

Abstract
Irinotecan and topotecan are the only camptothecin analogues approved by the FDA for cancer treatment. However, inherent and/or acquired irinotecan and topotecan resistance is a challenging issue in clinical practice. In this report, we showed that FL118, a novel camptothecin analogue, effectively obliterated human xenograft tumors that acquire irinotecan and topotecan resistance. Consistent with this finding, Pharmacokinetics studies indicated that FL118 rapidly clears from circulation, while effectively accumulating in tumors with a long elimination half-life. Consistent with our previous studies on irinotecan, FL118 exhibited ≥25 fold more effectiveness than topotecan at inhibiting cancer cell growth and colony formation; we further showed that although topotecan can inhibit the expression of survivin, Mcl-1, XIAP or cIAP2, its effectiveness is about 10-100 fold weaker than FL118. Lastly, in contrast to both SN-38 (active metabolite of irinotecan) and topotecan are substrates of the efflux pump proteins P-gp/MDR1 and ABCG2/BCRP, FL118 is not a substrate of P-gp and ABCG2. Consistently, sildenafil, a multiple efflux pump inhibitor, sensitized SN-38 much more than these of the ABCG2-selective inhibitor KO143 in growth inhibition of SW620 and HCT-8 cells. In contrast, both inhibitors showed no effect on FL118 efficacy. Given that both P-gp and ABCG2 express in SW620 and HCT-8 cells and FL118 is not a substrate for P-gp and ABCG2, this suggests that FL118 appears to bypass multiple efflux pump protein-induced resistance, which may contribute to FL118 overcoming irinotecan and topotecan resistance in vivo. These new findings provide renewed perspectives for further development of FL118 for clinical applications.
Keywords: FL118, irinotecan, topotecan, treatment resistance, antitumor activity, animal models of human tumor xenografts
Introduction
Camptothecin, a cytotoxic quinoline alkaloid (Figure 1), was discovered by Drs. Monroe Wall and Mansukh Wani together with their colleagues about fifty years ago in 1966 [1]. Over the past five decades, although several thousand camptothecin analogues have been synthesized and tested, most of them are highly toxic to normal tissues or have other disadvantages making them unsuitable candidates for cancer treatment. To date, only two camptothecin analogues (i.e. irinotecan and topotecan, Figure 1) are approved by the FDA for cancer treatment in the clinic [2,3]. However, treatment resistance to irinotecan [4,5] and topotecan [6,7] is often observed in clinic practice, especially in patients who use these drugs for an extended period of time [8,9].
Figure 1.

Comparison of FL118 chemical structure with camptothecin, irinotecan, SN-38 (active metabolite of irinotecan) and topotecan: So far, irinotecan and topotecan are the only camptothecin analogues that have been approved by the FDA for cancer treatment.
In an attempt to overcome resistance, we recently identified a novel camptothecin analogue (FL118) with exceptional antitumor activity [10]. Currently the known mechanism of action for camptothecin compounds is the ability to inhibit DNA topoisomerase 1 (Top1) activity, which thereby induces DNA damage and cancer cell death due to the inability of cells to complete DNA replication [11-13]. In contrast, previous studies on FL118 using the in vitro Top1 enzyme inhibition assay indicate that FL118 has less ability to inhibit Top1-mediated nick of supercoiled DNA than SN-38 at a dose as high as 1 μM, but FL118 can effectively inhibit cancer cell growth and induces apoptosis at ≤ nM levels [10]. FL118 is capable of eliminating colon and head-and-neck tumor xenografts at its maximum tolerated dose (MTD) [10,14], and even at sub-MTD levels a high percentage of human xenograft tumors can be eliminated [15]. Furthermore, while mutation of Top1 in prostate cancer cells strikingly increases resistance to camptothecin, SN-38 (active form of irinotecan), and topotecan, FL118 is much less affected [16], suggesting that inhibition of Top1 by FL118 may not play a major mechanism of action in FL118 function. Additionally, it is known that SN-38 [17,18] and topotecan [19-21] are substrates of the drug efflux pump ABCG2. In contrast, FL118 is a poor substrate of ABCG2, and overexpression of ABCG2 in cancer cells induces no resistance to FL118 [22]. FL118 selectively inhibits the expression of multiple antiapoptotic proteins (survivin, Mcl-1, XIAP and cIAP2) from the inhibitor of apoptosis (IAP) and Bcl-2 families, and effectively inhibits cancer cell growth regardless of p53 status (wild type, mutant or null) [10,23]. Intriguingly, cancer cells with null or mutated p53 are more sensitive to FL118 treatment than cancer cells with wild type p53 [23], implying that FL118 may be more effective in treatment of advanced cancers that lose wild type p53.
Although our previous studies indicate that FL118 is a novel camptothecin analogue with distinct mechanism of action and shows superior antitumor activity in comparison with irinotecan and topotecan, it is unclear whether FL118 could overcome human tumors that acquire irinotecan and topotecan resistance. In this report, we showed that FL118 is able to eliminate human tumors that acquire irinotecan and topotecan resistance. Consistent with this finding, intravenous administration of FL118 into human tumor xenograft models showed a favorable pharmacokinetic (PK) profile. FL118 was rapidly cleared from the blood stream and effectively accumulated in xenograft tumors. Furthermore, FL118 was able to bypass multiple ABC transporter efflux protein-induced drug resistance. These observations at least partially explain the favorable PK and toxicity profile of FL118 in xenograft model systems.
Materials and methods
Mouse models used in this study
Seven to ten-week-old female severe combined immunodeficiency (SCID) mice (body weight 20-25 g) were purchased from the Division of Laboratory Animal Resources (DLAR), Roswell Park Cancer Institute. Mice were housed at 5 mice per cage with water and food ad libitum. All animal experiments were performed in accordance with the Institute Animal Care and Use Committee (IACUC) approved animal protocols.
Formulation of the anticancer agent FL118 and its mock control solution (vehicle)
FL118 formulation for intravenous (IV) administration: The formulation for IV administration is described in detail in our patent (PCT/US2011/058558) [24]. Briefly, the IV formulation for FL118 in the current study used the basic formulation recipe, which contains FL118 (0.1-0.25 mg/ml), DMSO (5%), and hydroxypropyl-β-cyclodextrin (0.05-0.125%, w/v) in saline. The corresponding vehicle solution in the basic formulation recipe contains DMSO (5%) and hydroxypropyl-β-cyclodextrin (0.05-0.125%, w/v) in saline without FL118. This formulation was used for either IV or intraperitoneal (IP) administration of FL118. In this study, the FL118 PK studies used IV administration of FL118; all other in vivo studies used IP administration of FL118, which is more technically convenient and time saving.
Irinotecan or topotecan resistant human xenograft tumor generation and treatment
Human colon (SW620) and head-and-neck (FaDu) cancer cells were treated with gradually increased low SN-38 (1-10 nM) or topotecan (1-10 nM) for approximately four weeks, respectively. Alive cells were collected and amplified for in vivo generation of xenograft tumor in SCID mice by subcutaneously injecting 2 × 106 cells on each site at the flank area of mice as we did before [10]. The established xenografts were sub-transplanted to new SCID mice. When tumors grew to about 1000 mm3, tumor mass was isolated and subcutaneously inoculating 30-50 mg tumor mass on each site for establishing experimental tumor models. Seven days after inoculating tumor mass on SCID mice, mice were randomly divided into required groups (≥5 mice per group) for treatment.
A group of mice with established SW620 (colon) or FaDu (head-&-neck) tumor xenografts were initially treated with irinotecan at its MTD (100 mg/kg) via IP administration weekly until xenograft tumors showed irinotecan resistance (defined as tumor growing during irinotecan treatment). Of note, a weekly schedule is the most clinically compatible schedule for irinotecan. Another group of mice with SW620 or FaDu xenograft tumors were initially treated with topotecan at its MTD (4 mg/kg, IP) via daily × 5 schedules until the xenograft tumors exhibited topotecan resistance (defined as tumor growing during topotecan treatment), similar to the situation previously reported in the Brca1/p53-deficient mouse mammary xenograft tumor models [25]. After xenograft tumors showed irinotecan resistance or topotecan resistance, mice were then treated five times (q2d × 5) with FL118 (0.75 mg/kg, 1 mg/kg, or 1.5 mg/kg in different experiments) via IP with a schedule of every other day for one or more treatment cycles.
