

Abstract
Metastatic melanoma is a life-threatening disease for which no effective treatment is currently available. In melanoma cells, Rho overexpression promotes invasion and metastasis. However, the effect of statins on spontaneous metastasis and tumor growth remains unclear. In the present study, we investigated the mechanism of statin-mediated tumor growth and metastasis inhibition in an in vivo model. We found that statins significantly inhibited spontaneous metastasis and tumor growth. Statins inhibited the mRNA expression and enzymatic activities of matrix metalloproteinases (MMPs) in vivo and also suppressed the mRNA and protein expression of very late antigens (VLAs). Moreover, statins inhibited the prenylation of Rho as well as the phosphorylation of LIM kinase, serum response factor (SRF), and c-Fos downstream of the Rho signaling pathway. In addition, statins enhanced p53, p21, and p27 expression and reduced phosphorylation of cyclin-dependent kinase and expression of cyclin D1 and E2. These results indicate that statins suppress Rho signaling pathways, thereby inhibiting tumor metastasis and growth. Furthermore, statins markedly improved the survival rate in a metastasis model, suggesting that statins have potential clinical applications for the treatment of metastatic cancers.
Keywords: Statins, Rho, metastasis, tumor growth, melanoma
Introduction
Metastatic melanoma is a highly aggressive and often fatal malignancy that exhibits resistance to all current therapeutic approaches. At the time of diagnosis, approximately 20% of melanoma patients already have metastatic disease. Once metastasis has occurred, the overall median survival duration is only 6-9 months [1]. The recent increase in the incidence of melanoma has highlighted the need for novel molecular approaches in the treatment of metastatic disease [2].
Rho GTPases are important regulators of actin and microtubule filament organization and have also been implicated in the control of cell adhesion, cell motility, cell cycle progression, gene expression, and apoptosis [3]. Because all of these functions are dysregulated in tumor cells, it is not surprising that Rho GTPases might also be implicated in tumor growth and metastasis. In fact, Rho GTPases have been linked to tumor cell migration and invasion, and elevated RhoA and RhoC expression levels have been frequently associated with metastasis [4,5].
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase is considered the major regulatory enzyme in the mevalonate metabolic pathway. HMG-CoA reductase inhibitors (statins) are reversible inhibitors of the rate-limiting step in cholesterol biosynthesis [6]. Most experimental studies of statins have focused on the effects of these drugs on tumor cell growth in vitro and in vivo [7-11]. However, limited information is available regarding the effects of these agents on tumor growth and metastasis [12,13]. Our previous study indicated that statins could inhibit Rho/Rho-associated protein kinase (ROCK) pathway-mediated cell migration, invasion, adhesion, and metastasis [14]. However, whether statins inhibit spontaneous metastasis and tumor growth is unknown. In addition, whether statins act to inhibit metastasis via a mechanism includes blocking the mevalonate pathway to inhibit the prenylation and downstream signaling of small GTPases remains unclear. Clinically, statins are widely used; therefore, if these agents are found to inhibit tumor growth and metastasis, they could have additional potential therapeutic uses. In the present study, we investigated the mechanism of statin-mediated inhibition of tumor growth and metastasis in an in vivo model.
Materials and methods
Materials
Simvastatin was purchased from Wako (Osaka, Japan), and fluvastatin was purchased from Calbiochem (San Diego, CA, USA). These reagents were dissolved in dimethyl sulfoxide (DMSO) and filtered through 0.45-μm syringe filters (IWAKI GLASS, Japan). The dissolved regents were resuspended in phosphate-buffered saline (PBS; pH 7.4) and used in the various assays described below.
Cell culture
B16 melanoma BL6 cells (B16BL6 cells) were supplied by Dr. Inufusa (Kinki University, Osaka, Japan) and cultured in RPMI 1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 μg/ml penicillin (Gibco), 100 U/ml streptomycin (Gibco), and 25 mM HEPES (pH 7.4; Wako, Tokyo, Japan) in an atmosphere containing 5% CO2.
Mice
Female C57BL/6J mice (age, 8 weeks) were purchased from Shimizu Laboratory Animals (Kyoto, Japan). The mice were maintained in a pathogen-free environment at 25°C under controlled lighting (12-h light/12-h dark cycles) and allowed free access to water and food pellets. All animal studies were performed in accordance with the Recommendations for Handling of Laboratory Animals for Biomedical Research compiled by the Committee on Safety and Ethical Handling Regulations for Laboratory Animal Experiments, Kinki University. The ethical procedures followed met the requirements of the UKCCCR guidelines.
Pulmonary metastasis mouse model
For the spontaneous pulmonary metastasis studies, 1 × 106 B16BL6 cells in 50 μL PBS were injected the right hind footpad of each mouse. By day 21, the complete primary tumors were surgically removed from each mouse. Oral administrated with 10 mg/kg simvastatin or fluvastatin began from then after removed primary tumors. Fourteen days after surgery, the mice were sacrificed, the lungs removed, rinsed with PBS and fixed in a neutral-buffered formaldehyde solution. Nodules visible as black forms in the lungs were then enumerated.
Subcutaneous tumor growth study
To induce a melanoma allograft model, B16BL6 cells were grown to 80% confluence and trypsinized. Cell viability was confirmed by trypan blue exclusion. Suspensions consisting of single cells with >90% viability were injected subcutaneously (s.c.) as a bolus of 1 × 106 cells in 50 µL of PBS into the right hind footpad of each mouse. Oral administrated with 10 mg/kg simvastatin or fluvastatin began from the day of inoculation. Tumors were measured daily with a caliper square and their volumes were calculated using the formula (a × b 2)/2, where a and b is the larger and smaller diameters, respectively.
