanying these macroscopic findings, histopathological analysis of the DSS group revealed a severely damaged colonic mucosa with extensive loss of crypt structures and epithelial cell denudation, large areas of ulceration, and extensive infiltration of inflammatory cells (Figure 1E). In contrast, treatment with Nec-1 markedly suppressed this colonic inflammation, which was also reflected in the pathological assessment of colitis severity scores (Figure 1F). In addition, Nec-1 treatment during DSS challenge resulted in a significant reduction of the proinflammatory cytokine IL-6 in serum compared with the DSS control mice (Figure 1G). These data suggest that treatment with Nec-1 caused a strong protection in the mouse model of DSS-induced colitis.
Effect of Nec-1 on changes of necroptosis-related protein expression in DSS-induced colitis
Nec-1, a chemical inhibitor of the kinase activity of RIP1, prevents the RIP1/RIP3 interaction and blocks necroptosis [26]. To investigate the effect of Nec-1 on expression of necroptosis-related protein in colitis, we first analyzed the protein expression levels of RIP1 and RIP3 kinase, which function as a necrosome that is involved in the initiation of necroptosis [12]. Western blotting analyses were performed on colonic nuclear extracts from DSS-induced colitis mice, Nec-1-treated mice and the control littermates. Densitometric analysis showed a significant upregulation of RIP1 and RIP3 in the inflamed colon of the DSS group compared with the controls. We then analyzed the expression of the 43 kDa active fragment of caspase-8, and the results showed a significant decrease of caspase-8 in the DSS group compared with the controls. However, in the DSS-induced inflamed colon, treatment with Nec-1 almost completely abrogated the large increases in the RIP1 and RIP3 proteins and prevented the decrease in the expression of caspase-8 (Figure 2A). In agreement with these findings, immunohistochemistry analysis of RIP1, RIP3 and caspase-8 revealed similar changes (Figure 2B). However, there were no significant differences in the expression of the MLKL protein (Figure 2A).
Figure 2.
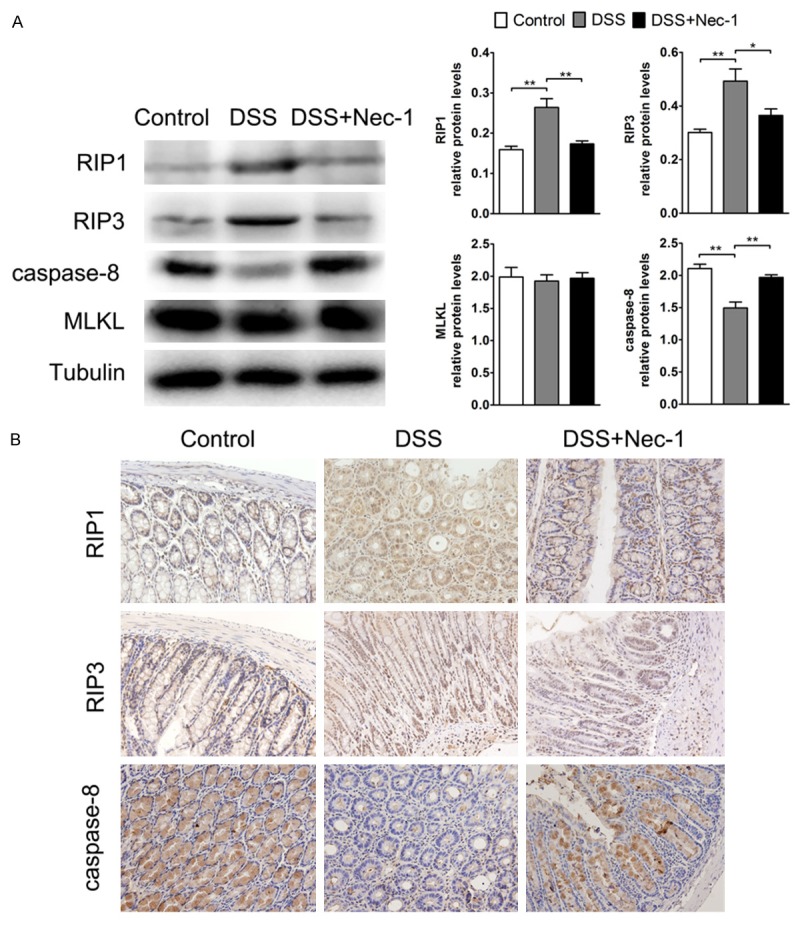
Effect of Nec-1 on expression of RIP1, RIP3, caspase-8 and MLKL in DSS-induced colitis. Nec-1 inhibited the upregulation of RIP1 and RIP3 and enhanced the expression of caspase-8 in colitis. However, Nec-1 showed no effect on MLKL. A. The protein levels of RIP1, RIP3, caspase-8, and MLKL were analyzed by western blot. The blots were probed for α-tubulin to ensure equal protein loading. Western blot densitometric analysis for three independent experiments. B. The expression of RIP1, RIP3, and caspase-8 was determined through immunohistochemistry. Original magnification, ×400. Data represent the mean (SEM). *P < 0.05, **P < 0.01.