Experimental human xenograft tumors in animal models, tumor measurement and analysis
Human tumor xenografts in mouse models were established from corresponding human cancer cell lines. Tumor xenografts were initially established from human cancer cell lines by subcutaneously injecting 1-3 × 106 cultured cancer cells in SCID mice as described previously [10]. Human tumor xenografts used in this study include human head & neck tumors established from FaDu cells, and human colon cancer tumors established from SW620 cell lines. Human xenograft tumors maintained in SCID mice were isolated and 30-50 mg tumor mass was inoculated into SCID mice to grow for 7 days before processing the mouse model experiments, at which (designated as day 0) the xenografted tumor sizes were approximately 100-200 mm3 depending on the initial tumor mass used. During experiments, tumor size was monitored three times a week for the first few weeks, and then twice a week thereafter. Two axes (mm) of a tumor (L, longest axis; W, shortest axis) were measured with a digital vernier caliper. Tumor volume (mm3) was calculated using a formula of tumor volume (mm3) = ½ (L × W2). If a tumor disappeared, we maintained the experimental mice for an additional period after the completion of drug treatment schedules for observation of tumor relapse. Data analysis and figures were made using Microsoft Excel. Tumor curve profiles from representative individual tumors after treatment were presented.
Specimen preparation for pharmacokinetics (PK) studies in tumor bearing mice
FL118 PK studies were performed as follows: SCID mice were first inoculated with human FaDu and SW620 tumors (of note, for PK data comparison, both tumors on individual mice at both left and right flank sites), when tumor size grew to approximate 800 mm3, a single injection of FL118 was given via IV (tail vein) at a dose of 1.5 mg/kg to six groups of SCID mice (3 mice per group) bearing human FaDu or SW620 tumors. Of note, based on the previous studies [14], 1.5 mg/kg is roughly the MTD of FL118 for the schedules of daily for five times (q1d × 5) and every other day for five times (q2d × 5). Tumor tissues and blood samples were collected at a series of time points of 10 minutes, 1 hour, 4 hours, 12 hours, 24 hours and 48 hours. Each tumor tissue was collected in a tube and immediately frozen with liquid nitrogen; each blood sample was collected in a Li-Heparin LH/1.3 tube (SARSTEDT), and plasma was recovered by centrifugation (1500 rpm × 2 minutes). The collected plasma from each blood sample was transferred into a new tube and immediately frozen in liquid nitrogen. Liquid nitrogen-frozen specimens were then transferred to a -80°C freezer for analysis.
PK assay development and FL118 samples PK analysis
For PK analysis of FL118 concentration in tumor tissue and plasma, FL118 was extracted with acidified methanol. An 800 µl aliquot of ice-cold acidified methanol was added to 200 µl plasma and vortexed for 15 seconds. In parallel, FL118 in human xenograft tumor tissue was first homogenized in 1× PBS (W/V = 1 g tissue/3 ml 1× PBS) and then extracted with acidified methanol. An 800µl aliquot of ice-cold acidified methanol was added to 200 µl homogenized tissue and then vortexed for 1 minute. The samples were then centrifuged at 13,000 rpm for 5 minutes. The supernatant was transferred to a clean 13 × 100 mm glass tube. Samples were dried under vacuum and stored at -20°C until analysis. For analysis, dried samples were reconstituted in 200 µl of mobile phase (80% 3% TEA and 20% acetonitrile pH 5.5) and 15 µl was used for injection. Analysis was carried out using an Acquity UPLC system with Fluorescence detection interfaced with Empower software (Waters, Milford, MA). Separation was carried out on an Acquity BEH Shield RP18 1.7 µm, 2.1 mm × 100 mm column (Waters). Irinotecan (also called CPT-11) was used as the internal standard (IS); a 10 µl aliquot of a 1.3 µg/ml solution was added to each 200 µl sample. The fluorescence detector was set at Excitation (Ex) 370 nm and Emissions (Em) 510 nm wavelengths.
The calibration standards were prepared by spiking plasma with FL118; the calibration curve range is 5 ng/ml-500 ng/ml. To ensure quality assurance, quality control samples were prepared in plasma at 25 and 250 ng/ml and stored at -20°C. The quality control (QC)’s samples were injected in duplicate at the beginning and end of the assay. Assay was validated via running twelve standard curves over the course of 5 days. QC samples were analyzed with each curve. The overall precision (%CV = 6.4) and overall accuracy (101%) of the assay calibrators was shown to be excellent. QC precision measured as %CV was equal to 7.5% and overall QC accuracy was 96%.
FL118 Caco-2 permeability assay and P-glycoprotein (P-gp)/MDR1 substrate assay
Caco-2 cells (clone C2BBe1) were obtained from American Type Culture Collection (Manassas, VA). Cell monolayers were grown to confluence on collagen-coated, microporous, polycarbonate membranes in 12-well Costar Transwell® plates. The permeability assay buffer was Hanks’ balanced salt solution (HBSS) containing 10 mM HEPES and 15 mM glucose at a pH of 7.4. The buffer in the receiver chamber also contained 1% bovine serum albumin. The dosing solution concentration of FL118 was 1 µM in the assay buffer with or without 1 µM valspodar, a P-gp inhibitor. Cells were first pre-incubated for 30 minutes with HBSS with or without 1 µM valspodar. Cell monolayers were dosed on the apical side (A-to-B) or basolateral side (B-to-A) and incubated at 37°C with 5% CO2 in a humidified incubator. Samples were taken from the donor and receiver chambers at 120 minutes. Each determination was performed in duplicate. The flux of co-dosed lucifer yellow was also measured for each monolayer to ensure no damage was inflicted to the cell monolayers during the flux period. All samples were assayed by LC-MS/MS using electrospray ionization for FL118. The apparent permeability (Papp) and percent recovery were calculated as follows: (1) Papp = (dCr/dt) × Vr/(A × CA); (2) Percent Recovery = 100 × ((Vr × Cr final) + (Vd × Cd final))/(Vd × CN). Where, dCr/dt is the slope of the cumulative concentration in the receiver compartment versus time in µM s-1; Vr is the volume of the receiver compartment in cm3; Vd is the volume of the donor compartment in cm3; A is the area of the insert (1.13 cm2 for 12-well Transwell); CA is the average of the nominal dosing concentration and the measured 120 minute donor concentration in µM; CN is the nominal concentration of the dosing solution in µM; Cr final is the cumulative receiver concentration in µM at the end of the incubation period; and Cdfinal is the concentration of the donor in µM at the end of the incubation period. Efflux ratio (ER) is defined as Papp (B-to-A)/Papp (A-to-B).
Drug dilution for in vitro studies
For in vitro studies, all drugs (topotecan, SN-38, FL118) were first dissolved in DMSO at a 1000× concentration as a stock solution, and then the corresponding stock solutions were directly diluted into cell cultural medium, for example, if the final concentration is 10 nM, we used 10 µM as a stock solution for 1000× dilution directly with cell cultural medium.
MTT cell viability assay
MTT assay was used to determine cancer cell growth and viability after drug treatment following our previously established procedure [26]. Briefly, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) was used as a colorimetric substrate for measuring cell viability after cancer cells were treated with or without drugs. After treatment, MTT was added to a final concentration of 0.5 mg/ml into treated cells in 96-well plates. Cells were then incubated in a 5% CO2 incubator at 37°C for 4 hours, followed by thoroughly lysed with lysis buffer (20% SDS, 50% N, N-dimethylformamide, pH 4.7, 100 µl/well). The absorbance in the relevant wells was measured at 570 nm using an Ultra Microplate Reader (Bio-Tek Instruments). Cell viability was calculated as a percentage.
Cancer cell colony formation assay
HCT-8 colon cancer cells were plated at 200 cells/well in 12-well plates overnight. Cells were then treated with and without a series of concentrations of FL118 and topotecan in parallel for 2 and 6 hours, respectively. Drugs were then washed out with PBS and cells were continuously cultured in complete medium with 10% serum in an incubator at 37°C, 5% CO2 for two weeks. Cells were then fixed and stained with crystal violet solution to present the colony formation profile.
Immunoblotting/western blotting analysis
Immunoblotting analysis of the expression of gene products of interest was performed following our previous protocols [27]. Briefly, cancer cells with or without treatment (FL118, topotecan) for a defined time, were washed with PBS and lysed on ice for 30 minutes in PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 µg/ml PMSF, and 20 µM leupeptin. After the lysates were cleared by centrifugation, the total protein concentration was determined. After denatured in 2 × SDS sample loading buffer for 5 minutes at 95°C, samples were separated on 10-15% SDS-PAGE gels, and electrotransferred to the Pure Nitrocellulose Membrane (Bio-Rad, Hercules, CA). After blocked with 5% skim milk in TBS-T for 3 hours at room temperature with constant shaking, the membranes were incubated in TBS-T containing the relevant primary antibody (1:500-1000) and 5% BSA overnight at 4°C. After washing with TBS-T three times, the membrane was incubated in 5% skim milk in TBS-T buffer containing the appropriate secondary anti-IgG antibody (1:5000) at room temperature for 1 hour with constant shaking. The protein of interest was detected using ECL plus (Perkin Elmer, Waltham, MA) and visualized by exposure on X-ray film for various times (5-60 seconds). For normalization of protein loading, the same membranes were stripped with stripping buffer and used for Western blots by the same procedure with actin antibody (1:1000 dilution).