Western blotting
Mice were sacrificed, and the tumors were quickly dissected and frozen on dry ice for analysis by Western blot. Briefly, tissues were homogenized in ice-cold buffer and centrifuged. Aliquots of supernatants were used to determine protein content using a BCA protein assay kit (Pierce, Rockford, IL, USA). Samples (40 μg of total protein) were separated by electrophoresis on a 10% sodium dodecyl sulfate-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Amersham, Arlington Heights, IL, USA). Membranes were blocked with a solution containing 3% skim milk and incubated overnight at 4°C with primary antibodies against anti-phospho-LIM kinase (LIMK) antibody, anti-LIMK antibody, anti-c-Fos antibody, anti-cyclin D1 antibody, anti-cyclin E2 antibody, anti-VLA-4 antibody, anti-VLA-5 antibody, anti-VLA-6 antibody (Cell Signaling Technology, Beverly, MA, USA), anti-MMP-14 antibody (Calbiochem), anti-VLA-1 antibody, anti-VLA-2 antibody, anti-VLA-3 antibody, anti-SRF antibody, anti-p53 antibody, anti-p27 antibody, anti-p21 antibody, anti-pan phospho-CDK antibody (SantaCruz Biotechnology, CA, USA), and anti-Rho antibody (Upstate Biology, Charlottesville, VA, USA). The membranes were subsequently incubated for 1 h at room temperature with anti-rabbit IgG sheep antibody coupled to horseradish peroxidase (Amersham). Proteins were visualized using a chemiluminescence kit (Amersham), according to the manufacturer’s instructions. The β-actin protein detected using a mouse monoclonal antibody (Sigma) was used as an internal standard.
Quantitative real-time polymerase chain reaction (PCR)
Total RNA was isolated using RNAiso (Takara Biomedical; Siga, Japan). One microgram of purified total RNA was used for the real-time PCR analysis with the PrimeScript RT reagent kit (Takara Biomedical). cDNA was subjected to quantitative real-time PCR by using SYBR Premix Ex Taq (Takara Biomedical) and the Thermal Cycler Dice Real Time system (Takara Biomedical) in a 96-well plate according to the manufacturer’s instructions. The PCR conditions for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), MMP-1, MMP-2, MMP-9, MMP-14, VLA-1, VLA-2, VLA-3, VLA-4, VLA-5, and VLA-6 were 94°C for 2 min; followed by 40 cycles of 94°C for 0.5 min, 50°C for 0.5 min, and 72°C for 0.5 min. The following primers were used: MMP-1, 5’-CGA CTC TAG AAA CAC AAG AGC AAG A-3’ (5’-primer) and 5’-AAG GTT AGC TTA CTG TCA CAC GCT T-3’ (3’-primer); MMP-2, 5’-TGT GTC TTC CCC TTC ACT TT-3’ (5’-primer) and 5’-GAT CTG AGC GAT GCC ATC AA-3’ (3’-primer); MMP-9, 5’-AGG CCT CTA CAG AGT CTT TG-3’ (5’-primer) and 5’-CAG TCC AAC AAG AAA GGA CG-3’ (3’-primer); MMP-14, 5’-ACA CCC TTT GAT GGT GAA GG-3’ (5’-primer) and 5’-TCG GAG GGA TCG TTA GAA TG-3’ (3’-primer); VLA-1, 5’-CCT GTA CTG TAC CCA ATT GGA TGG-3’ (5’-primer) and 5’-GTG CTC TTA TGA AAG TCG GTT TCC-3’ (3’-primer); VLA-2, 5’-TCT GCG TGT GGA CAT CAG TTT GGA-3’ (5’-primer) and 5’-GAT AAC CCC TGT CGG TAC TTC TGC-3’ (3’-primer); VLA-3, 5’-ATT GAC TCA GAG CTG GTG GAG GAG-3’ (5’-primer) and 5’-TAC TTG GGC ATA ATC CGG TAG TAG-3’ (3’-primer); VLA-4, 5’-GTC TTC ATG CTC CCA ACA GC-3’ (5’-primer) and 5’-ACT TCT GAC GTG ATT ACA GGA AGC-3’ (3’-primer); VLA-5, 5’-CTG CAG CTG CAT TTC CGA GTC TGG-3’ (5’-primer) and 5’-GAA GCC GAG CTT GTA GAG GAC GTA-3’ (3’-primer); VLA-6, 5’-GAG GAA TAT TCC AAA CTG AAC TAC-3’ (5’-primer) and 5’-GGA ATG CTG TCA TCG TAC CTA GAG-3’ (3’-primer); GAPDH, 5’-ACT TTG TCA AGC TCA TTT-3’ (5’-primer) and 5’-TGC AGC GAA CTT TAT TG-3’ (3’-primer). As an internal control for each sample, the GAPDH gene was used for standardization. Cycle threshold (Ct) values were established, and the relative difference in expression from GAPDH expression was determined according to the 2-∆∆Ct method of analysis and compared to the expression in vehicles.
Procedure for all survival studies
B16BL6 cells (1 × 105 cells in 0.2 ml) were injected into the tail vein of syngeneic C57BL/6J mice, after viable cells were counted with trypan blue exclusion. In the experiment, the B16BL6-inoculated mice were randomly divided into 3 groups comprising 20 mice each. Of these 3 groups, 1 was orally administered 0.1% DMSO and defined as the control (vehicle) group, whereas the other groups were orally administered 10 mg/kg/day of simvastatin or fluvastatin for end from the day of inoculation.
Statistical analysis
All results are expressed as means and S.D. of several independent experiments. Multiple comparisons of the data were done by ANOVA with Dunnet’s test. P values less than 5% were regarded as significant. To assess survival rates, the Kaplan-Meier model was used, followed by the long-rank test for pairwise group comparisons.
Results
Statins inhibited tumor growth and spontaneous metastasis by suppressing the expression of matrix metalloproteinases (MMPs) and very late antigens (VLAs)
We investigated the effects of simvastatin and fluvastatin in a spontaneous metastasis model. The number of lung metastatic nodules was significantly reduced (P<0.01) after 14 days of treatment with simvastatin or fluvastatin in comparison with vehicle (Figure 1A). We also investigated whether statins could suppress tumor growth in an in vivo model. B16BL6 melanoma cells were subcutaneously injected into C57BL/6J mice, which were treated with simvastatin, fluvastatin, or vehicle for 21 days. We observed a significant inhibition (P<0.01) in tumor growth in the simvastatin- and fluvastatin-treated groups relative to the vehicle-treated group (Figure 1B). The effects of statins on morbidity (as assessed by changes in body weight) and mortality were also evaluated. Mice treated with statins did not exhibit any weight loss (data not shown).
Figure 1.