Nec-1 attenuates inflammation in HT-29 cells after treatment with TNF-α + Smac + z-VAD
Necroptosis can be induced in HT-29 cells after treatment with TNF-α, Smac mimetic, and z-VAD-fmk [27,28]. To confirm in vitro that Nec-1 attenuates mucosal inflammation, we analyzed the expression of three potent inflammatory mediators, IL-8, IL-1β and IL-6, after treating HT-29 cells with TNF-α + Smac + z-VAD. The results showed that the three cytokines were markedly increased (P < 0.001) compared with cells treated with TNF-α alone. However, this effect was significantly reverted after the co-treatment with Nec-1, indicating that necroptosis strengthens inflammation, and inhibition of necroptosis causes a strong reduction (P < 0.001) of inflammation (Figure 3A-C).
Figure 3.
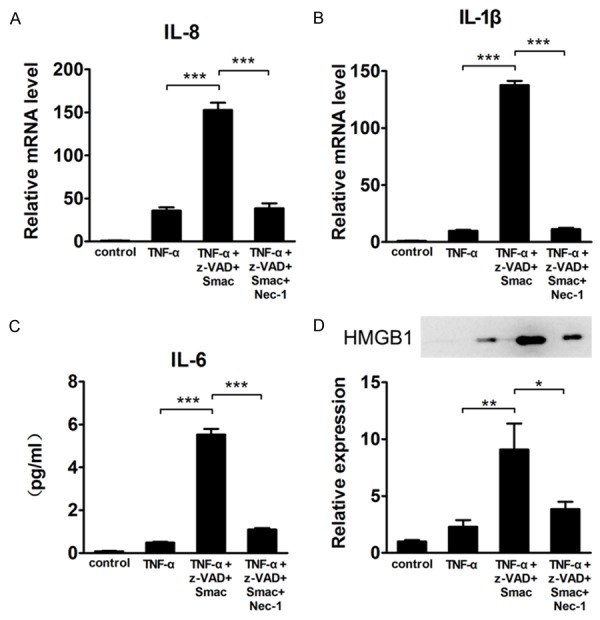
Treatment with Nec-1 significantly prevents the increases in IL-8, IL-1β, and IL-6 levels and extracellular HMGB1 release in HT-29 cells during necroptosis. mRNA expression of (A) IL-8 and (B) IL-1β in HT-29 cells after treatment with TNF-α/Smac/z-VAD or TNF-α/Smac/z-VAD/Nec-1. (C) IL-6 levels were measured via ELISA in the supernatants of HT-29 cells treated with TNF-α/Smac/z-VAD or TNF-α/Smac/z-VAD/Nec-1. (D) Extracellular HMGB1 protein expression in HT-29 cells after treatment with TNF-α/Smac/z-VAD or TNF-α/Smac/z-VAD/Nec-1. Data represent the mean (SEM). *P < 0.05, **P < 0.01, ***P < 0.001.
In addition, recent literature demonstrated that induction of necroptosis in the intestine provokes a strong inflammatory response through the release of intracellular components known as damage-associated molecular patterns (DAMPs) [29]. One of these components is represented by high mobility group box 1 (HMGB1), a non-histone nuclear protein that is passively released by necrotic cells, which can serve as an alarmin to drive the pathogenesis of inflammation [30]. We analyzed the supernatants of HT-29 cells treated with TNF-α + Smac + z-VAD and found that extracellular HMGB1 was significantly augmented (P < 0.01) as compared with TNF-α-treated cells. However, the exposure of cells to Nec-1 significantly decreased (P < 0.05) the amount of secreted HMGB1, suggesting that Nec-1 can attenuate inflammation by reducing the release of HMGB1 (Figure 3D).