Cytotoxicity assay for acquiring images
SW620 colon cancer cells were seeded at about 30% confluence overnight in 6-well plates. Cells were then treated with SN-38, topotecan and FL118 at 1 µM for 72 hours. Cancer cell images for each treatment were taken with a digital camera under a phase-contrast microscope.
Data analysis
Data curves shown in Figures 2, 3, 4B, 5A and 7 were plotted using Microsoft Excel. The values of individual time points presented in the Figures 4B, 5A and 7 are the mean ± standard deviation (SD) from at least three independent assays.
Figure 2.
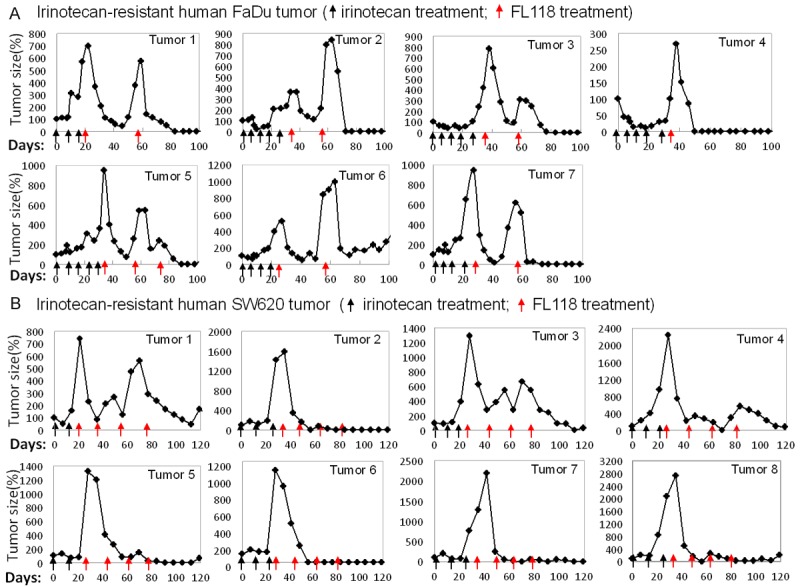
FL118 regresses human tumor that acquired irinotecan resistance: (A) Human head-&-neck FaDu tumors were subcutaneously inoculated on SCID mice. Treatment was initiated 7 days after the transplanted tumors reached 100-200 mm3 (designated day 0). Mice with FaDu tumor xenografts were first treated with irinotecan at its MTD (100 mg/kg, IP) weekly for 3-5 times (black arrows) until tumor acquired irinotecan resistance. After tumor acquired irinotecan resistance (tumor grows while treating), mice were then treated with FL118 (1.5 mg/kg, IP) with every other day for five times (q2d × 5) as one cycle. If tumors relapse, mice were treated with FL118 for second or third cycles (red arrows). Tumor curves are shown as individual tumor profiles after treatment. Of note, 2 FaDu tumors did not acquire irinotecan-resistance after 5 time irinotecan treatment (data not shown). (B) Mouse models of human colon SW620 tumors were established same as in (A). Irinotecan treatment (black arrows) as in (A) until resistant tumor appears (tumor grows while treating), followed by FL118 treatment (red arrows) as in (A). In this experiment, mice were treated with FL118 for total 4 cycles (red arrows). Tumor curves are shown as individual tumor profiles after treatment. Tumor mice without drug treatment were sacrificed within two weeks due to tumor size over 1500 mm3.
Figure 3.
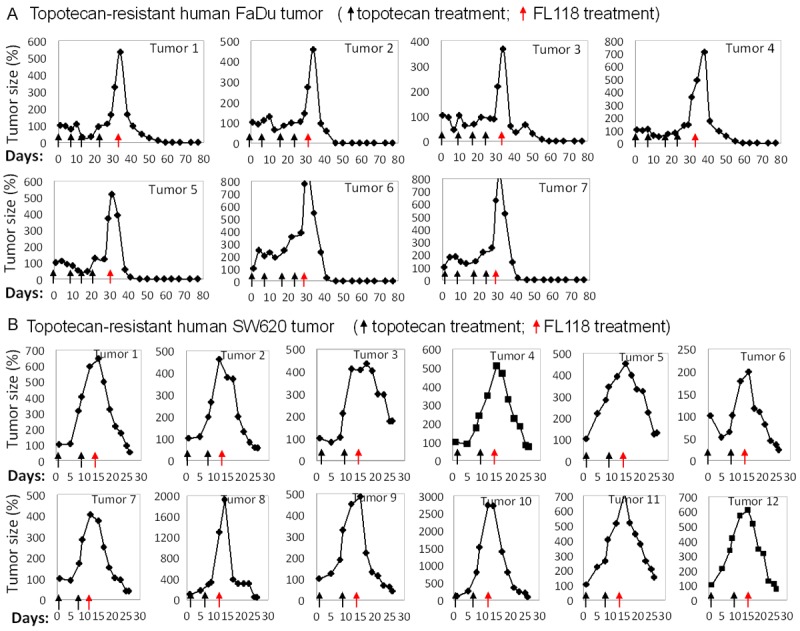
FL118 effectively regresses human tumor that acquired topotecan-resistance: (A) Human FaDu tumor animal model establishment and treatment starting day same as in Figure 2. Tumor mice were first treated with topotecan at its MTD (4 mg/kg, IP) with daily × 5 schedules for 4 cycles (each black arrow is one cycle). After tumors acquired topotecan resistance, mice were then treated with FL118 as in Figure 2 (q2d × 5, red arrow). Tumor curves are shown as individual tumor profiles after drug treatment. Of note, 2 FaDu tumors did not acquire topotecan-resistance after 4-cycle topotecan treatment (data not shown). (B) Mouse models of SW620 tumors were established as in Figure 2. Topotecan treatment (black arrow) was performed as in (A) until obtaining resistant tumor (tumor grows while treating), followed by FL118 treatment (q2d × 5, red arrows) at 1 mg/kg (2/3 MTD, top panel) and 0.75 mg/kg (1/2 MTD, bottom panel). Tumor curves were derived from individual tumor profiles after drug treatment. Tumor mice without drug treatment were sacrificed within two weeks due to tumor size over 1500 mm3.
Figure 4.
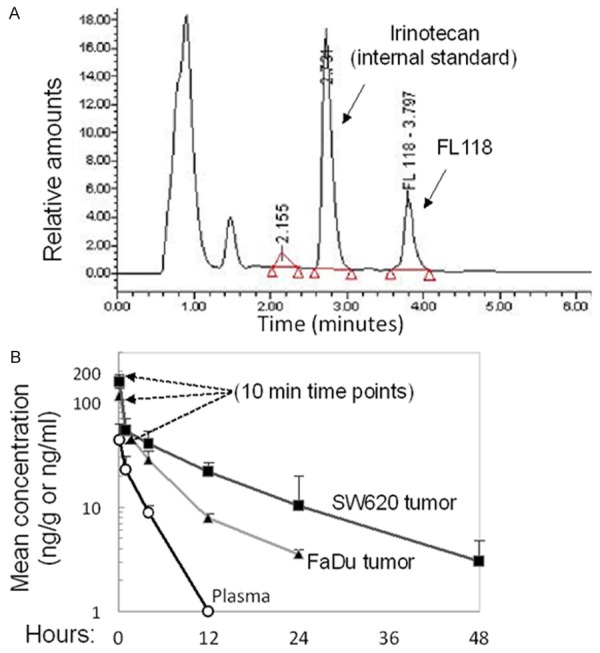
FL118 exhibits favorable pharmacokinetics (PK) profiles after IV injection: A. The eluting profile for standard FL118 and internal standard control irinotecan with the established gradient method are shown. B. FL118 IV PK results: SCID mice were subcutaneously implanted with human FaDu (head & neck) and SW620 (colon) tumor. After the implanted tumor grew to 800-1000 mm3, FL118 was IV-injected for one time at 1.5 mg/kg. Then, blood and tumor tissues were collected at 10 min, 1 h, 4 h, 12 h, 24, and 48 h. Three SCID mice at each time point were used. Standard deviation (SD) was analyzed using Excel software.
Figure 5.
