

Abstract
Progesterone induces proliferation of breast cancer cells and contributes to the development of breast cancer. The effects of progesterone are mediated by progesterone receptors (PRs). However, it is still not fully understood how the proliferative effects of PR is regulated in vivo. Increasing amount of evidence strongly suggests that dysregulation of ubiquitin-proteasome system is closely associated with cancer pathogenesis. Speckle-type POZ protein (SPOP) is an adaptor protein of the CUL3-based E3 ubiquitin ligase complexes. SPOP represents one of the highest loci for loss of heterozygosity (LOH) in breast cancer. SPOP downregulation contributes to breast cancer cell growth and invasion. In this study, we revealed PR as a bona fide substrate for SPOP. SPOP interacts with PR in vivo and targets PR for ubiquitin-dependent proteasomal degradation. Moreover, SPOP suppresses progesteroneinduced PR transactivation, S phase entry, and Erk1/2 activation. Our study revealed novel molecular mechanisms underlying the regulation of PR protein homeostasis in breast cancer cells, and provided insights in understanding the relationship between SPOP inactivation and the development of breast cancer.
Keywords: Breast cancer, SPOP, progesterone receptor, degradation, ubiquitination
Introduction
Breast cancer is the most frequently diagnosed malignant neoplasia and is a leading cause of cancer death in females worldwide. Progesterone and progesterone receptors (PRs) are essential for the development and cyclical regulation of hormone-responsive tissues including the breast and reproductive tract [1]. Progesterone and PR are increasing gaining attention for their merging roles as critical regulators of breast cancer [1,2].
PR is a member of the sex steroid receptors family that ligand-dependently regulates the functions of the sexual organs [3]. Other sex steroid receptors include the androgen receptor (AR), the estrogen receptor α/β (ER α/β) [3]. All sex steroid receptors are built with similar modular structure, including a with a nuclear location signal (NLS), and a ligand-binding domain (LBD) [3]. PR exists primarily as two coexpressed isoforms, PRA and PRB, encoded by the same gene downstream of distinct promoters. PRB, the full-length receptor, contains 164 amino acids at the N-terminus, not present in PR-A [4]. Unliganded PR rapidly shuttles between the cytoplasm and the nucleus. After binding to progesterone, PR undergoes a series of conformational changes, dimerizes and translocates into the nucleus, where it recruits co-activators or co-repressors, as well as chromatin-remodeling factors, to progesterone response elements (PREs) on target gene promoters to activate or repress transcription [5]. Moreover, it has been described a proportion of PR can be localized in the cytoplasm and at the cell membrane, where it triggers nongenomic or membrane-initiated signaling pathway [2].
The biological activity of PR is tightly controlled by posttranslational modifications such as phosphorylation, ubiquitination, sumoylation, methylation, and acetylation [1]. These modifications significantly alter receptor stability, localization, transcriptional activity and target gene selectivity. For example, upon ligand binding, PR becomes hyperphosphorylated at multiple sites, and then PR is subsequently polyubiquitinated and degraded by the proteasome [6,7]. However, the detailed mechanism of hormone-dependent or independent degradation of PR is still poorly understood.
SPOP (Speckle-type POZ Protein) is an adaptor protein of the CUL3-RBX1 E3 ubiquitin ligase complex. It selectively recruits substrates via its N-terminal MATH domain, whereas its BTB domain mediates dimerization and interaction with CUL3 [8]. SPOP has been linked to the ubiquitination and degradation of several substrates, including the AR, ERα, steroid receptor coactivator SRC-3, CHOP, Daxx, and Gli, and several others [9-13]. SPOP was identified as one of the most frequently affected genes by somatic point mutations in prostate and endometrial cancers [14,15]. However, SPOP mutation is rare in breast cancers. But a previous study showed that SPOP represented one of the highest loci for loss of heterozygosity (LOH) in breast cancer samples [11]. Moreover, SPOP depletion in breast cancer cell lines substantially reduced cell invasion and anchorage-independent growth in vitro and xenograft tumor in vivo [11], suggesting SPOP functions as a tumor suppressor in breast cancer.
In this study, we demonstrated that SPOP forms a complex with PR and targets PR for ubiquitination and proteasomal degradation. Moreover, SPOP suppresses progesterone-induced PR transactivation, S phase entry, and ERK activation. Therefore, our results propose that SPOP is a novel negative regulator of PR stability, and provide a functional insight into the function of SPOP in breast cancer proliferation.