Nec-1 suppresses the activation of JNK signaling during DSS-induced colitis
Activation of JNK signaling was recently described as a downstream mediator of RIP3-driven necroptosis [17], and JNK signaling has been strongly associated with intestinal inflammation and colitis-induced tumorigenesis in murine models of colitis and CAC [31,32]. To examine the effects of Nec-1 on JNK signaling pathway, we analyzed the levels of phosphorylated JNK and its main substrate, phosphorylated c-Jun, during DSS-induced colitis. Elevated levels of phosphorylated JNK were observed in the inflamed colon during DSS challenge. However, treatment with Nec-1 dramatically inhibited the activation of JNK. Concomitantly, Nec-1 administration also prevented the increased phosphorylated c-Jun levels in the colon induced by DSS treatment (Figure 4).
Figure 4.
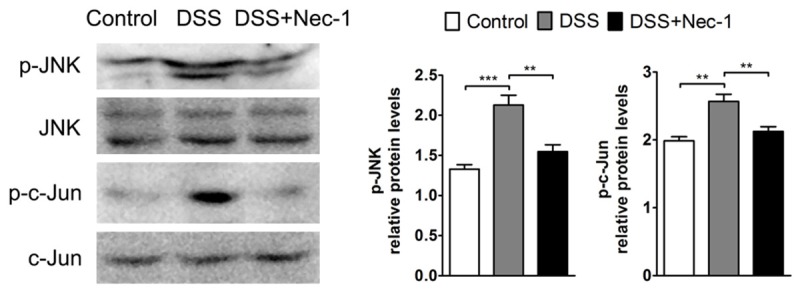
Nec-1 suppresses JNK/c-Jun signaling activation during acute colitis. The colonic expression of phospho-JNK, JNK, phospho-c-Jun, and c-Jun was detected by western blotting. Quantitative analysis of the phosphorylated fraction relative to the total fraction is shown. Bars indicate the mean (SEM). **P < 0.01, ***P < 0.001.
Treatment with Nec-1 prevents colitis-associated tumorigenesis
The following experiments directly investigated whether Nec-1 protects against colitis-associated tumorigenesis in the colon. We injected mice with the procarcinogen AOM, followed by three circles of DSS exposure to elicit colitis (Figure 5A). Compared with the vehicle-treated group, Nec-1 treatment significantly decreased the number and size of macroscopical tumors, with a concomitant decrease in tumor load (Figure 5B-E). There was also a notable reduction in adenomas with a high grade of dysplasia in Nec-1-treated mice compared with vehicle-treated mice (Figure 5F). To evaluate the in vivo antiproliferative properties of Nec-1, colon sections were stained for Ki-67. Robust proliferation was observed in vehicle-treated AOM-DSS mice, whereas there was a marked decrease in the number of Ki-67 positive cells in AOM-DSS Nec-1-treated animals (Figure 5G and 5H). These results suggested that Nec-1 also affects tumor growth and development.
This prompted us to examine the effect of Nec-1 on JNK/c-Jun pathway activation, which has been shown to accelerate tumorigenesis in the mouse CAC model [32]. Immunohistochemical analysis revealed that phosphorylated JNK was strongly expressed in nuclei of the intestinal epithelial cells (IECs) of CAC adenomas. Nec-1 administration significantly abolished nuclear p-JNK staining in mouse tumors (Figure 5I). Similarly, the increased p-c-Jun staining observed in IECs of CAC adenomas was reduced after treatment with Nec-1 (Figure 5J). These findings demonstrated that Nec-1 decreases the activation of JNK/c-Jun signaling and suppresses the development of CAC.
Discussion
IBD is an increased risk of developing colon cancer [6]. Previous reports have demonstrated that activation of necroptosis contributes to the pathogenesis of IBD in humans [15], and mouse-model studies have revealed significant functions for necroptosis in intestinal inflammation [12]. However, little is known about the inhibition of necroptosis in mouse models of colitis and CAC. The present study provided evidence that inhibition of necroptosis in vivo combines anti-inflammatory and antitumorigenic effects that attenuate intestinal inflammation and colitis-associated tumor growth in mice.
The in vivo anti-inflammatory efficacy of Nec-1 was evaluated in models of DSS-induced colitis. DSS-induced colitis is a widely used preclinical model for IBD and is extremely beneficial for investigating the therapeutic efficacy of pharmaceutically active compounds [33]. Our work showed that treatment with Nec-1 suppressed colitis symptoms in mice, including weight loss, colon shortening, colonic mucosa damage, and excessive proinflammatory cytokine production (particularly IL-6). The suppression of intestinal inflammation suggested that necroptosis is strongly involved in DSS-induced acute colitis.