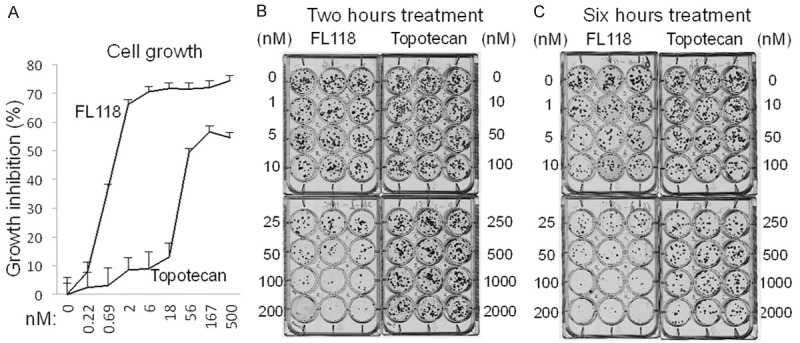
FL118 is ~25× more potent than topotecan in cancer cell growth inhibition and colony formation: (A) Cell growth: HCT-8 cells were treated with either FL118 or topotecan for 72 hours as shown. Cell growth was measured with MTT assay and the data were plotted as growth inhibition percentage. (B and C) Colony formation: After seeding HCT-8 cells in 12-well plates at 100 cells per well overnight, cells were then treated with either FL118 or topotecan for 2 (B) or 6 (C) hours at a series of concentrations as shown. Drugs were then washed out and cells were allowed to grow in new complete medium without drugs for 2 weeks. Cell colonies were stained with Crystal violet solution; images were digitally taken. Triplicate results are shown.
Figure 7.
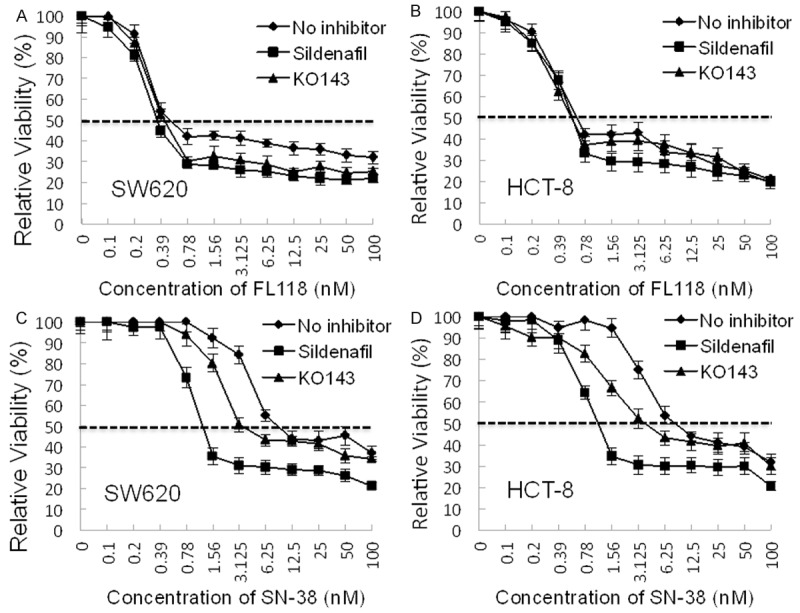
FL118 appears to bypasses not only ABCG2-induced treatment resistance but also other ABC transporters-induced resistance: Subconfluent SW620 and HCT-8 colon cancer cells were treated with FL118 (A, B) or with SN-38 (C, D) at a series of concentrations as shown in the presence or absence of sildenafil (50 µM, a multiple ABC transporter inhibitor) or KO143 (2 µM, an ABCG2-specific inhibitor) for three days. Cell growth/viability was then analyzed by MTT assays. Each time point in the individual curves is the mean ± DS from at least three independent assays. Of note, pre-experiment for testing sildenafil and KO143 toxicity showed that sildenafil at 50 µM or KO143 at 2 µM exhibit no toxicity to cells.
Results
FL118 effectively eliminates human colon and head-and-neck tumors that acquire irinotecan or topotecan resistance
Previous studies have demonstrated that FL118 exhibits better antitumor activity than irinotecan and topotecan in animal models of human colon and head-and-neck cancers, and can eliminate these tumors [10]. However, it is unclear whether FL118 is able to control colon and head-and-neck tumors that have acquired irinotecan or topotecan resistance. To address this, by following the procedure described in the Method section, we first treated mice bearing xenograft tumors with either irinotecan or topotecan until the tumors acquired resistance (tumor growing during drug treatment). The tumors acquired irinotecan and topotecan resistance were then treated with FL118 to assess its efficacy. Our studies demonstrated that FL118 effectively eliminated human tumor xenografts that acquired irinotecan resistance (Figure 2) or topotecan resistance (Figure 3) in both head-&-neck FaDu tumor models (Figures 2A, 3A) and colorectal SW620 tumor models (Figures 2B, 3B). Equally important, after FL118 treatment, some regressed irinotecan-resistant tumors exhibited regrowth; a second cycle of FL118 treatment was also highly effective against relapsed tumors (Figure 2A, 2B). This observation suggests that FL118 is not only able to eliminate human xenograft tumors that acquired irinotecan or topotecan resistance but is also effective after multiple cycles of treatment without the generation of FL118 resistance.
FL118 exhibits favorable pharmacokinetics (PK) profiles: rapidly cleared from blood stream and effectively accumulated in tumor tissue with a long retention half-life
Next, we determined FL118 PK profiles to see whether availability of FL118 in tumors contributes to the high efficacy of FL118. A single dose of FL118 (1.5 mg/kg) was injected IV via the tail vein of SCID mice bearing human SW620 (colon) and FaDu (head-and-neck) xenograft tumors. Blood and tumor were collected at 10 min, 1, 4, 12, 24, and 48 hours for PK profile determination. The established assay described in the Method section showed that FL118 and its internal standard (IS) could be resolved in less than 5 minutes with the following relative retention rates: FL118 at ~3.8 minutes and irinotecan at ~2.7 minutes as shown in Figure 4A. Analysis of all collected specimens demonstrated that FL118 exhibits a highly favorable PK profile. Specifically, FL118 rapidly accumulated in tumor tissues with a long half-life, but was quickly cleared from the circulation (Figure 4B, Table 1). For example, 10 minutes after IV injection, FL118 concentration in tumor tissue was >2.5 fold higher than plasma concentrations (Figure 4B). Similarly, FL118 was cleared within 12 hours from the plasma but retained for over 24 hours in tumors following a single IV injection (Figure 4B). The long time retention of FL118 in human xenograft tumor tissues versus in animal blood stream is consistent with the previous observation of FL118 showing favorable toxicity profiles and antitumor effects lasting well beyond the final dosing [10,14,15].
Table 1.
Pharmacokinetics parameters of FL118 in human tumor and mouse plasma following a single IV administration
| Sample | T1/2 (hr) | Tmax (hr) | Cmax (ng/g, ml) | AUC (hr*ng/g) | AUC∞ (hr*ng/g) | AUC% Extrap (%) | Vz (g/kg) (ml/kg) | Cl (g/hr/kg) (ml/hr/kg) |
|---|---|---|---|---|---|---|---|---|
| FaDu | 6.852 | 0.167 | 115 | 413 | 448 | 7.74 | 33052 | 3343 |
| SW620 | 12.75 | 0.167 | 158 | 842 | 897 | 6.17 | 30742 | 1671 |
| Plasma | 1.788 | 0.167 | 43 | 82 | 104 | 21.7 | 36849 | 14287 |
FL118 is ~25 fold more potent than topotecan in terms of in vitro inhibition of cancer cell growth and colony formation
In order to look for additional in vitro evidence to support the in vivo observation, we performed FL118 and topotecan comparison studies on the inhibition of cell growth and clonogenic potential. Irinotecan is a pro-drug and is largely inactive for cancer cell-based studies since it needs to be metabolized into its active form (SN-38). However, the ability to convert irinotecan into SN-38 shows substantial variations among individuals (animals or humans). Thus, variable drug activation presents a confounded uncertainty when assessing efficacy and toxicity outcomes in clinical use. Therefore, we chose to compare topotecan to FL118 in in vitro assays measuring the inhibition of cancer cell growth and colony formation. Consistent with animal model results that demonstrated FL118 can overcome tumor-acquired irinotecan and topotecan resistance (Figures 2, 3), FL118 was approximately 25 fold more effective at inhibiting colon cancer cell growth in comparison with topotecan (Figure 5A). Similarly, FL118 was about 25 fold more effective at inhibiting colon cancer cell colony formation than topotecan (Figure 5B). Importantly, while topotecan exhibited poor effectiveness at inhibiting colony formation after 2 hours of topotecan treatment compared to its 6 hours of treatment (Figure 5B and 5C), FL118 was equally effective at both time points (Figure 5B and 5C). This suggests FL118 has a faster uptake by the cancer cells than topotecan, which is consistent with the observation from the in vivo PK analysis showing that FL118 was rapidly accumulated and retained in tumor tissue (Figure 4B). Rapid uptake and retention are also consistent with our recent findings showing that FL118 is not a substrate of the efflux pump ABCG2, while both SN-38 (active metabolite of irinotecan) and topotecan are substrates of ABCG2 [22].