Materials and methods
Cell culture and treatments and transfection
293T and T47D cells were obtained from the American Type Culture Collection (ATCC). All cells were maintained in DMEM medium with 10% FBS. Cells were transiently transfected using Lipofectamine RNAiMAX (for siRNA transfection) or 3000 (for plasmids transfection) (Invitrogen) according to the manufacturer’s instructions. In experiments involving treatment with progesterone (Selleck), or vehicle ethanol (EtOH), cells were placed in phenol-red-free medium with 10% dextran-coated, charcoal-stripped FBS for 48 hr prior to treatment with hormone or vehicle.
Expression constructs
The PRA and PRB cDNAs were kindly provided by Dr. Bert W. O’Malley (Baylor College of Medicine) and subcloned into pCIN4-FLAG-HA and pCMV-Myc/Flag expression vectors. The SPOP constructs were described previously [9].
RNA interference
Non-specific control siRNA and siRNAs for human SPOP were purchased from GenePharma. siRNA transfection of cells was performed following the manufacturer’s instructions. The siRNA oligos sequences for SPOP are: si-SPOP1#1: 5’-GGAUGAUGUAAAUGAGCAA-3’. si-SPOP#2: 5’-GGACAGCGACTCTGAATCT-3’. The sequence of negative control is: si-Control: 5’-ACAGACUUCGGAGUACCUG-3’.
Antibodies
The following antibodies were used: SPOP (ab137537; Abcam), SPOP (16750-1-AP; Proteintech), Cyclin D1 (ab134175; Abcam), PR (8644; CST), Myc (9E10; Sigma), Flag (M2; Sigma), HA (MM5-101R; Millipore), and Actin (AC-74; Sigma).
Immunoprecipitation
To immunoprecipitate the ectopically expressed Flag-tagged proteins, transfected cells were lysed 24 hr post-transfection in BC100 buffer. The whole-cell lysates were immunoprecipitated with the monoclonal anti-Flag antibody-conjugated M2 agarose beads (Sigma) at 4°C overnight. After three washes with Flag lysis buffer, followed by two washes with BC100 buffer, the bound proteins were eluted using Flag-Peptide (Sigma)/BC100 for 3 hr at 4°C. The eluted material was resolved by SDS-PAGE. To immunoprecipitate the endogenous proteins, cells were lysed with 1 × cell lysis buffer (Cell Signaling), and the lysate was centrifuged. The supernatant was precleared with protein A/G beads (Sigma) and incubated with indicated antibody overnight. Thereafter, protein A/G beads were applied, all at 4°C. After 2 hr of incubation, pellets were washed five times with lysis buffer and resuspended in sample buffer and analyzed by SDS-PAGE.
Western blot
Cell lysates or immunoprecipitates were subjected to SDS-PAGE and proteins were transferred to nitrocellulose membranes (GE Healthcare). The membranes were blocked in Tris-buffered saline (TBS, pH 7.4) containing 5% non-fat milk and 0.1% Tween-20, washed twice in TBS containing 0.1% Tween-20, and incubated with primary antibody for 2 hr and followed by secondary antibody for 1 hour at room temperature. The proteins of interest were visualized using ECL chemiluminescence system (Santa Cruz).
Quantitative RT-PCR
Total RNA was isolated from transiently transfected cells using the TRIzol reagent (Tiangen), and cDNA was reversed-transcribed using the Superscript RT kit (TOYOBO), according to the manufacturer’s instructions. PCR amplification was performed using the SYBR Green PCR master mix Kit (TOYOBO). All quantization were normalized to the level of endogenous control GAPDH. The primer sequences for SYBR green are as follows: 11βHSD2_fwd: CAA GGG GCC GCA TCG TGA CT-3’; 11βHSD2_rev: GCA GCA GCT CTT GAG GCA GGT TG-3’; FKBP5_fwd: 5’-CCC CCG CGG CGA CAG GTT CTC TAC-3’; FKBP5_rev: 5’-CCA ATC ATC GGC GTT TCC TCA CCA-3’; Cyclin D1_fwd: 5’-GAA CAC GGC TCA CGC TTA CCT C-3’; Cyclin D1_rev: 5’-ACT TGT GCC CTT GCC CCA TC-3’; MUC1_fwd: 5’-AGA CGT CAG CGT GAG TGA TG-3’; MUC1_rev: 5’-CAG CTG CCC GTA GTT CTT TC-3’; PGR-fwd: 5’-CGC GCT CTA CCC TGC ACT C-3’; PGR-rev: 5’-TGA ATC CGG CCT CAG GTA GTT-3’; SPOP-fwd: 5’-TGAAGCCAGAGAGCGGTATGC-3’; SPOP-rev: 5’-GAT TGC TTC AGG CGT TTG CGT G-3’.