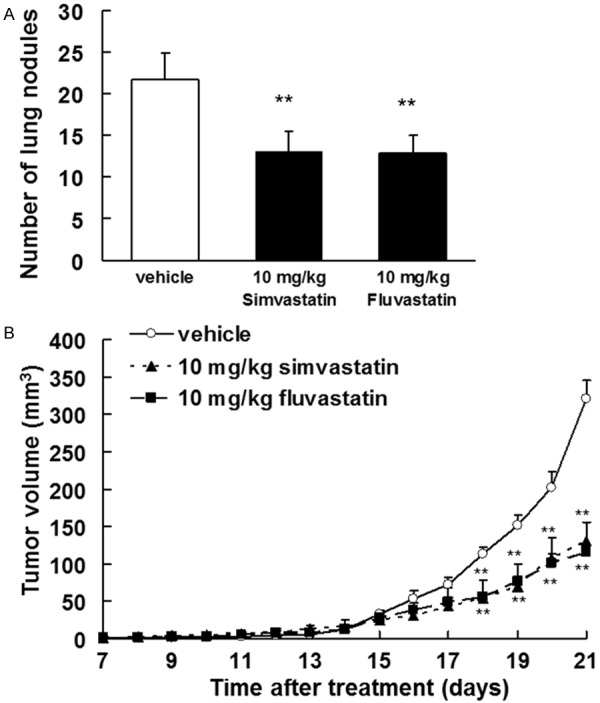
Statins inhibited spontaneous metastasis and tumor growth in vivo. A. The number of lung metastases was quantified 14 days after removal of the primary tumor. Treatment with 10 mg/kg of simvastatin or fluvastatin began 1 day after removal of the primary tumors. The results are expressed as the means ± SD of 9 mice per group. **P<0.01 vs. vehicle (0.1% DMSO-treated; ANOVA with Dunnett’s test). B. B16BL6 melanoma cells (1 × 106 cells/mouse) were injected subcutaneously into the right hind footpad of female C57BL/6J mice. Treatment with 10 mg/kg of simvastatin or fluvastatin began 1 day after inoculation. The results are expressed as the means ± SD of 9 mice per group. **P<0.01 vs. vehicle (ANOVA with Dunnett’s test).
We next investigated the inhibitory effects of statins on MMP and VLA expression in whole tumor lysates. These proteins are presumed to be critically involved in tumor invasion and metastasis [15-21]. Statins markedly inhibited the mRNA expression and enzymatic activities of MMP-1, MMP-2, MMP-9, and MMP-14 (Figure 2) as well as the mRNA and protein expression of VLA-2, VLA-4, and VLA-5 (Figure 3).
Figure 2.
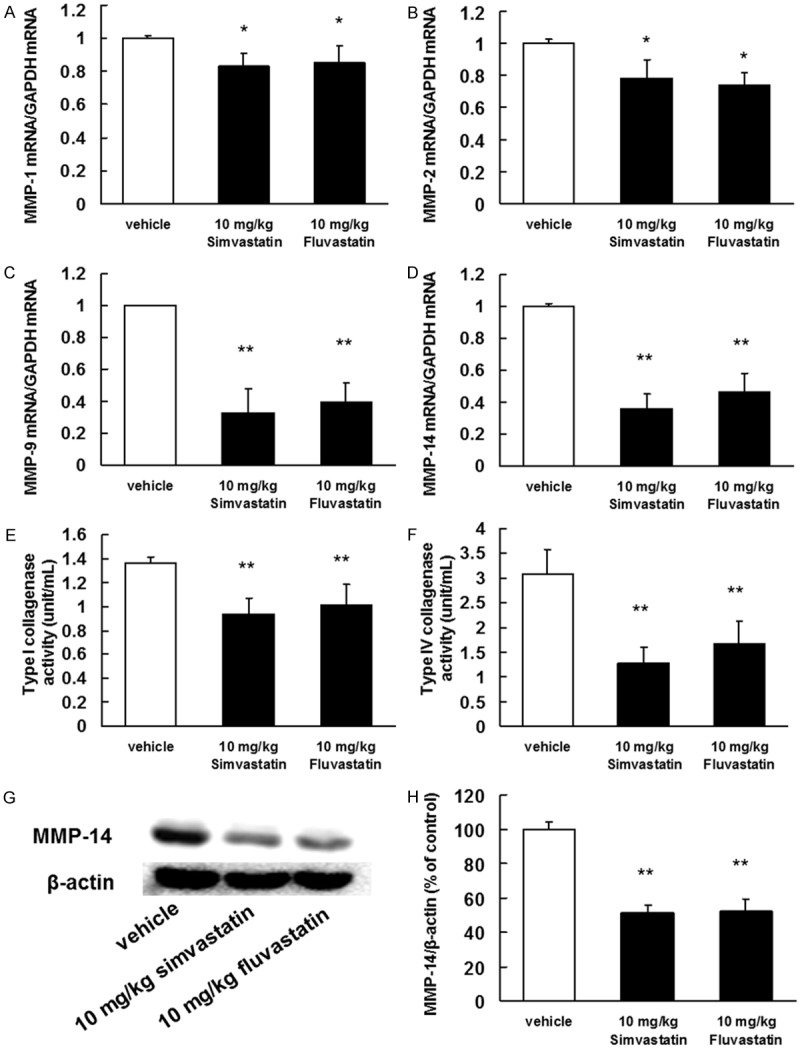
Inhibitory effects of statins on MMP mRNA expression and protein activity. B16BL6 melanoma cells (1 × 106 cells/mouse) were injected subcutaneously into the right hind footpad of female C57BL/6J mice. Treatment with 10 mg/kg of simvastatin or fluvastatin began 1 day after inoculation. After 21 days, the primary tumors were harvested. (A-D) Total RNA was extracted, and the MMP-1, MMP-2, MMP-9, and MMP-14 mRNA levels were determined by real-time polymerase chain reaction. The results are expressed as test: control ratios after correction according to the glyceraldehyde 3-phosphate dehydrogenase mRNA levels. The results are representative of 5 independent experiments. *P<0.01 vs. vehicle (ANOVA with Dunnett’s test). Activity levels of (E) type I collagenase (MMP-1) and (F) the type IV collagenases (MMP-2 and MMP-9) in the primary tumors. (G) Image of a western blot showing MMP-14 protein expression. (H) Quantification of the amount of MMP-14 after normalization to the amount of total β-actin. The results are representative of 5 independent experiments. **P<0.01 vs. vehicle (ANOVA with Dunnett’s test).
