

Abstract
Metalloproteinase (MMP)9 plays a pivotal role in ischemic stroke induced blood brain barrier (BBB) disruption. Correlation between HSP90 and MMP9 in several diseases prompted us to evaluate the efficacy of HSP90 inhibition as a novel approach to protect BBB integrity in ischemic stroke. ELISA was used to detect HSP90α and MMP9 in serum samples of stroke patients, which showed that HSP90α significantly correlated with MMP9 among 63 serum samples of stroke patients. Male C57/BL6 mice were pretreated with 17-Dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) or vehicle before being subjected to transient occlusion of middle cerebral artery and reperfusion (MCAO). Infarction, neurological scores, Evans blue (EB) extravasation, inflammatory responses and tight junction protein expression were examined 24 h after MCAO. We also investigated if 17-DMAG protected BBB integrity by suppressing inflammation and MMP9 activation. Oxygen glucose deprivation (OGD) was performed on bEnd.3 cells to explore the mechanisms of HSP90 inhibition in inhibiting MMP9. The results demonstrated that infarct volume was reduced in 17-DMAG-treated mice compared to control group following MCAO. Neurological outcomes were greatly improved in 17-DMAG-treated mice. Inflammatory responses, MMP9 activity and EB extravasation were decreased by 17-DMAG. In addition, 17-DMAG inhibited nuclear factor kappa B (NF-κB) activation following MCAO. Furthermore, HSP90 inhibition decreased NF-κB dependent MMP9 expression in bEnd.3 after OGD /reoxygenation. These findings suggested that HSP90 could be a novel therapeutic target in BBB breakdown during ischemic stroke. As several HSP90 inhibitors are in clinical trials for cancer, these findings have translational implications.
Keywords: Blood brain barrier, inflammation, ischemic stroke, heat shock protein 90
Introduction
Ischemic stroke is the second most common cause of death and the most frequent cause of disability worldwide [1]. Tissue plasminogen activator (tPA) remains the only US Food and Drug approved treatment for ischemic stroke. Due to an increased risk of hemorrhagic transformation, only 5% patients are eligible for tPA treatment [2]. Moreover, tPA may cause injury to blood-brain barrier (BBB) through activating matrix metalloproteinases (MMPs) [3]. Therefore, it is imperative to develop new combination therapies for ischemic stroke.
Disruption of BBB is a hallmark of stroke pathogenesis, which results in vasogenic edema formation and further brain damage [4]. Increased expression of MMPs has been reported to play an important role in stroke-induced BBB leakage through degrading the TJ proteins and basal proteins [5,6]. In particular, an increase in serum MMP9 was observed and related to more severe stroke in patients [7]. MMP9 KO or pharmacological inhibition provides strong neuroprotection and attenuates proteolysis of BBB in mouse model of transient middle cerebral artery occlusion (MCAO) [8-10]. Although the exact cell sources for the MMP9 generation during focal cerebral ischemia are not yet defined in vivo, microglial activation has been evidenced to potentiate damage to components of the BBB under ischemia-like conditions [11]. Thus, inhibition of inflammation may decrease MMPs and attenuate the disruption of BBB.
The heat shock protein (HSP) 90 is an important molecular chaperone for protein folding, intracellular disposition and assembly of many proteins, including key mediators of signal transduction and transcriptional regulation. Previous study has demonstrated that HSP90α is over expressed in animal model of cerebral ischemia [12,13]. In other diseases, the levels of HSP90 and MMP9 have been demonstrated to be correlated with each other [14,15]. However, no studies have been conducted on HSP90α level in human stroke and the association with MMP9.
17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) is a selective HSP90 inhibitor currently undergoing clinical trials against cancers, which block the ATP-binding site of HSP90 and exert pleiotropic functions, which include induction of the heat shock response and degradation of some client proteins. Moreover, many studies have found that HSP90 inhibition by 17-DMAG protects the brain against ischemic injury through inhibiting inflammation and reducing MMP9 after ischemia [16].
Therefore, the aim of our study is to demonstrate the relationship of HSP90α level with MMP9 in stroke patients, and examine the protective effect of HSP90 inhibition by 17-DMAG on BBB in mouse model of MCAO. As several HSP90 inhibitors have been demonstrated a favorable safety profile in clinical trials in cancer patients, our results may have translational implications.