Transcriptional reporter assay
Luciferase assay (Promega) was conducted in cells that were transiently transfected with the reporter (MMTV-Luc), pTK-galactosidase construct and the PRA/B expression constructs and/or with the Myc-SPOP. Luciferase activity in cell lysates was measured using the luciferase assay system in a Berthold Lumat LB 9507 luminometer (Promega). Luciferase activity was normalized to galactosidase activity as an internal control. Each assay was performed in triplicate and results were confirmed by at least three individually repeating experiments.
Results
SPOP interacts with PR in breast cancer cells
We and others previously reported that SPOP regulates AR and ERα protein stability in prostate and endometrial cancers, respectively [9,10]. Since PR shares many common regulators with AR and ERα [5], we ask whether SPOP could regulate PR protein stability as seen in AR and ERα. Accordingly, we first examined whether SPOP interacts with PR in cells. To do this, FLAG-HA (FH)-PRA or PRB and Myc-SPOP constructs were co-expressed in 293T cells. Cell lysates were subsequently prepared for co-immunoprecipitation (co-IP) with anti-Flag antibody. As shown in Figure 1A, Myc-SPOP was immunoprecipitated by FH-PRA or PRB, suggesting SPOP can interact with PRA or PRB. Next, we decided to extend our analysis by investigating whether endogenous SPOP and PR can interact with each other. In this case, we chose T47D cells, a PR-positive breast cancer cell line, for subsequent study. Immunoprecipitation using anti-PR antibody was performed using cell lysates prepared from T47D cells. As shown in Figure 1B, endogenous SPOP was able to be immunoprecipitated by PR, suggesting these two proteins can also interact with each other at endogenous levels. SPOP contains two structural domains: a substrate-binding MATH domain at the N-terminus and a CUL3-binding BTB domain at the C-terminus. To determine which domain may mediate its interaction with PR, we generated two deletion mutants of SPOP (SPOP-ΔBTB and ΔMATH), corresponding to the deletion of these two domains respectively (Figure 1C). co-IP assay was performed to test the binding affinity of the full length SPOP (SPOP-WT) and the two deletion mutants with overexpressed PRA in 293T cells. As shown in Figure 1D, while SPOP-WT and SPOP-ΔBTB interacted efficiently with PRA, the interaction between SPOP-ΔMATH and PRA was totally abolished. Therefore, this result suggests that the MATH domain is responsible for the interaction between SPOP and PR. Taken together, our findings demonstrate that SPOP interacts with PR in cells through the MATH domain.
Figure 1.