Multiple mechanisms might be involved in and contribute to FL118 anticancer efficacy
Previous studies demonstrated that inhibition of multiple cancer-associated anti-apoptotic gene products (survivin, Mcl-1, XIAP, and cIAP2) by FL118 play important roles in FL118-mediated cancer cell growth inhibition and apoptosis induction [10,15]. Inhibition of survivin expression by FL118 is at least 10 fold greater than the inhibition produced by SN-38 [10]. Following this line of studies, we here provide new data showing that inhibition of the expression of survivin, Mcl-1, XIAP, and cIAP2 by FL118 was about 10-100 fold more effective than topotecan (Figure 6A). These observations are consistent with previous findings: 1) survivin, Mcl-1, XIAP and cIAP2 are involved in FL118 function [10,15], while these FL118 downstream targets may only minimally contribute to the function of irinotecan/SN-38 and topotecan; and 2) antitumor activity of FL118 is superior to irinotecan and topotecan [10,15].
Figure 6.
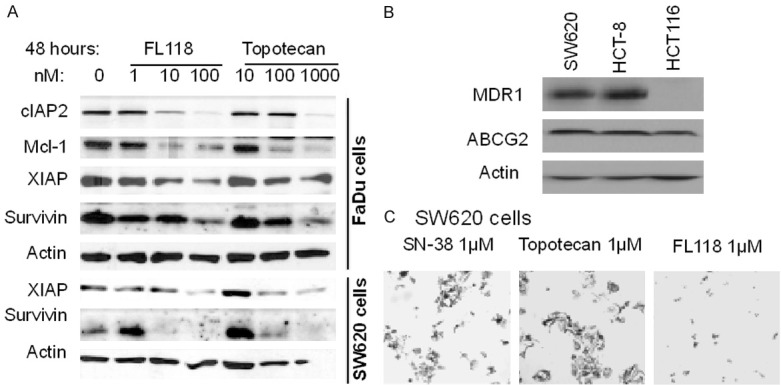
A. FL118 exhibits superior ability to inhibit survivin, Mcl-1, XIAP, or cIAP2 than topotecan. Subconfluent FaDu and SW620 cancer cells were treated with FL118 or topotecan as shown. Cells were then analyzed for FL118 target gene expression using western blots. Actin was used as an internal control for protein loading. B. Expression of MDR1 and ABCG2 in colon cancer cells. Subconfluent SW620, HCT-8 and HCT-116 cancer cells were lysed and analyzed for the expression of MDR1 and ABCG2 using western blots. Actin is the internal control. C. Comparison of the effectiveness of SN-38, topotecan and FL118 in cancer growth inhibition and killing. Subconfluent SW620 cancer cells were treated with SN-38, topotecan and FL118 for 72 hours at 1 µM as shown. Representative cancer cell images were shown.
Most recently, our studies revealed that while both SN-38 (active metabolite of irinotecan) and topotecan are substrates of the ATP-binding cassette (ABC) transporter ABCG2 (also called breast cancer resistance protein-BCRP), FL118 is not a substrate of ABCG2 and ABCG2 is not a resistance marker for FL118 [22]. However, an unanswered question is whether FL118 is or is not a substrate of other ABC transporters. In this regard, we found that both SW620 and HCT-8 colon cancer cells express both P-glycoprotein (P-gp, also called MDR1 or ABCB1) and ABCG2 (BCRP) (Figure 6B). Accordingly, similar to the topotecan situation shown in Figure 5, SN-38 needs a very high concentration to induce cancer cell growth inhibition (Table 2, Figure 6C), suggesting that ABC transporter efflux pump proteins play a resistance role for these drugs. Alternatively, Caco-2 permeability assay with and without P-gp/MDR1 inhibitor valspodar indicated that FL118 exhibits high permeability to pass through Caco-2 confluence monolayers and importantly, in the presence or absence of P-gp/MDR1 inhibitor valspondar, FL118 exhibits a similar, very low efflux ratio (1.2 versus 1.0, Table 3), indicating that FL118 is not a substrate of P-gp/MDR1.
Table 2.
FL118 but not SN-38 can inhibit colon caner cell growth in a relative low concentration
| Determined in SW620 cells | Determined in HCT-8 cells | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| SN-38 | FL118 | SN-38/FL118 | SN-38 | FL118 | SN-38/FL118 | |
| IC25 | ND | ND | NA | 0.187 | 0.047 | 3.958 |
| IC50 | 14.55 | 5.867 | 2.48 | 4.229 | 0.529 | 7.994 |
| IC75 | 157.5 | 12.53 | 12.57 | 95.80 | 5.95 | 16.10 |
| IC90 | 1795.5 | 27.00 | 66.50 | 2172.2 | 66.80 | 32.52 |
| IC95 | 3811.5 | 45.33 | 84.08 | 18143.4 | 346.11 | 52.42 |
Table 3.
FL118 is highly permeable in Caco-2 permeability assay and not a P-gp substrate
| Test Article | Direction | Recovery (%) | Papp value (10-6 cm/s) | Efflux Ratio | ||
|---|---|---|---|---|---|---|
|
| ||||||
| R1* | R2 | AVG | ||||
| FL118 (1 µM) | A-to-B | 43 | 7.27 | 6.67 | 6.97 | 1.2 |
| B-to-A | 48 | 7.35 | 8.86 | 8.10 | ||
| FL118 (1 µM) + Valspodar (1 µM) | A-to-B | 46 | 9.26 | 7.48 | 8.37 | 1.0 |
| B-to-A | 48 | 8.29 | 8.65 | 8.47 | ||
Papp: apparent permeability; R1: replicate 1; R2: replicate 2; AVG: average.
Consistent with this notion, sildenafil, a multiple ABC transporter inhibitor, can selectively reverse ABC transporter-mediated drug resistance associated with multiple pumps, including ABCB1 (P-gp/MDR1) and ABCG2 (BCRP) [28], ABCC10 (MRP7) [29] and possibly others such as ABCC4 (MRP4) [30] and ABCC5 (MRP5) [31]. Therefore, we compared the relative effectiveness of FL118 and SN-38 to inhibit cell growth in both SW620 and HCT-8 colon cancer cells in the presence and absence of efflux pump inhibitors. In these studies, FL118 showed similar anti-cancer cell growth/viability in the presence or absence of sildenafil or KO143, an ABCG2-selective inhibitor (Figure 7A, 7B and Table 4). In contrast, sildenafil sensitized SW620 and HCT-8 colon cancer cells to SN-38 treatment much more than KO142 can do (Figure 7A, 7B and Table 4). Given the fact that sildenafil can inhibit multiple efflux pump protein [28-31], these observations suggest that FL118 is likely not only able to bypass ABCG2 and P-gp/MDR1 resistance, but also bypass resistance conferred by other ABC transporter proteins inhibited by sildenafil.
Table 4.
Comparison of IC50 of FL118 to SN-38 with and without ATP-binding cassette transporter inhibitors
| Drugs | Cell Types | No inhibitor | Ko143 | Sildenafil |
|---|---|---|---|---|
| FL118 (nM) | SW620 | 0.54 | 0.45 | 0.36 |
| HCT-8 | 0.67 | 0.58 | 0.59 | |
| SN-38 (nM) | SW620 | 10.07 | 3.78 | 1.25 |
| HCT-8 | 8.92 | 3.82 | 1.15 |
Discussion
A series of experiments with distinct approaches demonstrated that although FL118 is structurally related to irinotecan and topotecan (Figure 1), FL118 is functionally superior and mechanistically distinct from those camptothecin analogues [10,14,15,22,23]. While we currently do not know detailed target-directed processes that account for this different selectivity, we recognize that FL118 possesses a unique methylenedioxy (MD) structure attached to the A ring at positions 10 and 11 of camptothecin (Figure 1). In appearance, the two oxygen atoms in the MD structure may substantially alter the status of overall electron distribution, motility and flow within the FL118 molecule throughout rings A to E, possibly making ring A more electronegative. However, we found that adding the MD structure did not increase electronegativity on the ring A side of FL118, and thus did not alter polarity of the molecule [22]. In contrast, there is a clear electronegativity and polarity increase at ring A of SN-38 and topotecan [22]. Furthermore, our studies indicate that the increase of electronegativity and polarity at the ring A or B site of the molecule plays a role for the molecule acting as a substrate of the drug efflux pump ABCG2; in turn ABCG2 expression becomes a resistance factor for these drugs with higher electronegativity at the rings A and B end [22]. In the current study, we showed that ABCG2 and P-gp/MDR1 are both expressed in SW620 and HCT-8 colon cancer cells (Figure 6B). We further demonstrated in the Caco-2 permeability assay that FL118 exhibits high Caco-2 permeability, and FL118 is not a P-gp/MDR1 substrate (Table 3). Based on the absorption potential classification, Papp (A-to-B) ≥1.0 × 10-6 cm/s is considered as high permeability, FL118 permeability value is 7-8 times of this number on average (Table 3). So FL118’s Caco-2 permeability is very high. Consistently, with and without P-gp inhibitor valspodar, efflux rate of FL118 in Caco-2 cells is 1 or close to 1 (1.2). This is an extreme low efflux number, which suggests that FL118 is absolutely not a P-gp/MDR1 substrate. In consideration of the fact that intestine epithelial cells highly express both ABCG2 and P-gp/MDR1 and given that FL118 is not a substrate of ABCG2 nor P-gp, FL118 would be a favorable drug candidate for oral administration.