Materials and methods
Study subject
Subjects were enrolled in the study from February 2012 to July 2013 and were approved by the institutional Review board of Shanghai Jiaotong University, and gave informed consent. The age and gender from the control group matched that of the stroke patients. Demographic changes, associated laboratory inspection, imaging information were also collected to analyze. Whether subjects taking medicines including antidiabets, hypotensor or platelet aggregation drugs was also recorded. The exclusion criteria included recurrent stroke, intracranial tumor, multiple trauma, hematological system diseases, renal or liver failure, acute infectious diseases and other diseases affecting the hemogram. If the time from the onset of stroke symptoms to blood sample collection was longer than 24 hours, the patient was excluded. The risk factors were defined as following: hypertension: blood pressure above 140/90 mmHg; hyperlipidemia: total cholesterol level ≥ 0.7 mmol/L, triglyceride level ≥ 1.8 mmol/L and HDL level < 1 mmol/L; Diabetes mellitus: fastingblood glucose level > 6.1 mmol/L or HbA1c ≥ 7%.
Transient middle cerebral artery occlusion model and drug administration
All animal protocols have been approved by Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, Shanghai, China. tMCAO was carried out as previously described [17]. 17-DMAG (0.2 mg/kg) or vehicle was intraperitoneally injected immediately after MCAO. The optimal dose is based on previous study [18].
Measurement of physiological parameters
Regional cerebral blood flow (rCBF) was measured by the probe of laser-Doppler flowmetry (MoorLDI2; Moor Co., Ltd., U.K.) as previously described [19]. In randomly selected animals, mean arterial pressure was monitored, and arterial blood pH, pCO2, and pO2 were measured before MCAO operation, during (10 min after MCAO), and 30 min after reperfusion (Rapid Lab 248, Bayer, Wuppertal, Germany). Core temperature as maintained at about 37°C ± 5°C during the surgery.
Infarct volume measurement
Infarct volume was measured using TTC staining as previously described [17]. The relative infarct size measure was calculated with the following equation: relative infarct size = (contralateral area-ipsilateral non-infarct area)/contralateral area.
Neurobehavioral assessments
Neurological assessments were performed by an experimenter blind to the treatment conditions at 24 h after MCAO. The neurological findings were scored on a 4-point scale: 0 (no neurological deficit); 1 (Horner’s syndrome); 2 (forelimb flexion); 3 (circling to right). Cumulative scores were calculated for each group.
Western blot analysis
Total proteins were extracted from cortex. Cytoplasmic and nuclear protein subfractions were prepared as described previously [20]. Protein concentration was measured by BCA kit (Thermo Scientific). Equal amounts of protein lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membrane and then immunoblotted with antibodies against HSP90α (1:1000 dilution; Cell Signaling Technology); HSP90 (1:1000 dilution; Cell Signaling Technology); HSP70 (1:1000 dilution; Cell Signaling Technology); p65 (1:1000 dilution; Cell Signaling Technology), ZO-1 (1:500; Cell Signaling Technology), occludin (1:500; Cell Signaling Technology), IKKα (1:1000 dilution; Cell Signaling Technology), IKKβ (1:1000 dilution; Cell Signaling Technology) and β-actin (1:1000 dilution; Cell Signaling Technology). Then, the membranes were incubated with IRDye800CW-conjugated secondary antibody. Protein bands were captured by Odyssey imagining system (LICOR).
Real-time PCR
Total RNA was extracted from cells or brain tissue using TRIZOL (Invitrogen). 1 μg total RNA was used to perform the reverse transcription with High Capacity cDNA Archive Kit (Applied Biosystem). Real-time quantitative polymerase chain reaction (PCR) analysis for interleukin (IL)-1β, MCP-1, MMP9 was performed using TaqMan gene expression assays and the ΔΔCt method with housekeeping gene 18S as the endogenous control. The primers used for the qRT-PCR are listed in Table 1.
Table 1.