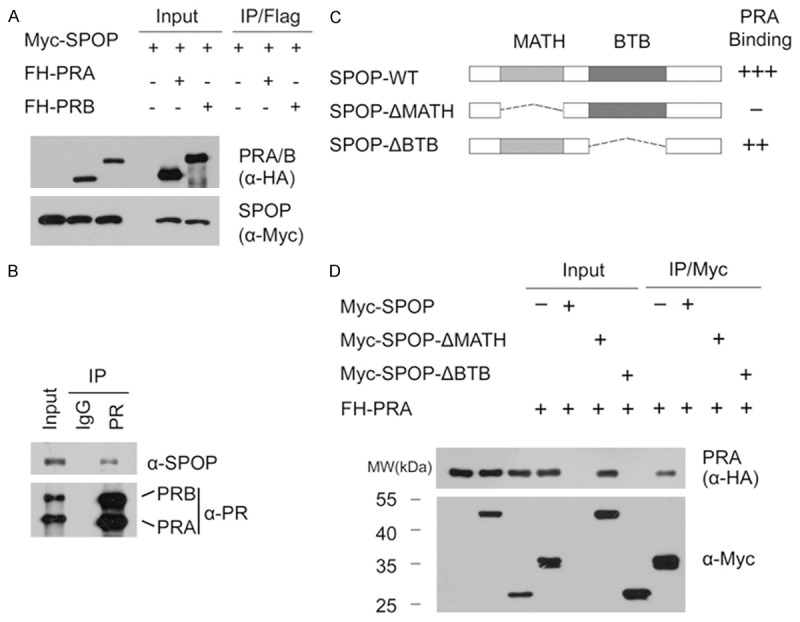
SPOP interacts with PR in cells. A. 293T cells were co-transfected with Myc-SPOP and FH-PRA or PRB constructs. After 24 hr, cell lysates were prepared for co-IP with anti-Flag antibody and WB analyses; B. After treated with 20 µM MG132 for 4 hr, T47D cell lysates were prepared for co-IP with anti-PR antibody and WB analyses with indicated antibodies; C. Schematic representation of SPOP deletion mutants. Binding capacity of SPOP to PR is indicated with the symbol; D. 293T cells were co-transfected with FH-PRA and Myc-SPOP-WT or deletion mutants (ΔMATH, ΔBTB) constructs. After 24 hr, cell lysates were prepared for co-IP assay with anti-Myc antibody and WB analyses.
SPOP targets PR for ubiquitin-dependent proteasomal degradation
Then we explored whether the SPOP could promote the ubiquitination and degradation of PR. As shown in Figure 2A and 2B, expression of SPOP decreased the ectopically co-expressed PRA or PRB protein level in 293T cells. However, this effect was completely blocked when cells were treated with the proteasome inhibitors MG132 or Bortezomib, indicating that SPOP downregulates PRA (or PRB) protein via the proteasomal degradation pathway. Moreover, SPOP-WT, but not the SPOP-ΔBTB or SPOP-ΔMATH mutant, promoted PRA (or PRB) degradation (Figure 2C), indicating that the BTB and MATH domains are both required for SPOP-mediated PR degradation. Next, we examined the effect of SPOP on the degradation of endogenous PR. Similarly, as shown in Figure 2D, overexpression of SPOP-WT, but not the SPOP-ΔBTB or SPOP-ΔMATH mutant in T47D cells resulted in a moderate decrease in the protein level of endogenous PR. Moreover, knockdown of endogenous SPOP using two gene-specific siRNAs increased PR protein levels in T47D cells (Figure 2E). We also performed qRT-PCR to measure the mRNA levels of SPOP and PR in SPOP-depleted T47D cells. In contrast to the significant decrease of SPOP mRNA level, the mRNA level of PGR in SPOP-depleted T47D cells was moderately increased when compared with control cells (Figure 2F), suggesting SPOP may play role in regulating PR mRNA level. This result may not be surprising, since PR is a transcriptional target of ERα, and SPOP negatively regulates ERα protein stability [10]. To demonstrate SPOP also regulates PR abundance at the post-translational level, we measured PR protein turnover. We inhibited new protein synthesis with cycloheximide (CHX) and followed the degradation of the PR over time. As shown in Figure 2G and 2H, knockdown of SPOP remarkably prolonged the half-life of endogenous PR protein in T47D cells. Therefore, SPOP may negatively modulate PR expression via both transcriptional and post-translational regulation.
Figure 2.

SPOP targets PR for ubiquitination and degradation. (A, B) 293T cells were transfected with FH-PRA (A) or PRB (B) in combination with or without Myc-SPOP constructs. After 24 hr, cells were treated with MG132 (20 µM), Bortezomib (200 nM), or DMSO for 4 hr before cell lysates were prepared for WB analyses; (C) FH-PRA/B and different amounts of Myc-SPOP-WT or deletion mutants (ΔMATH, ΔBTB) constructs were transfected into 293T cells. After 24 hr, cell lysates were prepared for WB analyses; (D) T47D cells were transfected with Myc-SPOP-WT, or deletion mutants (ΔMATH, ΔBTB) constructs. After 36 hr, cell lysates were prepared for WB analyses; (E) T47D cells were transfected with control siRNAs or two SPOP-specific siRNAs. After 48 hr, cell lysates were prepared for WB analyses; (F) Quantitative RT-PCR measurement of the mRNA levels of SPOP and PGR in SPOP-depleted T47D cells. The mRNA level of GAPDH was used for normalization. The mean values (S.D.) of three independent experiments are shown. (G, H) T47D cells were transfected with control or SPOP-specific siRNAs. After 48 hr, cells were chased with 30 μM cycloheximide (CHX). At the indicated time points, cell lysates were prepared for WB analyses; (G) At each time point, the intensity of PR was first normalized to the intensity of Actin (loading control) and then to the value of the 0-hr time point; (H) The mean values (S.D.) of three independent experiments are shown; (I) SPOP promotes PRA polyubiquitination in vivo. Flag-PRA, HA-Ub and Myc-SPOP-WT or ΔBTB mutant constructs were co-transfected into 293T cells. After 24 hr, cells were treated with 20 µM MG132 for 6 hr. PRA proteins were immunoprecipitated with anti-Flag antibody and resolved by SDS/PAGE. The ubiquitinated forms of PRA were analyzed by WB with anti-HA antibody.