Figure 3.
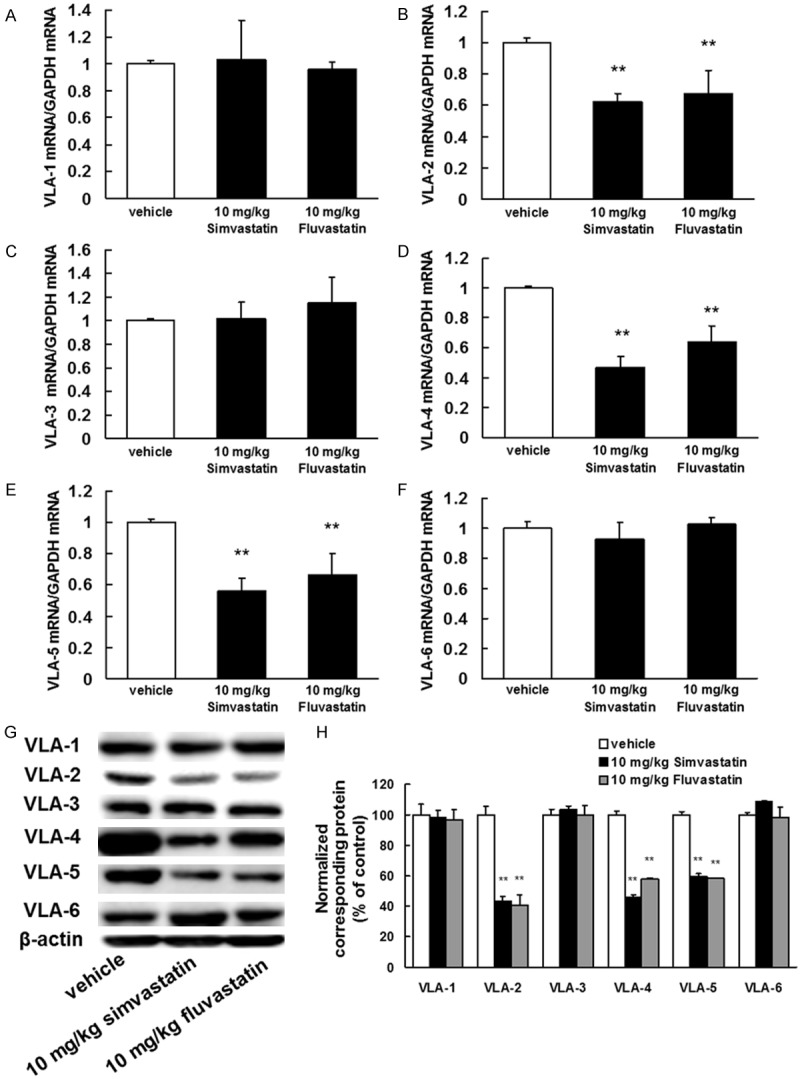
Inhibitory effects of statins on VLA mRNA and protein expression. B16BL6 melanoma cells (1 × 106 cells/mouse) were injected subcutaneously into the right hind footpad of female C57BL/6J mice. Treatment with 10 mg/kg of simvastatin or fluvastatin began 1 day after inoculation. After 21 days, the primary tumors were harvested. A-F. Total RNA was extracted, and the VLA-1, VLA-2, VLA-3, VLA-4, VLA-5, and VLA-6 mRNA levels were determined by real-time polymerase chain reaction. The results are expressed as test: control ratios after correction according to the glyceraldehyde 3-phosphate dehydrogenase mRNA levels. The results are representative of 5 independent experiments. *P<0.01 vs. vehicle (ANOVA with Dunnett’s test). G. Image of a western blot showing VLA-1, VLA-2, VLA-3, VLA-4, VLA-5, and VLA-6 protein expression. Primary tumor lysates were generated and immunoblotted with antibodies against VLA-1, VLA-2, VLA-3, VLA-4, VLA-5, VLA-6, and β-actin (internal standard). H. Quantification of the amounts of VLA-1, VLA-2, VLA-3, VLA-4, VLA-5, or VLA-6 after normalization to the amount of total β-actin. **P<0.01 vs. vehicle (ANOVA with Dunnett’s test).
Inhibitory effects of statins on the Rho/LIM kinase (LIMK)/serum response factor (SRF)/c-Fos signaling pathway in vivo
To test whether statins inhibited the function of Rho by suppressing its prenylation, whole tumor lysates were subjected to a standard western blot assay to detect the presence of small GTPases. A significant decrease in prenylated Rho proteins was observed in tumor lysates from statin-treated mice compared with lysates from vehicle-treated mice. The reverse was true for unprenylated Rho proteins (Figure 4A, 4D). Moreover, statins inhibited phosphorylation of LIMK and expression of SRF and c-Fos, which are signaling pathway components located downstream of Rho (Figure 4B, 4E). Thus, our experimental results suggested that the Rho/LIMK/SRF/c-Fos signaling pathway is inhibited by statins.
Figure 4.