Primer lists
| Mouse MMP-9 promoter | 5’-GCGACAAAGGGTCTGTTTTTG-3’ and |
| 5’-TGACTCAGCTTCCTCTCCTGG-3’ for p65 site | |
| Mouse MMP-9 | 5’-GATCCCCAGAGCGTCATTC-3’ |
| 5’-CCACCTTGTTCACCTCATTTTG-3’ | |
| Mouse IL-1β | 5’-CCCTGCAGCTGGAGAGTGTGGA-3’ |
| 5’-TGTGCTCTGCTTGTGAGGTGCTG-3’ | |
| Mouse MCP-1 | 5’-GTCCCTGTCATGCTTCTGG-3’ |
| 5’-GCTCTCCAGCCTACTCATTG-3’ | |
| 18S rRNA | 5’-TCAAGAACGAAAGTCGGAGG-3’ |
| 5’-GGACATCTAAGGGCATCAC-3’ |
Evans blue extravasation
Evans blue extravasation was measured as previously described [21]. Briefly, EVANS blue (2% in saline, 4 ml/kg; Sigma) was administered intraperitoneally at the onset of reperfusion. Mice were transcardially perfused with saline to remove the intravascular dye at 22 hours after reperfusion. Each hemisphere was weighed rapidly, homogenized in 1 ml of 50% trichloroacetic acid (wt/vol), and then centrifuged (12,000*g for 20 minutes). The supernatant was collected and mixed with ethanol (1:3).The concentration of Evans blue was determined by measuring the 610 nm absorbance with spectrophotometry. The results were expressed as Evans blue (μg)/tissue (g) calculated against a liner standard curve.
Gelatin zymography for MMP9
The evaluation of MMP9 activity in conditioned media form cells cultures or homogenates of brain tissue was performed by zymography as described previously [22].
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were drawn from peripheral veins and were clotted for 30 min. After centrifuging at 4°C for 10 min, serum was stored at -20°C until assayed. The levels of human plasma HSP90αand MMP9 were measured by ELISA kits (Westang Bio-Tech, Shanghai, China) according to the manufacturer’s protocols.
Cell cultures
A transformed mouse brain endothelial cell line, bEnd.3 cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured in medium (DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine, and 1 × antibiotic/antimycotic; Invitrogen, Carlsbad, CA) at 37°C in a humidified incubator (10% CO2, 90% air).
Oxygen-glucose deprivation
To simulate ischemic conditions, cultures were subjected to oxygen glucose deprivation (OGD). Before induction of OGD, serum-containing media were removed from the cell cultures before adding low-glucose medium (1.0 g/L, supplemented DMEM). The cultures containing low-glucose medium were placed in a hypoxia chamber (Billups-Rothenburg, Del Mar, CA, USA), which was flushed through with a mixture of 95% N2 and 5% CO2 for 1 hour, and then closed for the duration of the experiment. O2 levels decreased to 0.1% to 0.4% at 4 hours, and were maintained throughout the experiment (18 hours total).
Transfection of small-interfering RNA
scrambled siRNA (Ambion) or HSP90 (Santa Cruz) siRNA was transfected with HiPerFect Transfection Reagent (Qiagen, Shanghai, China). The ability of the siRNA to inhibit HSP90 expression was assessed 48 h post-transfection by western blot.
Chromatin immunoprecipitation (CHIP) assay for NF-κB and MMP9 promoters binding
ChIP assays were performed with a chip kit (Millipore, Billerica, MA) according to manufacturer’s instructions. Briefly, Chromatin was cross-linked by adding formaldehyde to cell culture medium for 10 min at room temperature. Cross-linking reaction was ceased by glycine solution. Cross-linked chromatin was sheared by supersonic at 4°C. Debris was removed by centrifugation and supernatants were collected. The purified chromatin was immunoprecipitated with p65 antibody from Santa Cruz Biotechnology and normal rabbit IgG from Millipore. The DNA/Protein complexes were collected by the use of Protein G-agarose beads. Beads were washed and bound DNA was eluted. After reverse cross-linking reaction and proteinase K digestion, the eluted DNA was used in 35 cycles of PCR amplification with the NF-κB binding site specific primers, which are listed in Table 1.