To further determine whether PR was degraded through SPOP-mediated polyubiquitination, HA-Ubiquitin and FH-PRA constructs were co-expressed in 293T cells with different doses of SPOP-WT or SPOP-ΔBTB mutant. As shown in Figure 2I, PRA protein was robustly polyubiquitinated by the co-expressed SPOP-WT in a dose-dependent manner. In contrast, little or no PRA polyubiquitination was observed in SPOP-ΔBTB expressing cells (Figure 2I). Taken together, these data demonstrate that the SPOP regulates PR stability through ubiquitin-dependent proteasomal degradation pathway in PR-positive breast cancer cell lines.
SPOP suppresses progesterone-induced PR transactivation
To determine whether SPOP-mediated PR degradation affects PR transcriptional activity, 293T cells were co-transfected with the mouse mammary tumor virus containing reporter (MMTV-Luc), Myc-PRA/B, and Myc-SPOP constructs. As shown in Figure 3A, PRA and PRB transcriptional activation was suppressed by SPOP in the presence of progesterone. To investigate the effect of endogenous SPOP in PR-mediated transcriptional activation, T47D cells were transfected with control siRNAs or SPOP siRNAs, and then treated with EtOH or progesterone. As shown in Figure 3B, progesterone-induced PR transactivation was significantly increased in SPOP-depleted cells.
Figure 3.
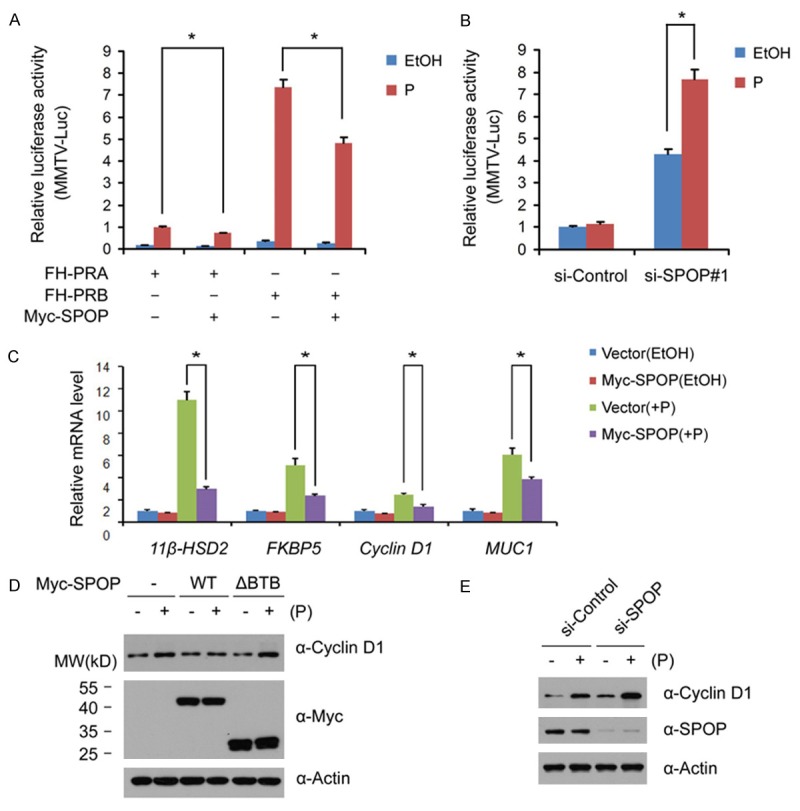
SPOP inhibits progesterone-induced transcription. A. 293T Cells were transfected with MMTV luciferase reporter, and Myc-PRA, or Myc-SPOP constructs, alone or in combination as indicated. After 24 hr, cells were treated with vehicle ethanol (EtOH) or progesterone (10 nM) for 24 hr, and the luciferase activities were measured by luminometer; B. T47D cells were transfected with control siRNAs or SPOP siRNAs, After 24 hr, cells were co-transfected with pMMTV luciferase reporter for 24 hr, and then treated with vehicle ethanol (EtOH) or progesterone (10 nM) for 24 hr, and the luciferase activities were measured by luminometer; C. T47D cells were transfected with control siRNAs or SPOP-specific siRNA. After 48 hr, cells were then treated with the vehicle ethanol (EtOH) or progesterone (10 nM) for 18 hr. The mRNA level of PR target genes (KFBP5, Cyclin D1 and HSD2) was measured by qRT-PCR methods. The mRNA level of GAPDH was used for normalization. The mean values (S.D.) of three independent experiments are shown. * indicates statistical significance (*, p < 0.01); D. T47D cells were transfected with control or Myc-SPOP or SPOP-ΔBTB mutant constructs. After 24 hr, cells were treated with vehicle ethanol (EtOH) or progesterone (10 nM) for 24 hr. Cell lysates were prepared for WB analyses; E. T47D cells were transfected with control siRNAs or SPOP-specific siRNAs. After 48 hr, cells were then treated with the vehicle ethanol (EtOH) or progesterone (10 nM) for 24 hr. Cell lysates were prepared for WB analyses.