Consistent with the findings summarized above, our study also showed that inhibition of 90% or 95% of SW620 or HCT-8 cell growth needs much higher SN-38 concentration than FL118 in comparison with the ratio of SN-38/FL118 at 25% or 50% cell inhibition by these drugs (Table 2). This observation suggests that efflux pump proteins likely play important roles in drug resistance by pumping SN-38 or topotecan out of cancer cells during dosing increase, which is consistent with the previous finding that irinotecan/SN-38 and topotecan are substrates of multiple ABC transporters including ABCG2 [17-21] and P-gp [19,20,32-37]. Actually, for many drugs, inhibition of ABCG2 and/or P-gp is an effective way to increase drug efficacy [38-42]. Thus, FL118 has inherent advantages over many other drugs for avoiding ABCG2 and P-gp-induced resistance. Overcoming ABCG2 and P-gp/MDR1 resistance by FL118 is further supported by other observations. First, while inhibition of HCT-8 colon cancer cell colony formation by FL118 treatment for 2 hours versus 6 hours showed very similar results (Figure 5B versus 5C, left panel), the inhibition of HCT-8 cell colony formation with 6-hour topotecan treatment was much more effective than 2-hour topotecan treatment (Figure 5B versus 5C, right panel). Second, sildenafil, a multiple ABC transporter inhibitor [28-31], is much more effective than the ABCG2 inhibitor KO143 for increasing the potent of SN-38 inhibition on SW620 and HCT-8 cell growth, while both sildenafil and KO143 do not significantly alter FL118 inhibition of SW620 and HCT-8 cell growth (Figure 7, Table 4). Thus, this provides strong evidence suggesting that FL118 is likely able to bypass resistance resulted from other efflux pump proteins in addition to ABCG2 and P-gp/MDR1. This interpretation is consistent with the fact that SW620 and HCT-8 cells express both MDR1 and ABCG2 (Figure 6B) and that the efflux pump proteins of ABCG2/BCRP, ABCB1/Pgp/MDR1, ABCC10, ABCC4 and ABCC5 were demonstrated to be inhibited by sildenafil [28-31]. Nevertheless, further studies will be required for confirmation of individual efflux pump molecules.
Here we want to point out that based on the fact that FL118 is highly superior to irinotecan/SN-38 and topotecan in vitro and in vivo, it is possible that FL118 possesses additional mechanism of action to overcome or bypass irinotecan/SN-38 and topotecan resistance. For example, it was reported that SN-38 activates MAPK14/p38alpha, which confers irinotecan resistance to p53-defective cells by inducing survival autophagy [43]. Therefore, it would be an interesting research area to explore whether a differential effect of FL118 versus SN-38 and topotecan on MAPK14/p38alpha and autophagy can be defined and thus, extending additional uniqueness of FL118 over irinotecan and topotecan. In any case, it is well known that drug resistance could be through multiple mechanisms [44]. For example, it was shown that BRCA1/p53-deficient mammary tumors initially respond to topotecan, but frequently acquire resistance by overexpression of the efflux transporter ABCG2 [25]. However, ABCG2 overexpression could not provide a full explanation, since topotecan can not eradicate ABCG2(-/-);BRCA1(-/-);p53(-/-) tumors, not even by combination of topotecan-olaparib. Instead, olaparib substantially increases topotecan toxicity [25], which is a common issue for combinational treatment. Additionally, tumor hosts may also be a factor for the time needed to produce tumor resistance to drug treatment. We observed that the same human xenograft tumor on different individual SCID mice do not produce irinotecan or topotecan resistance at the same time after treatment. Instead, certain mice may produce resistance much faster than others, and still others may not produce irinotecan or topotecan resistance in the defined irinotecan and topotecan treatment period (e.g. 4 weeks, not shown). Such observation is actually consistent with the situation in clinical practice for cancer treatment. Nevertheless, in consideration of our finding that FL118 is not a substrate of the efflux pump ABCG2 [22], possibly not a substrate of other efflux pump proteins as well (e.g. P-gp/MDR1, this study), and that it is known that P-gp and ABCG2 are major efflux pump proteins in the gastrointestinal tract epithelial cells, FL118 may exhibit favorable bioavailability via oral administration, which is an active research area in our research group.
While a great deal of studies in the literature have reported the combination of irinotecan/CPT-11 [28-31] or topotecan [45-53] with other therapeutic agents as a common strategy for cancer treatment, there is no easy approach that could resolve either the inherent or acquired resistance of human tumors to irinotecan, topotecan, or other therapeutic agents. As mentioned above, the current approach to overcome irinotecan and topotecan resistance is to combine one or more different chemotherapeutic agents. This approach usually results in three consequences: 1) partially overcome drug resistance with tolerated toxicity in most reported cases; 2) well tolerated but no benefit for patients; 3) over toxicity without benefit to patients. Therefore, even in the best situation, the irinotecan/topotecan resistance issue may not be well resolved [54]. As mentioned above, toxicity from multiple drug combinations usually has the toxicity issue during treatment. In this regard, our previous study showed that the antitumor efficacy of FL118 is highly superior to irinotecan and topotecan [10], and our current studies demonstrated that FL118 overcomes human xenograft tumors which acquired irinotecan and topotecan resistance. Furthermore, consistent with these findings, the PK data obtained in this study at least in part explained the favorable toxicity and high efficacy of FL118. However, for the follow-up studies toward filing an investigational new drug (IND) at the FDA, we will need to investigate FL118 absorption, distribution, metabolism and excretion (ADME) in animal models in a more comprehensive approach. When comparing the result from Figure 2, with the result from Figure 3 there is an interesting observation, it appears that FL118 tended to be more effective in eliminating topotecan-resistant tumors than eliminating irinotecan-resistant tumors, although this trend requires further study to confirm. Nevertheless, given the challenging issue of irinotecan and topotecan resistance in the current clinical practice, application of our finding reported here may result in significant consequences for solving the challenging issue of acquirement of irinotecan or topotecan resistance during cancer treatment.
In conclusion, irinotecan and topotecan resistance is a challenging issue for cancer treatment, especially for the therapeutics of advanced cancer. We demonstrated that FL118 effectively overcomes irinotecan and topotecan resistance without inducing its own drug resistance in human tumor xenograft models during its multiple time usage. Mechanistically, FL118 not only inhibits the expression of survivin, Mcl-1, XIAP, cIAP2 [10] and HdmX/Hdm4 [23], but also at least bypasses ABCG2 [22] and P-pg/MDR1 resistance (this study) and possible other efflux pump protein-induced drug resistance (suggested from the Figure 7 data of this study), which explains the favorable toxicity and PK profile of FL118; rapidly cleared in blood circulation while accumulated in xenograft tumors. Together, these findings provide new perspectives of the significance of FL118 for its further clinical development for treatment of cancer.
Acknowledgements
This work was sponsored in part by grants from NIH/NCI (R44CA176937), NCI (R21CA180764), US Army Department of Defense (W81XWH-12-1-0305), and by shared resources supported by NCI Cancer Center Support Grant to Roswell Park Cancer Institute (P30CA016056). The authors would like to thank relevant staff scientists including the former PK facility Director Dr. Jerry Fetterly from the Pharmacokinetics (PK) Facility at Roswell Park Cancer Institute for their cooperation during development of FL118 assay and the measurement of FL118 concentration in tumor and plasma specimen. We would also like to thank our department writing group for critical review of this manuscript and Mrs. Katherine Plante for her editorial check of the manuscript before submission. Additionally, we thank the CRO Absorption System for their service of the Caco-2 experiment, and the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program (DTP), Division of Cancer Treatment and Diagnosis, National Cancer Institute (NCI) for initially providing the FL118 drug before we synthesize our own.
Disclosure of conflict of interest
FL118 is currently being developed in Canget BioTekpharma LLC (www.canget-biotek.com), a Roswell Park Cancer Institute-spinoff company. XL, XJL and FL are investors in Canget.