Statistical analysis
Spearman’s analysis was used for bivariate correlations. Quantitative data are presented as mean ± SEM. Parameters between two groups were compared by 2-tailed Student’s t test. Comparisons of parameters among multiple groups were made by one-way analysis of variance, and comparisons of different parameters between each group were made by Bonferroni’s post-hoc test. A p value < 0.05 was considered to be statistically significant.
Results
Patients and clinical data
The study was approved by the Institutional Review Board and Human Ethics Committee of Shanghai Jiaotong University. Written consent for using the samples for research purposes was obtained from all patients prior to research. The clinical characteristics of ischemic stroke patients and healthy control are listed in Table 2. On admission, the ages of patients were 66.7 ± 9.28 and the ages of healthy control were 64.1 ± 6.91 (p > 0.05). There is no significant differences in gender between control and patients. To eliminate unmatched factor impaction on HSP90α expreesion levels between healthy subjects and stroke patients, a logistic regression was performed, which suggested the HSP90α levels was a stroke risk factor.
Table 2.
Patients characteristics
| Control | Acute ischemic stroke | P | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| LA | CE | SA | Total | |||
| N | 50 | 20 | 23 | 21 | 64 | 1.00 |
| Race (Asia, %) | 100% | 100% | 100% | 100% | 100% | 1.00 |
| Ethnicity (Han, %) | 100% | 100% | 100% | 100% | 100% | 1.00 |
| Age (years, mean ± SD) | 64.1 ± 6.91 | 68.1 ± 8.23 | 62.1 ± 9.56 | 65.4 ± 7.97 | 66.7 ± 9.28 | > 0.05 |
| Sex (male/female, N) | 8/7 | 12/8 | 13/10 | 11/10 | 36/28 | > 0.05 |
| Hypertension (N, %) | 5 (33.3%) | 16 (80%) | 15 (65%) | 14 (67%) | 45 (70%) | < 0.05 |
| Diabetes (N, %) | 2 (13.3%) | 14 (70%) | 8 (35%) | 6 (29%) | 28 (44%) | < 0.05 |
| Hyperlipidemia (N, %) | 3 (20%) | 12 (60%) | 10 (43%) | 14 (67%) | 36 (56%) | < 0.05 |
| Cardiopathy (N, %) | 5 (10%) | 0 (0%) | 23 (100%) | 4 (19%) | 27 (42%) | < 0.05 |
| NHISS (Mean, Min, Max) | NA | 4.89 (1, 11) | 5.01 (0, 19) | 4.73 (1, 10) | 4.88 (0, 19) | NA |
The levels of blood HSP90α in acute ischemic stroke patients
ELISA for HSP90α showed that HSP90α level in serum from 64 patients is higher than serum from 50 healthy subjects (Figure 1A). No difference was observed between subtypes. In addition, we also analyzed the difference between patients with/without antidiabetes, hypotensor or platelet aggregation. No significant difference of the expression levels of HSP90α was found.
Figure 1.
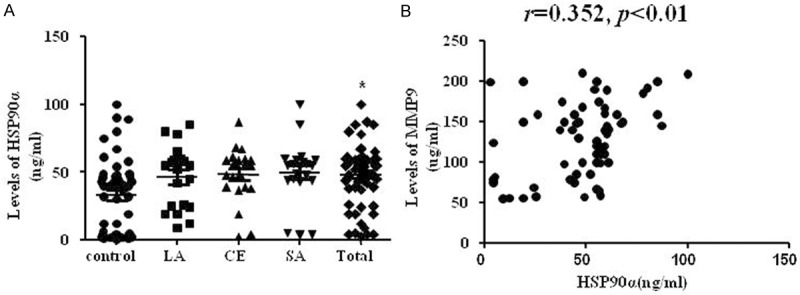
Blood level of HSP90α and the correlation analysis between HSP90α and MMP9 in acute ischemic stroke. A. HSP90α was highly expressed in acute stroke patients. *p < 0.05, vs. control. n = 50 in control group, n = 64 in total patients. B. The scatterplot showed MMP9 levels were positively associated with HSP90α level. R = 0.352, p < 0.01.