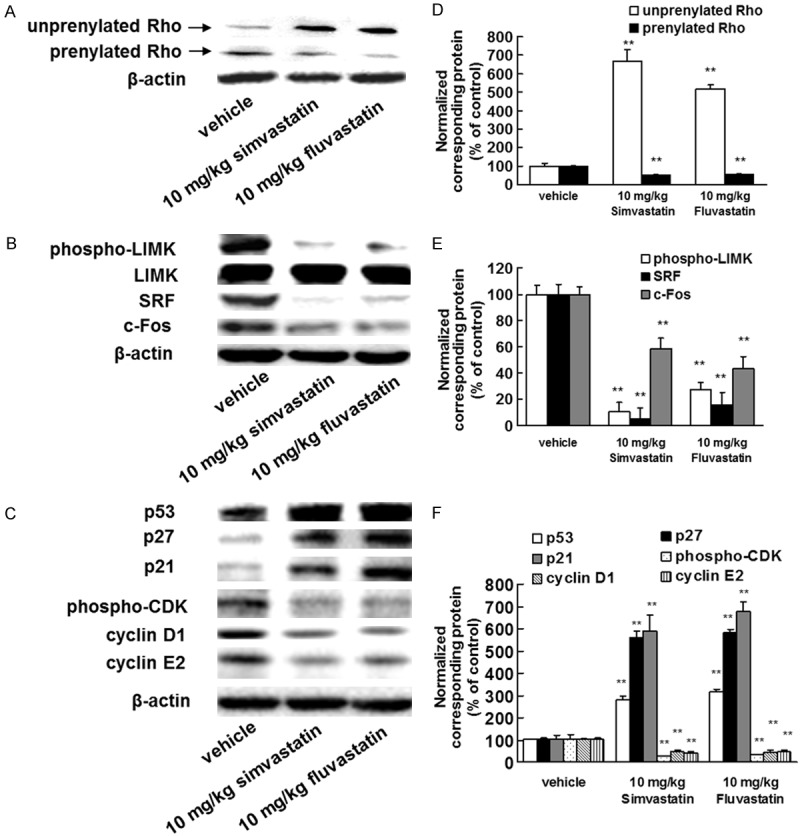
Statins specifically suppress the Rho/LIMK/SRF/c-Fos pathway. B16BL6 melanoma cells (1 × 106 cells/mouse) were injected subcutaneously into the right hind footpad of female C57BL/6J mice. Treatment with 10 mg/kg of simvastatin or fluvastatin began 1 day after inoculation. After 21 days, the primary tumors were harvested. A-C. Images of western blots for the Rho, phospho-LIMK, LIMK, SRF, c-Fos, p53, p27, p21, pan phospho-CDK, cyclin D1, and cyclin E2 proteins. Primary tumor lysates were generated and immunoblotted with antibodies against Rho and β-actin (internal standard). D-F. Quantification of the amounts of Rho, phospho-LIMK, SRF, c-Fos, p53, p27, p21, phospho-CDK, cyclin D1, or cyclin E2 after normalization to the amounts of total LIMK (phospho-LIMK) or β-actin (all other proteins). **P<0.01 vs. vehicle (ANOVA with Dunnett’s test).
Statins enhance cyclin-dependent kinase (CDK) inhibitor expression and reduce CDK phosphorylation and cyclin D1 and E2 expression
To elucidate the mechanisms by which statins inhibit tumor growth, we investigated the downstream targets of Rho. Rho has been suggested to target CDK inhibitors [22]. In addition, SRF and c-Fos regulate the expression of cyclin D1 and cyclin E2 [23,24]. We therefore investigated the levels of CDK inhibitors, cyclin D1, cyclin E2, and pan phosphorylated CDK in whole tumor lysates. Statin treatment enhanced the expression of p53, p27, and p21 (Figure 4C, 4F). Moreover, statins decreased the levels of pan phosphorylated CDK and expression of cyclin D1 and cyclin E2 (Figure 4C, 4F). These results indicated that statins inhibited tumor growth by enhancing expression of CDK inhibitors, reducing CDK phosphorylation, and decreasing levels of cyclin D1 and E2.
Statins improve the survival rate in a metastasis model
To determine whether statins improve the survival rate in vivo, an experimental metastasis model was generated by injecting B16BL6 cells into the tail veins of C57BL/6J mice to elicit lung metastases. Treatment with simvastatin or fluvastatin significantly improved the survival rates of the mice (P<0.01, log-rank test; Figure 5). The median survival durations of the vehicle, simvastatin, and fluvastatin-treated groups were 27, 38, and 36 days, respectively. All mice in the vehicle group died within 29 days post-injection, whereas mice in the simvastatin and fluvastatin groups remained alive at days 44 or 40, respectively. These results indicated that statin treatment improved the survival rate of lung metastasis-bearing mice.
Figure 5.
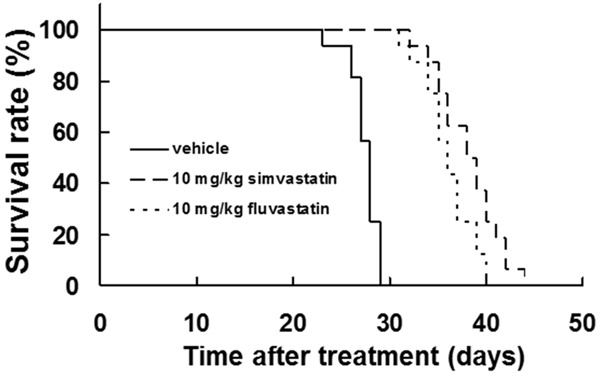
Statins prolonged the lifespan in a metastatic mouse model. B16BL6 melanoma cells (1 × 105 cells) were injected into the tail veins of syngeneic C57BL/6J mice. Treatment with 10 mg/kg of simvastatin or fluvastatin began 1 day after inoculation. The survival duration was significantly longer in the statin-treated group than in the vehicle-treated group (P<0.01, log-rank test).
Discussion
Although most melanomas are diagnosed at early stages, later stage metastatic disease is associated with a dismal prognosis and a median survival rate of less than a year. Our previous study indicated that statins inhibit tumor cell migration, invasion, and adhesion in vitro and reduce metastasis by suppressing the Rho/ROCK pathway signaling [14]. However, whether statins can inhibit spontaneous metastasis and tumor growth remained unclear. In this study, we demonstrated that statins inhibit spontaneous metastasis and tumor growth in a mouse model. RhoC expression has been shown to play an important role in tumor growth; for example, increased RhoC expression in primary cutaneous melanoma was strongly associated with larger, ulcerated tumors [5]. Plexin B1-mediated suppression of Rho activity inhibited integrin-dependent migration in melanoma cells [25]. In addition, RhoC has been shown to play an important role in metastasis and tumor cell invasion [26]. These findings suggest that Rho activation might play an important role in tumor metastasis and growth.