To corroborate the results of the luciferase reporter assay, the effects of SPOP on the expression of PR target genes were examined. T47D cells were transiently transfected with control or Myc-SPOP constructs in the absence or presence of progesterone for 24 hr, and the mRNA levels of four PR transcriptional targets (11β-HSD2, FKBP5, Cyclin D1, and MUC1) were assessed by qRT-PCR. As shown in Figure 3C, progesterone-induced transcription of targets mRNAs (11β-HSD2, FKBP5, Cyclin D1, and MUC1) was significantly suppressed by Myc-SPOP overexpression. To further demonstrate SPOP regulates progesterone-induced transactivation of PR target genes, the protein level of Cyclin D1 in progesterone-treated T47D cells were assessed by Western blot. Consistent with qRT-PCR results, progesterone-induced Cyclin D1 protein expression in T47D cells was greatly comprised by SPOP overexpression (Figure 3D). In contrast, SPOP-ΔBTB mutant had no such effect (Figure 3D). Accordingly, SPOP depletion in T47D cells enhanced progesterone-induced Cyclin D1 protein expression (Figure 3E). Taken together, these results indicate that SPOP negatively regulates progesterone-induced transactivation.
SPOP suppresses progesterone-induced S phase entry, Erk1/2 activation
Progesterone can promote cell proliferation by inducing S-Phase entry in PR-positive breast cancer cells [16,17]. To investigate the effects of SPOP on the cell cycle, T47D cells were transfected with control or Myc-SPOP constructs in the absence or presence of progesterone. The cell cycle was analyzed by propidium iodide (PI) flow cytometric assay. Consistent with previous studies, there was a significant increase in S-phase percentage in T47D cells upon progesterone treatment (Figure 4A). However, the increase of the cells in S phase was largely suppressed by SPOP overexpression (Figure 4A). To further test the role of endogenous SPOP on cell cycle, T47D cells were transfected with control siRNAs or SPOP siRNAs. As shown in Figure 4B, the cells in S phase were increased when SPOP was depleted. These results suggest that SPOP inhibits progesterone-induced cell cycle progression.
Figure 4.