Abbreviations
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- ABC
ATP-binding cassette
- BCRP
ABC transporter G2 (ABCG2)/breast cancer resistance protein
- IV
intravenous
- IP
intraperitoneal
- MTD
maximum tolerated doses
- PK
pharmacokinetics
- MDR1
permeability glycoprotein (P-gp)/multi-drug resistance 1
- SCID
severe combined immunodeficiency
- Top1
topoisomerase 1
References
- 1.Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminate. J Am Chem Soc. 1966;88:3888–3890. [Google Scholar]
- 2.Liew ST, Yang LX. Design, synthesis and development of novel camptothecin drugs. Curr Pharm Des. 2008;14:1078–1097. doi: 10.2174/138161208784246180. [DOI] [PubMed] [Google Scholar]
- 3.Li QY, Zu YG, Shi RZ, Yao LP. Review camptothecin: current perspectives. Curr Med Chem. 2006;13:2021–2039. doi: 10.2174/092986706777585004. [DOI] [PubMed] [Google Scholar]
- 4.Chen MC, Lee NH, Ho TJ, Hsu HH, Kuo CH, Kuo WW, Lin YM, Tsai FJ, Tsai CH, Huang CY. Resistance to irinotecan (CPT-11) activates epidermal growth factor receptor/nuclear factor kappa B and increases cellular metastasis and autophagy in LoVo colon cancer cells. Cancer Lett. 2014;349:51–60. doi: 10.1016/j.canlet.2014.03.023. [DOI] [PubMed] [Google Scholar]
- 5.Bessho Y, Oguri T, Achiwa H, Muramatsu H, Maeda H, Niimi T, Sato S, Ueda R. Role of ABCG2 as a biomarker for predicting resistance to CPT-11/SN-38 in lung cancer. Cancer Sci. 2006;97:192–198. doi: 10.1111/j.1349-7006.2006.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, Mercer KE, Zhuang Y, Panetta JC, Johnston B, Scheper RJ, Stewart CF, Schuetz JD. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612–7621. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz-Munoz W, Di Desidero T, Man S, Xu P, Jaramillo ML, Hashimoto K, Collins C, Banville M, O’Connor-McCourt MD, Kerbel RS. Analysis of acquired resistance to metronomic oral topotecan chemotherapy plus pazopanib after prolonged preclinical potent responsiveness in advanced ovarian cancer. Angiogenesis. 2014;17:661–673. doi: 10.1007/s10456-014-9422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemunaitis J, Cox J, Meyer W, Courtney A, Mues G. Irinotecan hydrochloride (CPT-11) resistance identified by K-ras mutation in patients with progressive colon cancer after treatment with 5-fluorouracil (5-FU) Am J Clin Oncol. 1997;20:527–529. doi: 10.1097/00000421-199710000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Saraiya B, Gounder M, Dutta J, Saleem A, Collazo C, Zimmerman L, Nazar A, Gharibo M, Schaar D, Lin Y, Shih W, Aisner J, Strair RK, Rubin EH. Sequential topoisomerase targeting and analysis of mechanisms of resistance to topotecan in patients with acute myelogenous leukemia. Anticancer Drugs. 2008;19:411–420. doi: 10.1097/CAD.0b013e3282f5218b. [DOI] [PubMed] [Google Scholar]
- 10.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A Novel Small Molecule FL118 That Selectively Inhibits Survivin, Mcl-1, XIAP and cIAP2 in a p53-Independent Manner, Shows Superior Antitumor Activity. PLoS One. 2012;7:e45571. doi: 10.1371/journal.pone.0045571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. Mechanism of action of camptothecin. Ann N Y Acad Sci. 2000;922:1–10. doi: 10.1111/j.1749-6632.2000.tb07020.x. [DOI] [PubMed] [Google Scholar]
- 12.Kang MR, Muller MT, Chung IK. Telomeric DNA damage by topoisomerase I. A possible mechanism for cell killing by camptothecin. J Biol Chem. 2004;279:12535–12541. doi: 10.1074/jbc.M309779200. [DOI] [PubMed] [Google Scholar]
- 13.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ling X, Li F. An intravenous (i. v.) route-compatible formulation of FL118, a survivin, Mcl-1, XIAP, and cIAP2 selective inhibitor, improves FL118 antitumor efficacy and therapeutic index (TI) Am J Transl Res. 2013;5:139–154. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Ling X, Cao S, Liu X, Wan S, Jiang T, Li F. Antitumor activity of FL118, a survivin, Mcl-1, XIAP, cIAP2 selective inhibitor, is highly dependent on its primary structure and steric configuration. Mol Pharm. 2014;11:457–467. doi: 10.1021/mp4004282. [DOI] [PubMed] [Google Scholar]
- 16.Li F. Anticancer drug FL118 is more than a survivin inhibitor: Where is the Achilles’ heel of cancer? Am J Cancer Res. 2014;4:304–311. [PMC free article] [PubMed] [Google Scholar]
- 17.Nakatomi K, Yoshikawa M, Oka M, Ikegami Y, Hayasaka S, Sano K, Shiozawa K, Kawabata S, Soda H, Ishikawa T, Tanabe S, Kohno S. Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem Biophys Res Commun. 2001;288:827–832. doi: 10.1006/bbrc.2001.5850. [DOI] [PubMed] [Google Scholar]
- 18.Kawabata S, Oka M, Shiozawa K, Tsukamoto K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y, Kamihira S, Doyle LA, Ross DD, Kohno S. Breast cancer resistance protein directly confers SN-38 resistance of lung cancer cells. Biochem Biophys Res Commun. 2001;280:1216–1223. doi: 10.1006/bbrc.2001.4267. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Jin HE, Kim W, Han YH, Kim DD, Chung SJ, Shim CK. Involvement of P-glycoprotein, multidrug resistance protein 2 and breast cancer resistance protein in the transport of belotecan and topotecan in Caco-2 and MDCKII cells. Pharm Res. 2008;25:2601–2612. doi: 10.1007/s11095-008-9678-0. [DOI] [PubMed] [Google Scholar]
- 20.de Vries NA, Zhao J, Kroon E, Buckle T, Beijnen JH, van Tellingen O. P-glycoprotein and breast cancer resistance protein: two dominant transporters working together in limiting the brain penetration of topotecan. Clin Cancer Res. 2007;13:6440–6449. doi: 10.1158/1078-0432.CCR-07-1335. [DOI] [PubMed] [Google Scholar]
- 21.Jia P, Wu S, Li F, Xu Q, Wu M, Chen G, Liao G, Wang S, Zhou J, Lu Y, Ma D. Breast cancer resistance protein-mediated topotecan resistance in ovarian cancer cells. Int J Gynecol Cancer. 2005;15:1042–1048. doi: 10.1111/j.1525-1438.2005.00260.x. [DOI] [PubMed] [Google Scholar]
- 22.Westover D, Ling X, Lam H, Welch J, Jin C, Gongora C, Del Rio M, Wani M, Li F. FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol Cancer. 2015;14:92. doi: 10.1186/s12943-015-0362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling X, Xu C, Fan C, Zhong K, Li F, Wang X. FL118 Induces p53-Dependent Senescence in Colorectal Cancer Cells by Promoting Degradation of MdmX. Cancer Res. 2014;74:7487–7497. doi: 10.1158/0008-5472.CAN-14-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li F, Ling X, Cao S, inventors. Roswell Park Cancer Institute, assignee. Novel Formulations of Water-Insoluble Chemical Compounds and Methods of Using a Formulation of Compound FL118 for Cancer Therapy. (PCT/US11/58558) USA. 2011
- 25.Zander SA, Kersbergen A, van der Burg E, de Water N, van Tellingen O, Gunnarsdottir S, Jaspers JE, Pajic M, Nygren AO, Jonkers J, Borst P, Rottenberg S. Sensitivity and acquired resistance of BRCA1;p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Res. 2010;70:1700–1710. doi: 10.1158/0008-5472.CAN-09-3367. [DOI] [PubMed] [Google Scholar]
- 26.Ling X, Yang J, Tan D, Ramnath N, Younis T, Bundy BN, Slocum HK, Yang L, Zhou M, Li F. Differential expression of survivin-2B and survivin-DeltaEx3 is inversely associated with disease relapse and patient survival in non-small-cell lung cancer (NSCLC) Lung Cancer. 2005;49:353–361. doi: 10.1016/j.lungcan.2005.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event which is independent on taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–15203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 28.Shi Z, Tiwari AK, Shukla S, Robey RW, Singh S, Kim IW, Bates SE, Peng X, Abraham I, Ambudkar SV, Talele TT, Fu LW, Chen ZS. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011;71:3029–3041. doi: 10.1158/0008-5472.CAN-10-3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JJ, Sun YL, Tiwari AK, Xiao ZJ, Sodani K, Yang DH, Vispute SG, Jiang WQ, Chen SD, Chen ZS. PDE5 inhibitors, sildenafil and vardenafil, reverse multidrug resistance by inhibiting the efflux function of multidrug resistance protein 7 (ATP-binding Cassette C10) transporter. Cancer Sci. 2012;103:1531–1537. doi: 10.1111/j.1349-7006.2012.02328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen ZS, Lee K, Kruh GD. Transport of cyclic nucleotides and estradiol 17-beta-D-glucuronide by multidrug resistance protein 4. Resistance to 6-mercaptopurine and 6-thioguanine. J Biol Chem. 2001;276:33747–33754. doi: 10.1074/jbc.M104833200. [DOI] [PubMed] [Google Scholar]