Rho promotes gene transcriptional regulation by modulating nuclear actin polymerization. The actin cytoskeleton is used as a signaling intermediate by SRF, thus culminating in the transcriptional regulation of growth-promoting genes [27]. SRF can control the induction of c-Fos, leading to the upregulation of MMPs and VLAs [28,29]. We observed that statins significantly inhibited the activation of LIMK and expression of SRF and c-Fos by suppressing Rho prenylation. We also found that statins suppressed the mRNA expression and enzymatic activities of MMP-1, MMP-2, MMP-9, and MMP-14 as well as the mRNA and protein levels of VLA-2, VLA-4, and VLA-5 in tumors. RhoA overexpression induces MMP expression and activity [30]. Additionally, the activation of small GTPases increases cell adhesion to collagen, fibronectin, and laminin [31]. Our previous report suggested that activation of Rho inhibit cell invasion and adhesion via the suppression of MMP and VLA mRNA expression [16]. These findings suggest that statins inhibit MMP and VLA expression by downregulating the Rho/LIMK/SRF/c-Fos pathway, thereby suppressing spontaneous metastasis.
Rho activation induces cell proliferation by enhancing the expression of cyclins and CDKs while suppressing CDK inhibitors [32]. In addition, CCG-1423, a Rho/SRF inhibitor, was shown to inhibit cell growth in RhoC-overexpressing melanoma cell lines [33]. Our present study clearly indicated that statins induced p53, p27, and p21 expression but suppressed CDK phosphorylation and cyclin D1 and E1 levels in tumor cells. It has been reported that the ubiquitin-mediated degradation of p53 protein enhanced the activation of RhoA and MDM2, and activation of MDM2 by RhoA induced cell proliferation, invasion, migration, and stress fiber formation [34]. Furthermore, inhibitor of growth-4, a tumor suppressor that promotes apoptosis by inhibiting the RhoA/ROCK kinase pathway, was significantly decreased in malignant melanoma cells, correlating with poor overall and disease-specific 5-year survival of primary melanoma patients [35]. It has also been shown that inhibition of protein geranylgeranylation and RhoA pathways induced apoptosis in HUVEC, and that induction of p53 or other proapoptotic proteins is required for this process [36]. Furthermore, inhibition of Rho activity suppressed cell proliferation via enhancing p27 expression [22]. These findings suggest that statins inhibit tumor cell growth by reducing phosphorylation of CDK and expression of cyclin D1 and E1, while enhancing p53, p27, and p21 levels, via suppression of the Rho/LIMK/SRF/c-Fos pathway.
In the present study, we found that statins improved survival rates in lung metastasis-bearing mice. Fluvastatin or simvastatin are usually administered orally at daily doses of 20-80 mg or 5-40 mg, respectively, to patients with hypercholesterolemia. The peak plasma concentrations of fluvastatin and simvastatin achieved with standard doses were ≤1 μM and 3.9 μM, respectively [37,38]. It has been reported that the peak plasma concentration of simvastatin following a 100 mg/kg oral dose was 1360 ng/mL (3.0 μM) in FVB mice [39]. In addition, the average plasma concentration of fluvastatin following oral administration at 40 mg/kg/day in chow was 60 ng/mL (0.14 μM) in mice [40]. These findings indicate that doses of 10 mg/kg of fluvastatin or simvastatin are within the peak plasma values in humans. Importantly, the oral statin dosages administered to patients with hypercholesterolemia would have prophylactic effects against tumor growth and metastasis [14]. These data suggest that statins are therapeutically useful for the treatment of various tumors.
Previous reports suggest that long-term use of statins is associated with lower risk of melanoma (relative risk = 0.79, 95% confidence interval [CI] = 0.66 to 0.96) [41]. Statin use is associated with a significantly reduced Breslow thickness at diagnosis (-19.2%, 95% CI = -33.2 to -2.3, P = 0.03), and average number of months to metastasis was shown to be significantly higher for statin users than for non-users (28.4 versus 16.5, P = 0.03) [42]. Moreover, a simvastatin dose of 40 mg or higher was associated with a statistically significant decrease in incidences of melanoma (hazard ratio [HR] = 0.64, 95% CI = 0.44 to 0.94) [43]. Simvastatin enhanced the sensitivity of gefitinib in advanced gefitinib-resistant non-small cell lung cancer patients, and statin use in patients with inflammatory breast cancer was associated with a significant improvement in disease-free survival (HR = 0.46, 95% CI 0.23 to 0.69; P<0.001), overall survival (HR = 0.50, 95% CI 0.31 to 0.80; P = 0.004) [44]. In the present study, we clearly demonstrated that statins inhibited tumor growth and metastasis and improved survival rates in lung metastasis-bearing mice. Therefore, these findings collectively suggest that statins may have an important role in the treatment of melanoma, either as single agents or in combination with chemotherapy. Our study is the first to identify the Rho/LIMK/SRF/c-Fos pathway as the mechanism through which statins inhibit tumor growth and metastasis, leading to improved survival rates in lung metastasis-bearing mice. These results clearly suggest that statins might act more effectively against Rho-variable tumors. Our findings support the further development of statins as potential anticancer agents.
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (Grant number 15K08116), Grant-in-Aid for Young Scientists (B) (Grant number 25860071) from the Japan Society for the Promotion of Science (JSPS) and by Ministry of Education, Culture, Sports, Science, and Technology (MEXT)-Supported Program for the Strategic Reseach Foundation at Private Universities, 2014-2018 (Grant number S1411037).
Disclosure of conflict of interest
None.