A recent study on the inflamed mucosa of IBD patients has shown that RIP3 and MLKL expression is upregulated and that caspase-8 expression is reduced [15]. The results of our research showed that the expression of RIP1 and RIP3 was upregulated, whereas caspase-8 expression was downregulated in the inflamed colon tissue of murine colitis models. However, Nec-1 significantly inhibited the upregulation of RIP1 and RIP3 and enhanced the expression of caspase-8 in colitis, suggesting the existence of active necroptosis in DSS-induced colitis. As necroptosis exhibits important functions in the pathogenesis of intestinal inflammation [12], our experimental results suggested that Nec-1 suppresses intestinal inflammation during DSS-induced colitis by inhibiting necroptosis. Interestingly, we found that there were no significant differences in the protein expression of MLKL among the three groups of mice during DSS challenge, which may have been due to the analysis of total MLKL protein and not phosphorylated MLKL protein. MLKL moved from the cytosol to the plasma membrane after phosphorylation by RIP3 during necroptosis. The level of phosphorylated MLKL protein is increased during necroptosis, however, the amount of total MLKL protein is not obviously altered [28,34].
We then set up in vitro experiments to further assess the anti-inflammatory potency of Nec-1. Exposure of the colonic adenocarcinoma HT-29 cell line to mixed treatment with TNF-α/Smac/z-VAD is often used as a model of induced necroptosis [27]. To investigate whether Nec-1 reduces mucosal inflammation, we first analyzed the expression of three potent inflammation markers, IL-8, IL-1β and IL-6, before and after treatment with Nec-1. The results showed that the expression of the three cytokines was significantly decreased in the presence of Nec-1, suggesting that Nec-1 resulted in a strong reduction of inflammation. Because necroptosis leads to rapid plasma membrane permeabilization and to the release of DAMPs, which contributes to the inflammatory response [29], we analyzed the amount of secreted HMGB1 protein, a prototype DAMP. The results showed that cells exposed to the mixed treatment (TNF-α/Smac/z-VAD) released more extracellular HMGB1 into the medium than cells treated with TNF-α alone. However, the exposure of cells to Nec-1 markedly decreased the amount of secreted HMGB1, suggesting that Nec-1 prevented necroptosis-induced inflammation by inhibiting HMGB1 release.
In ulcerative colitis, the risk of developing colorectal cancer significantly increases after a disease duration of > 10 years in patients with pancolitis [35], in accord with the concept that chronic inflammation under certain circumstances represents a precancerous state [36]. We have chosen the AOM-DSS model of CAC to evaluate inflammation-mediated tumor growth [37]. AOM is a colonic genotoxic carcinogen that is extensively used for the investigation of colon carcinogenesis in rodents. Although AOM alone does not cause dysplasia or tumors in C57/BL6 mice, in combination with repeated cycles of DSS treatment, it increases the incidence of dysplastic lesions [38]. The attenuated tumor growth observed in CAC models treated with Nec-1 raises the question of what the contributing mechanisms downstream of Nec-1 inhibition may be.
Treatment with Nec-1 leads to suppression of proinflammatory cytokines in vivo, such as IL-6, which has been described as a critical tumor promoter during early CAC tumorigenesis [23,39]. The inhibition of proinflammatory IL-6 at the site of inflammation certainly contributes to the reduced tumorigenesis observed when Nec-1 is applied intraperitoneally.
A recent study strongly has suggested a second, though related, mechanism being involved. Activation of the JNK pathway is thought to act as a downstream mediator of RIP3-driven necroptosis [17]. Another study has also elegantly demonstrated the presence of a positive feedback loop between RIP3 and JNK-signaling. RIP3-dependent JNK activation promotes the release of proinflammatory mediators [40]. In addition, the specific JNK inhibitor SP600125 significantly reduces inflammation in DSS-induced colitis, suggesting that the JNK signaling pathway contributes to intestinal inflammation [31].
Furthermore, the JNK pathway has been implicated in oncogenic transformation and cell proliferation during the pathogenesis of cancer in various tissues [41]. The role of the JNK pathway in the promotion of colon carcinogenesis has been described recently. The JNK pathway and its main substrate, the c-Jun transcription factor, have been associated with increases in intestinal cell numbers through regulating the proliferation of intestinal progenitor cells. Activation of JNK/c-Jun signaling accelerates colitis-induced tumorigenesis, and inactivation of c-Jun leads to decreased progenitor cell proliferation and delays tumorigenesis in mice [32]. Our work showed that the expression of both p-JNK and p-c-Jun in mouse models of colitis and CAC is significantly reduced in the presence of Nec-1. Thus, inhibition of the JNK signaling pathway via Nec-1 treatment could contribute to the suppression of inflammation and tumor growth.