- 31.Jedlitschky G, Burchell B, Keppler D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J Biol Chem. 2000;275:30069–30074. doi: 10.1074/jbc.M005463200. [DOI] [PubMed] [Google Scholar]
- 32.Miettinen S, Grenman S, Ylikomi T. Inhibition of P-glycoprotein-mediated docetaxel efflux sensitizes ovarian cancer cells to concomitant docetaxel and SN-38 exposure. Anticancer Drugs. 2009;20:267–276. doi: 10.1097/CAD.0b013e328329977f. [DOI] [PubMed] [Google Scholar]
- 33.Arimori K, Kuroki N, Hidaka M, Iwakiri T, Yamsaki K, Okumura M, Ono H, Takamura N, Kikuchi M, Nakano M. Effect of P-glycoprotein modulator, cyclosporin A, on the gastrointestinal excretion of irinotecan and its metabolite SN-38 in rats. Pharm Res. 2003;20:910–917. doi: 10.1023/a:1023847521767. [DOI] [PubMed] [Google Scholar]
- 34.Takeba Y, Sekine S, Kumai T, Matsumoto N, Nakaya S, Tsuzuki Y, Yanagida Y, Nakano H, Asakura T, Ohtsubo T, Kobayashi S. Irinotecan-induced apoptosis is inhibited by increased P-glycoprotein expression and decreased p53 in human hepatocellular carcinoma cells. Biol Pharm Bull. 2007;30:1400–1406. doi: 10.1248/bpb.30.1400. [DOI] [PubMed] [Google Scholar]
- 35.Tagen M, Zhuang Y, Zhang F, Harstead KE, Shen J, Schaiquevich P, Fraga CH, Panetta JC, Waters CM, Stewart CF. P-glycoprotein, but not multidrug resistance protein 4, plays a role in the systemic clearance of irinotecan and SN-38 in mice. Drug Metab Lett. 2010;4:195–201. doi: 10.2174/187231210792928251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J. Clin. Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 37.Yamasaki T, Fujinaga M, Kawamura K, Hatori A, Yui J, Nengaki N, Ogawa M, Yoshida Y, Wakizaka H, Yanamoto K, Fukumura T, Zhang MR. Evaluation of the P-glycoproteinand breast cancer resistance protein-mediated brain penetration of 11C-labeled topotecan using small-animal positron emission tomography. Nucl Med Biol. 2011;38:707–714. doi: 10.1016/j.nucmedbio.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Gardner ER, Smith NF, Figg WD, Sparreboom A. Influence of the dual ABCB1 and ABCG2 inhibitor tariquidar on the disposition of oral imatinib in mice. J Exp Clin Cancer Res. 2009;28:99. doi: 10.1186/1756-9966-28-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He Y, Bi Y, Hua Y, Liu D, Wen S, Wang Q, Li M, Zhu J, Lin T, He D, Li X, Wang Z, Wei G. Ultrasound microbubble-mediated delivery of the siRNAs targeting MDR1 reduces drug resistance of yolk sac carcinoma L2 cells. J Exp Clin Cancer Res. 2011;30:104. doi: 10.1186/1756-9966-30-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cordo Russo RI, Garcia MG, Alaniz L, Blanco G, Alvarez E, Hajos SE. Hyaluronan oligosaccharides sensitize lymphoma resistant cell lines to vincristine by modulating P-glycoprotein activity and PI3K/Akt pathway. Int J Cancer. 2008;122:1012–1018. doi: 10.1002/ijc.23122. [DOI] [PubMed] [Google Scholar]
- 41.Peng H, Qi J, Dong Z, Zhang JT. Dynamic vs static ABCG2 inhibitors to sensitize drug resistant cancer cells. PLoS One. 2010;5:e15276. doi: 10.1371/journal.pone.0015276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seo SB, Hur JG, Kim MJ, Lee JW, Kim HB, Bae JH, Kim DW, Kang CD, Kim SH. TRAIL sensitize MDR cells to MDR-related drugs by downregulation of P-glycoprotein through inhibition of DNA-PKcs/Akt/GSK-3beta pathway and activation of caspases. Mol Cancer. 2010;9:199. doi: 10.1186/1476-4598-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paillas S, Causse A, Marzi L, de Medina P, Poirot M, Denis V, Vezzio-Vie N, Espert L, Arzouk H, Coquelle A, Martineau P, Del Rio M, Pattingre S, Gongora C. MAPK14/p38alpha confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy. 2012;8:1098–1112. doi: 10.4161/auto.20268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 45.Kawano M, Mabuchi S, Kishimoto T, Hisamatsu T, Matsumoto Y, Sasano T, Takahashi R, Sawada K, Takahashi K, Takahashi T, Hamasaki T, Kimura T. Combination treatment with trabectedin and irinotecan or topotecan has synergistic effects against ovarian clear cell carcinoma cells. Int J Gynecol Cancer. 2014;24:829–837. doi: 10.1097/IGC.0000000000000143. [DOI] [PubMed] [Google Scholar]
- 46.Spigel DR, Waterhouse DM, Lane S, Legenne P, Bhatt K. Efficacy and safety of oral topotecan and bevacizumab combination as secondline treatment for relapsed small-cell lung cancer: an open-label multicenter single-arm phase II study. Clin Lung Cancer. 2013;14:356–363. doi: 10.1016/j.cllc.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Trafalis DT, Alifieris C, Krikelis D, Tzogkas N, Stathopoulos GP, Athanassiou AE, Sitaras NM. Topotecan and pegylated liposomal doxorubicin combination as palliative treatment in patients with pretreated advanced malignant pleural mesothelioma. Int J Clin Pharm Ther. 2012;50:490–499. doi: 10.5414/CP201688. [DOI] [PubMed] [Google Scholar]
- 48.Zarogoulidis K, Mylonaki E, Kakavelas P, Zarogoulidis P, Tsiouda T, Rapti E, Lithoxopoulou H, Zarogoulidou V, Kontakiotis T. Topotecancarboplatin-etoposide combination as 1st line treatment in patients with small cell lung cancer. Lung Cancer. 2009;66:226–230. doi: 10.1016/j.lungcan.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 49.Choi HJ, Cho BC, Shin SJ, Cheon SH, Jung JY, Chang J, Kim SK, Sohn JH, Kim JH. Combination of topotecan and etoposide as a salvage treatment for patients with recurrent small cell lung cancer following irinotecan and platinum first-line chemotherapy. Cancer Chemother Pharmacol. 2008;61:309–313. doi: 10.1007/s00280-007-0505-9. [DOI] [PubMed] [Google Scholar]
- 50.Brave M, Dagher R, Farrell A, Abraham S, Ramchandani R, Gobburu J, Booth B, Jiang X, Sridhara R, Justice R, Pazdur R. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. Oncology. 2006;20:1401–1404. 1410. discussion 1410-1411, 1415-1406. [PubMed] [Google Scholar]
- 51.Ardizzoni A, Manegold C, Debruyne C, Gaafar R, Buchholz E, Smit EF, Lianes P, ten Velde G, Bosquee L, Legrand C, Neumaier C, King K. European organization for research and treatment of cancer (EORTC) 08957 phase II study of topotecan in combination with cisplatin as second-line treatment of refractory and sensitive small cell lung cancer. Clin Cancer Res. 2003;9:143–150. [PubMed] [Google Scholar]
- 52.Simpson AB, Calvert PM, Sludden JA, Boddy AV, Griffin MJ, Schatzlein A, Wilson P, Fishwick K, Wheatley A, Ross GA, Calvert AH, Twelves CJ. Topotecan in combination with carboplatin: phase I trial evaluation of two treatment schedules. Ann Oncol. 2002;13:399–402. doi: 10.1093/annonc/mdf041. [DOI] [PubMed] [Google Scholar]
- 53.Thompson J, George EO, Poquette CA, Cheshire PJ, Richmond LB, de Graaf SS, Ma M, Stewart CF, Houghton PJ. Synergy of topotecan in combination with vincristine for treatment of pediatric solid tumor xenografts. Clin Cancer Res. 1999;5:3617–3631. [PubMed] [Google Scholar]
- 54.Gholam D, Giacchetti S, Brezault-Bonnet C, Bouchahda M, Hauteville D, Adam R, Ducot B, Ghemard O, Kustlinger F, Jasmin C, Levi F. Chronomodulated irinotecan, oxaliplatin, and leucovorin-modulated 5-Fluorouracil as ambulatory salvage therapy in patients with irinotecan- and oxaliplatin-resistant metastatic colorectal cancer. Oncologist. 2006;11:1072–1080. doi: 10.1634/theoncologist.11-10-1072. [DOI] [PubMed] [Google Scholar]