References
- 1.Tarhini AA, Agarwala SS. Cutaneous melanoma: available therapy for metastatic disease. Dermatol Ther. 2006;19:19–25. doi: 10.1111/j.1529-8019.2005.00052.x. [DOI] [PubMed] [Google Scholar]
- 2.Howe HL, Wingo PA, Thun MJ, Ries LA, Rosenberg HM, Feigal EG, Edwards BK. Annual report to the nation on the status of cancer (1973 through 1998), featuring cancers with recent increasing trends. J Natl Cancer Inst. 2001;93:824–842. doi: 10.1093/jnci/93.11.824. [DOI] [PubMed] [Google Scholar]
- 3.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 4.Collisson EA, Carranza DC, Chen IY, Kolodney MS. Isoprenylation is necessary for the full invasive potential of RhoA overexpression in human melanoma cells. J Invest Dermatol. 2002;119:1172–1176. doi: 10.1046/j.1523-1747.2002.19519.x. [DOI] [PubMed] [Google Scholar]
- 5.Boone B, Van Gele M, Lambert J, Haspeslagh M, Brochez L. The role of RhoC in growth and metastatic capacity of melanoma. J Cutan Pathol. 2009;36:629–636. doi: 10.1111/j.1600-0560.2008.01117.x. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 7.Nonaka M, Uota S, Saitoh Y, Takahashi M, Sugimoto H, Amet T, Arai A, Miura O, Yamamoto N, Yamaoka S. Role for protein geranylgeranylation in adult T-cell leukemia cell survival. Exp Cell Res. 2009;315:141–150. doi: 10.1016/j.yexcr.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Nishida S, Matsuoka H, Tsubaki M, Tanimori Y, Yanae M, Fujii Y, Iwaki M. Mevastatin induces apoptosis in HL60 cells dependently on decrease in phosphorylated ERK. Mol Cell Biochem. 2005;269:109–114. doi: 10.1007/s11010-005-3086-0. [DOI] [PubMed] [Google Scholar]
- 9.Lu G, Xiao H, You H, Lin Y, Jin H, Snagaski B, Yang CS. Synergistic inhibition of lung tumorigenesis by a combination of green tea polyphenols and atorvastatin. Clin Cancer Res. 2008;14:4981–4988. doi: 10.1158/1078-0432.CCR-07-1860. [DOI] [PubMed] [Google Scholar]
- 10.Tsubaki M, Yamazoe Y, Yanae M, Satou T, Itoh T, Kaneko J, Kidera Y, Moriyama K, Nishida S. Blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathways by statins reduces the expression of bFGF, HGF, and TGF-β as angiogenic factors in mouse osteosarcoma. Cytokine. 2011;54:100–107. doi: 10.1016/j.cyto.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Yanae M, Tsubaki M, Satou T, Itoh T, Imano M, Yamazoe Y, Nishida S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J Exp Clin Cancer Res. 2011;30:74. doi: 10.1186/1756-9966-30-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alonso DF, Farina HG, Skilton G, Gabri MR, De Lorenzo MS, Gomez DE. Reduction of mouse mammary tumor formation and metastasis by lovastatin, an inhibitor of the mevalonate pathway of cholesterol synthesis. Breast Cancer Res Treat. 1998;50:83–93. doi: 10.1023/a:1006058409974. [DOI] [PubMed] [Google Scholar]
- 13.Zhong WB, Liang YC, Wang CY, Chang TC, Lee WS. Lovastatin suppresses invasiveness of anaplastic thyroid cancer cells by inhibiting Rho geranylgeranylation and RhoA/ROCK signaling. Endocr Relat Cancer. 2005;12:615–629. doi: 10.1677/erc.1.01012. [DOI] [PubMed] [Google Scholar]
- 14.Kidera Y, Tsubaki M, Yamazoe Y, Shoji K, Nakamura H, Ogaki M, Satou T, Itoh T, Isozaki M, Kaneko J, Tanimori Y, Yanae M, Nishida S. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J Exp Clin Cancer Res. 2010;29:127. doi: 10.1186/1756-9966-29-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsubaki M, Satou T, Itoh T, Imano M, Ogaki M, Yanae M, Nishida S. Reduction of metastasis, cell invasion, and adhesion in mouse osteosarcoma by YM529/ONO-5920-induced blockade of the Ras/MEK/ERK and Ras/PI3K/Akt pathway. Toxicol Appl Pharmacol. 2012;259:402–410. doi: 10.1016/j.taap.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 16.Tanimori Y, Tsubaki M, Yamazoe Y, Satou T, Itoh T, Kidera Y, Yanae M, Yamamoto C, Kaneko J, Nishida S. Nitrogen-containing bisphosphonate, YM529/ONO-5920, inhibits tumor metastasis in mouse melanoma through suppression of the Rho/ROCK pathway. Clin Exp Metastasis. 2010;27:529–538. doi: 10.1007/s10585-010-9342-z. [DOI] [PubMed] [Google Scholar]
- 17.Yamazoe Y, Tsubaki M, Matsuoka H, Satou T, Itoh T, Kusunoki T, Kidera Y, Tanimori Y, Shoji K, Nakamura H, Ogaki M, Nishiura S, Nishida S. Dimethylfumarate inhibits tumor cell invasion and metastasis by suppressing the expression and activities of matrix metalloproteinases in melanoma cells. Cell Biol Int. 2009;33:1087–1094. doi: 10.1016/j.cellbi.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 18.Matsuoka H, Tsubaki M, Yamazoe Y, Ogaki M, Satou T, Itoh T, Kusunoki T, Nishida S. Tamoxifen inhibits tumor cell invasion and metastasis in mouse melanoma through suppression of PKC/MEK/ERK and PKC/PI3K/Akt pathways. Exp Cell Res. 2009;315:2022–2032. doi: 10.1016/j.yexcr.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Tsubaki M, Matsuoka H, Yamamoto C, Kato C, Ogaki M, Satou T, Itoh T, Kusunoki T, Tanimori Y, Nishida S. The protein kinase C inhibitor, H7, inhibits tumor cell invasion and metastasis in mouse melanoma via suppression of ERK1/2. Clin Exp Metastasis. 2007;24:431–438. doi: 10.1007/s10585-007-9080-z. [DOI] [PubMed] [Google Scholar]
- 20.Eble JA, Haier J. Integrins in cancer treatment. Curr Cancer Drug Targets. 2006;6:89–105. doi: 10.2174/156800906776056518. [DOI] [PubMed] [Google Scholar]
- 21.Stricker TP, Dumin JA, Dickeson SK, Chung L, Nagase H, Parks WC, Santoro SA. Structural analysis of the alpha(2) integrin I domain/procollagenase-1 (matrix metalloproteinase-1) interaction. J Biol Chem. 2001;276:29375–29381. doi: 10.1074/jbc.M102217200. [DOI] [PubMed] [Google Scholar]
- 22.Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27 (Kip1) J Biol Chem. 1999;274:21926–21931. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- 23.Babu RL, Naveen Kumar M, Patil RH, Devaraju KS, Ramesh GT, Sharma SC. Effect of estrogen and tamoxifen on the expression pattern of AP-1 factors in MCF-7 cells: role of c-Jun, c-Fos, and Fra-1 in cell cycle regulation. Mol Cell Biochem. 2013;380:143–151. doi: 10.1007/s11010-013-1667-x. [DOI] [PubMed] [Google Scholar]