In summary, the present study demonstrated that necrostatin-1, a specific inhibitor of necroptosis, shows both anti-inflammatory and anti-tumorigenic effects in murine models of colitis and CAC. However, further studies are required to reveal the detailed mechanism underlying inflammation-induced tumor growth to evaluate the relevance of Nec-1 as a therapeutic option for the treatment of colitis-associated colorectal cancer in patients with IBD.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81472700 and 31101005), and the National HighTechnology Research and Development Program (“863” Program) of China (2014AA021605).
Disclosure of conflict of interest
None.
References
- 1.Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006;66:7395–7400. doi: 10.1158/0008-5472.CAN-06-0558. [DOI] [PubMed] [Google Scholar]
- 2.Sin DD, Man SF, McWilliams A, Lam S. Progression of airway dysplasia and C-reactive protein in smokers at high risk of lung cancer. Am J Respir Crit Care Med. 2006;173:535–539. doi: 10.1164/rccm.200508-1305OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunter MJ, Stolzenberg-Solomon R, Cross AJ, Leitzmann MF, Weinstein S, Wood RJ, Virtamo J, Taylor PR, Albanes D, Sinha R. A prospective study of serum C-reactive protein and colorectal cancer risk in men. Cancer Res. 2006;66:2483–2487. doi: 10.1158/0008-5472.CAN-05-3631. [DOI] [PubMed] [Google Scholar]
- 4.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 5.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 6.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–1816. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 7.Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- 8.Tait SWG, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 2008;27:6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nuñez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G Nomenclature Committee on Cell Death 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 13.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 15.Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, Gough PJ, Aloi M, Cucchiara S. Necroptosis Is Active in Children With Inflammatory Bowel Disease and Contributes to Heighten Intestinal Inflammation. Am J Gastroenterol. 2014;109:279–287. doi: 10.1038/ajg.2013.403. [DOI] [PubMed] [Google Scholar]
- 16.Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci. 2010;35:434–441. doi: 10.1016/j.tibs.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 19.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 20.Dong K, Zhu H, Song Z, Gong Y, Wang F, Wang W, Zheng Z, Yu Z, Gu Q, Xu X, Sun X. Necrostatin-1 protects photoreceptors from cell death and improves functional outcome after experimental retinal detachment. Am J Pathol. 2012;181:1634–1641. doi: 10.1016/j.ajpath.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 21.King MD, Whitaker-Lea WA, Campbell JM, Alleyne CH Jr, Dhandapani KM. Necrostatin-1 reduces neurovascular injury after intracerebral hemorrhage. Int J Cell Biol. 2014;2014:495817. doi: 10.1155/2014/495817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Wang J, Yang H, Zhou J, Feng X, Wang H, Tao Y. Necrostatin-1 mitigates mitochondrial dysfunction post-spinal cord injury. Neuroscience. 2015;289:224–232. doi: 10.1016/j.neuroscience.2014.12.061. [DOI] [PubMed] [Google Scholar]
- 23.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. 2002;122:2011–2025. doi: 10.1053/gast.2002.33631. [DOI] [PubMed] [Google Scholar]
- 26.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 27.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNFalpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 31.Assi K, Pillai R, Gomez-Munoz A, Owen D, Salh B. The specific JNK inhibitor SP600125 targets tumour necrosis factor-alpha production and epithelial cell apoptosis in acute murine colitis. Immunology. 2006;118:112–121. doi: 10.1111/j.1365-2567.2006.02349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sancho R, Nateri AS, de Vinuesa AG, Aguilera C, Nye E, Spencer-Dene B, Behrens A. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009;28:1843–1854. doi: 10.1038/emboj.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegmund B, Rieder F, Albrich S, Wolf K, Bidlingmaier C, Firestein GS, Boyle D, Lehr HA, Loher F, Hartmann G, Endres S, Eigler A. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. J Pharmacol Exp Ther. 2001;296:99–105. [PubMed] [Google Scholar]
- 34.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 37.Cooper HS, Murthy S, Kido K, Yoshitake H, Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757–768. doi: 10.1093/carcin/21.4.757. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis. 2006;27:162–169. doi: 10.1093/carcin/bgi205. [DOI] [PubMed] [Google Scholar]
- 39.Kim S, Keku TO, Martin C, Galanko J, Woosley JT, Schroeder JC, Satia JA, Halabi S, Sandler RS. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 2008;68:323–328. doi: 10.1158/0008-5472.CAN-07-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, Kreggenwinkel K, Schneider AT, Bartneck M, Neumann UP, Canbay A, Reeves HL, Luedde M, Tacke F, Trautwein C, Heikenwalder M, Luedde T. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. 2014;6:1062–1074. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]