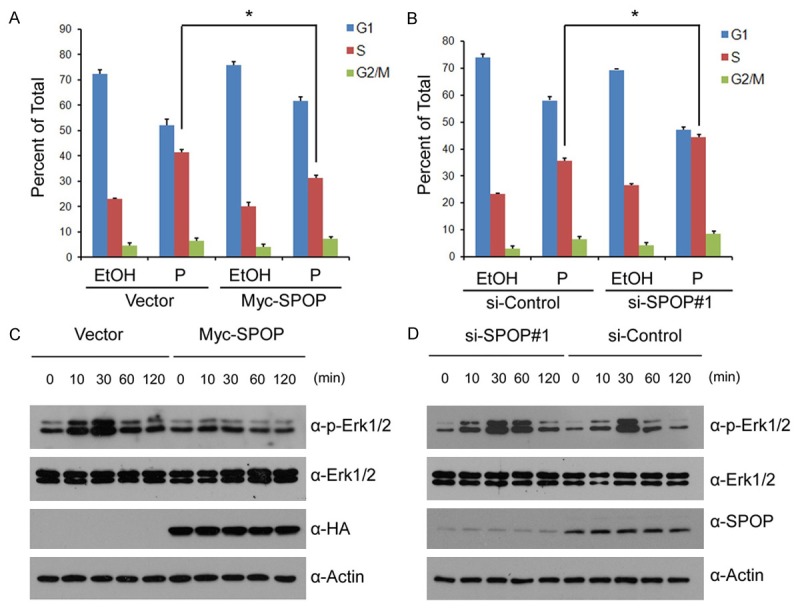
SPOP inhibits progesterone-induced S phase entry and Erk1/2 activation. (A) T47D cells were transfected with control or Myc-SPOP constructs. After 24 hr, cells were treated with the vehicle ethanol (EtOH) or progesterone (10 nM) for 24 hr. The cell cycle percentages was determined by PI staining and FACS; (B) T47D cells were transfected with control siRNAs or SPOP-specific siRNAs. After 48 hr, cells were treated and analyzed as in (A); (C) T47D cells were transfected with control or Myc-SPOP constructs. After 24 hr, cells were treated with the vehicle ethanol (EtOH) or progesterone (10 nM) for indicated times. Cell lysates were prepared for WB analyses; (D) T47D cells were transfected with control or SPOP-specific siRNA. After 48 hr, cells were treated and analyzed as in (C).
It is reported that PR induces cell cycle progression via activation of MAPK-Erk1/2 pathway [16]. To determine whether SPOP inhibits Erk1/2 activity, we transfected Myc-SPOP into T47D cells and detected the phosphorylation levels of Erk1/2. As shown in Figure 4C, SPOP overexpression decreased the progesterone-induced Erk1/2 phosphorylation. Accordingly, SPOP depletion in T47D cells increased the progesterone-induced ERK1/2 phosphorylation (Figure 4D). Taken together, these results suggest SPOP suppresses progesterone-mediated proliferative signaling in breast cancer cells, as measured by S-phase entry and ERK1/2 activation.
Discussion
Increasing amounts of epidemiological and clinical findings support the notion that uncontrolled PR action in pre-neoplastic breast tissues contributes to breast cancer development [1]. PRs are valid targets for breast cancer therapy, and antiprogestins, such as mifepristone and Lonaprisan, have already been clinically used in patients who have failed to other therapies [2]. However, how PR was dysregulated in breast cancer was less understood. In this study, we present evidence that PR is a bona fide substrate of ubiquitin ligase SPOP. SPOP promotes PR ubiquitination and proteasomal degradation, and suppresses progesterone-induced PR transactivation, targets transcription, S phase entry and Erk1/2 activation. A systematic analysis of the SPOP genomic locus revealed that a high percentage of genomic loss or loss of heterozygosity occurs at this locus in breast cancers [11]. Moreover, SPOP negatively regulates breast cell proliferation and invasion [11]. Our results suggest a possible mechanism that SPOP down-regulation leads to hyperactivity of PR signal, which promote breast cancer initiation and progression.
In addition to PR, SPOP may suppress PR signal through degrading PR co-activators. PR-regulated gene transcription is mediated through interaction with steroid receptor coregulators, such as SRC-3 [18]. SRC-3 can interact with the ligand binding domain of PR in a ligand-dependent manner. These interactions recruit SRC-3 to the promoter/enhancer region of PR target genes and facilitate hormonal regulation of transcription [18]. A previous study demonstrated SPOP interacts directly with SRC-3 in a phosphorylation-dependent manner, and target SRC-3 for ubiquitination and proteolysis [11]. In addition to PR and its co-activators, SPOP may exert its tumor suppressor in breast tissues through regulating multiple substrates. We revealed that SPOP specifically recognizes multiple S/T-rich degrons located in the AF2 domain of ERα, and triggers ERα degradation via the ubiquitin-proteasome pathway [2,10]. More than 70% of breast cancers express ERα and respond to antiestrogen therapies [2]. Most of these ERα-positive tumors also express of PR, the expression of which has been considered as a reliable marker of a functional ERα [19]. Our results also showed SPOP depletion in T47D cells moderately elevated PR mRNA level. It is not surprising, since SPOP regulates ERα stability and PR is a transcriptional target of ERα. Thus, SPOP can regulate PR at both translational and post-translational level.