- 24.He X, Xu H, Zhao M, Wang S. Serum response factor is overexpressed in esophageal squamous cell carcinoma and promotes Eca-109 cell proliferation and invasion. Oncol Lett. 2013;5:819–824. doi: 10.3892/ol.2013.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spangler B, Vardimon L, Bosserhoff AK, Kuphal S. Post-transcriptional regulation controlled by E-cadherin is important for c-Jun activity in melanoma. Pigment Cell Melanoma Res. 2011;24:148–164. doi: 10.1111/j.1755-148X.2010.00787.x. [DOI] [PubMed] [Google Scholar]
- 26.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 27.Wojnacki J, Quassollo G, Marzolo MP, Cáceres A. Rho GTPases at the crossroad of signaling networks in mammals: impact of Rho-GTPases on microtubule organization and dynamics. Small GTPases. 2014;5:e28430. doi: 10.4161/sgtp.28430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzniewska B, Rejmak E, Malik AR, Jaworski J, Kaczmarek L, Kalita K. Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol Cell Biol. 2013;33:2149–2162. doi: 10.1128/MCB.00008-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksson M, Arminen L, Karjalainen-Lindsberg ML, Leppä S. AP-1 regulates alpha2beta1 integrin expression by ERK-dependent signals during megakaryocytic differentiation of K562 cells. Exp Cell Res. 2005;304:175–186. doi: 10.1016/j.yexcr.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 30.Cáceres M, Guerrero J, Martínez J. Overexpression of RhoA-GTP induces activation of the Epidermal Growth Factor Receptor, dephosphorylation of focal adhesion kinase and increased motility in breast cancer cells. Exp Cell Res. 2005;309:229–238. doi: 10.1016/j.yexcr.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 31.Danker K, Mechai N, Lucka L, Reutter W, Horstkorte R. The small Gtpase ras is involved in growth factor-regulated expression of the alpha1 integrin subunit in PC12 cells. Biol Chem. 2001;382:969–972. doi: 10.1515/BC.2001.121. [DOI] [PubMed] [Google Scholar]
- 32.Xie S, Zhu M, Lv G, Geng Y, Chen G, Ma J, Wang G. Overexpression of Ras homologous C (RhoC) induces malignant transformation of hepatocytes in vitro and in nude mouse xenografts. PLoS One. 2013;8:e54493. doi: 10.1371/journal.pone.0054493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez-Lluhí JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6:2249–2260. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 34.Ma J, Xue Y, Cui W, Li Y, Zhao Q, Ye W, Zheng J, Cheng Y, Ma Y, Li S, Han T, Miao L, Yao L, Zhang J, Liu W. Ras homolog gene family, member A promotes p53 degradation and vascular endothelial growth factor-dependent angiogenesis through an interaction with murine double minute 2 under hypoxic conditions. Cancer. 2012;118:4105–4116. doi: 10.1002/cncr.27393. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Martinka M, Li G. Role of ING4 in human melanoma cell migration, invasion and patient survival. Carcinogenesis. 2008;29:1373–1379. doi: 10.1093/carcin/bgn086. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Liu L, Tupper JC, Bannerman DD, Winn RK, Sebti SM, Hamilton AD, Harlan JM. Inhibition of protein geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis in human endothelial cells. J Biol Chem. 2002;277:15309–15316. doi: 10.1074/jbc.M201253200. [DOI] [PubMed] [Google Scholar]
- 37.Horiguchi A, Sumitomo M, Asakuma J, Asano T, Asano T, Hayakawa M. 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitor, fluvastatin, as a novel agent for prophylaxis of renal cancer metastasis. Clin Cancer Res. 2004;10:8648–8655. doi: 10.1158/1078-0432.CCR-04-1568. [DOI] [PubMed] [Google Scholar]
- 38.van der Weide K, de Jonge-Peeters S, Huls G, Fehrmann RS, Schuringa JJ, Kuipers F, de Vries EG, Vellenga E. Treatment with high-dose simvastatin inhibits geranylgeranylation in AML blast cells in a subset of AML patients. Exp Hematol. 2012;40:177–186. doi: 10.1016/j.exphem.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 39.Higgins JW, Bao JQ, Ke AB, Manro JR, Fallon JK, Smith PC, Zamek-Gliszczynski MJ. Utility of Oatp1a/1b-knockout and OATP1B1/3-humanized mice in the study of OATP-mediated pharmacokinetics and tissue distribution: case studies with pravastatin, atorvastatin, simvastatin, and carboxydichlorofluorescein. Drug Metab Dispos. 2014;42:182–92. doi: 10.1124/dmd.113.054783. [DOI] [PubMed] [Google Scholar]
- 40.Iida Y, Xu B, Schultz GM, Chow V, White JJ, Sulaimon S, Hezi-Yamit A, Peterson SR, Dalman RL. Efficacy and mechanism of angiotensin II receptor blocker treatment in experimental abdominal aortic aneurysms. PLoS One. 2012;7:e49642. doi: 10.1371/journal.pone.0049642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobs EJ, Newton CC, Thun MJ, Gapstur SM. Long-term use of cholesterol-lowering drugs and cancer incidence in a large United States cohort. Cancer Res. 2011;71:1763–1771. doi: 10.1158/0008-5472.CAN-10-2953. [DOI] [PubMed] [Google Scholar]
- 42.Koomen ER, Joosse A, Herings RM, Casparie MK, Bergman W, Nijsten T, Guchelaar HJ. Is statin use associated with a reduced incidence, a reduced Breslow thickness or delayed metastasis of melanoma of the skin? Eur J Cancer. 2007;43:2580–2589. doi: 10.1016/j.ejca.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Farwell WR, Scranton RE, Lawler EV, Lew RA, Brophy MT, Fiore LD, Gaziano JM. The association between statins and cancer incidence in a veterans population. J Natl Cancer Inst. 2008;100:134–139. doi: 10.1093/jnci/djm286. [DOI] [PubMed] [Google Scholar]
- 44.Han JY, Lee SH, Yoo NJ, Hyung LS, Moon YJ, Yun T, Kim HT, Lee JS. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2011;17:1553–1560. doi: 10.1158/1078-0432.CCR-10-2525. [DOI] [PubMed] [Google Scholar]