For future studies, it will be useful to generate mice models of conditional breast-specific SPOP knockout to further characterize the phenotype of SPOP function in vivo, and determine whether PR pathway is dysregulated in SPOP down-regulated breast cancer.
Acknowledgements
This work was in part supported by the National Natural Science Foundation of China (81472567 to L.Y., 30872947 to P.Z., 31400753 to K.G. and 81171964 to C.J.).
References
- 1.Diep CH, Daniel AR, Mauro LJ, Knutson TP, Lange CA. Progesterone action in breast, uterine, and ovarian cancers. J Mol Endocrinol. 2015;54:R31–53. doi: 10.1530/JME-14-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanari C, Wargon V, Rojas P, Molinolo AA. Antiprogestins in breast cancer treatment: are we ready? Endocr Relat Cancer. 2012;19:R35–50. doi: 10.1530/ERC-11-0378. [DOI] [PubMed] [Google Scholar]
- 3.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 4.Hill KK, Roemer SC, Churchill ME, Edwards DP. Structural and functional analysis of domains of the progesterone receptor. Mol Cell Endocrinol. 2012;348:418–429. doi: 10.1016/j.mce.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 6.Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci U S A. 2000;97:1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen T, Horwitz KB, Lange CA. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol. 2001;21:6122–6131. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32:2307–2320. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014;6:657–669. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang P, Gao K, Jin X, Ma J, Peng J, Wumaier R, Tang Y, Zhang Y, An J, Yan Q, Dong Y, Huang H, Yu L, Wang C. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-alpha protein turnover. Cell Death Dis. 2015;6:e1687. doi: 10.1038/cddis.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, O’Malley BW. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene. 2011;30:4350–4364. doi: 10.1038/onc.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang P, Gao K, Tang Y, Jin X, An J, Yu H, Wang H, Zhang Y, Wang D, Huang H, Yu L, Wang C. Destruction of DDIT3/CHOP protein by wildtype SPOP but not prostate cancer-associated mutants. Hum Mutat. 2014;35:1142–1151. doi: 10.1002/humu.22614. [DOI] [PubMed] [Google Scholar]
- 13.Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH, Baek SH, Chiba T, Tanaka K, Bang OS, Joe CO, Chung CH. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem. 2006;281:12664–12672. doi: 10.1074/jbc.M600204200. [DOI] [PubMed] [Google Scholar]
- 14.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T, Guiducci C, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Hussain WM, Ramos AH, Winckler W, Redman MC, Ardlie K, Tewari AK, Mosquera JM, Rupp N, Wild PJ, Moch H, Morrissey C, Nelson PS, Kantoff PW, Gabriel SB, Golub TR, Meyerson M, Lander ES, Getz G, Rubin MA, Garraway LA. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC NIH Intramural Sequencing Center (NISC) Comparative Sequencing Program. Hieter P, Mullikin JC, Merino MJ, Bell DW. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol. 2005;19:327–339. doi: 10.1210/me.2004-0306. [DOI] [PubMed] [Google Scholar]
- 17.Groshong SD, Owen GI, Grimison B, Schauer IE, Todd MC, Langan TA, Sclafani RA, Lange CA, Horwitz KB. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1) Mol Endocrinol. 1997;11:1593–1607. doi: 10.1210/mend.11.11.0006. [DOI] [PubMed] [Google Scholar]
- 18.Han SJ, DeMayo FJ, Xu J, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol Endocrinol. 2006;20:45–55. doi: 10.1210/me.2005-0310. [DOI] [PubMed] [Google Scholar]
- 19.Brisken C. Progesterone signalling in breast cancer: a neglected hormone coming into the limelight. Nat Rev Cancer. 2013;13:385–396. doi: 10.1038/nrc3518. [DOI] [PubMed] [Google Scholar]