The correlation between HSP90α and MMP9 level of stroke patients’ serum
HSP90α protein expression was reported to be significantly associated with MMP9 expression in human gastric carcinoma tissues and chronic kidney disease [14,15]. Therefore, we investigated the relationship of HSP90α and MMP9 protein level in stroke. The level of serum MMP9 was positively associated with HSP90α level (Figure 1B, r=0.352, p<0.01).
Effects of 17-DMAG on physiological parameters
The effects of 17-DMAG on regional cerebral blood flow, arterial blood pressure, pH, pO2, and pCO2 were evaluated in mice after MCAO. No significant difference was observed between 17-DMAG- and vehicle-treated groups in any of the respiratory and cardiovascular parameters tested (Table 3).
Table 3.
Physiological parameters
| Parameter | Group | Before | During | After |
|---|---|---|---|---|
| rCBF (%) | vehicle | 100 | 12.7 ± 3.3 | 95.7 ± 8.6 |
| DM | 100 | 10.4 ± 3.9 | 93.2 ± 9.4 | |
| MABP (mmHg) | vehicle | 94.3 ± 4.9 | 92.5 ± 9.8 | 88.7 ± 11.2 |
| DM | 98.4 ± 7.6 | 94.3 ± 7.9 | 91.3 ± 8.1 | |
| PCO2 (mmHg) | vehicle | 46.9 ± 6.4 | 46.4 ± 4.9 | 47.9 ± 6.3 |
| DM | 49.8 ± 7.3 | 49.9 ± 7.1 | 46.8 ± 5.9 | |
| PO2 (mmHg) | vehicle | 130.5 ± 19.9 | 119.5 ± 16.4 | 135.4 ± 25.9 |
| DM | 129.6 ± 21.8 | 124.8 ± 19.5 | 131.9 ± 19.5 | |
| Temperature (°C) | vehicle | 36.8 ± 0.3 | 36.9 ± 0.2 | 37.0 ± 0.4 |
| DM | 37.2 ± 0.2 | 36.8 ± 0.1 | 37.2 ± 0.3 |
The effect of 17-DMAG on HSP90 and HSP70 expression
The heat shock response is a powerful cellular defense mehanism against a variety of insults. As shown in Figure 2, HSP90α and HSP70 expression was significantly increased in the mouse ischemic cortex. 17-DMAG and its analogues have been shown to disassociate the interaction of HSP90 with HSF1, which subsequently induces the expression of HSP70. Therefore, assessment of HSP70 induction has been used as a useful pharmacodynamic marker of HSP90 inhibition.Indeed, 17-DMAG treatment markedly increased HSP70 expression in ischemic cortex, but without effect on HSP90.
Figure 2.
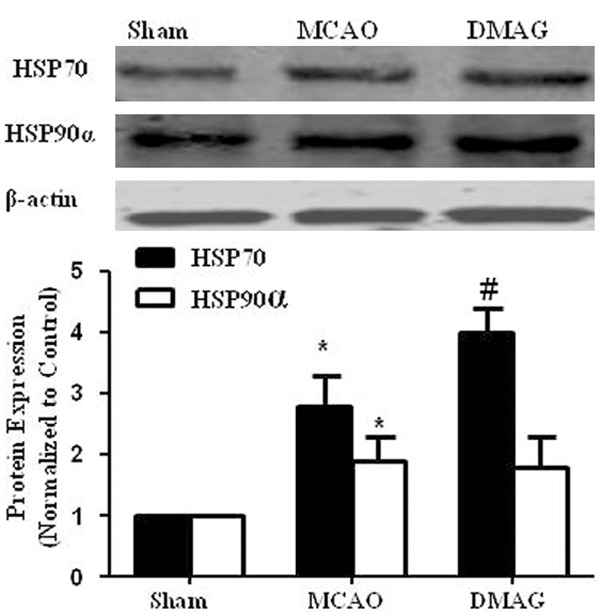
The effect of 17-DMAG on HSP90α and HSP70 expressions in mouse cortex. Mice were given either vehicle or 17-DMAG (0.2 mg/kg, IP) immediately after MCAO. 23 hr after injection of 17-DMAG, expressions of Hsp70 and HSP90 in mice cortex was determined by western blot. HSP90 and HSP70 expression were then quantitated by densitometric analysis. *p < 0.05 compared to sham group; #p < 0.05 compared to MCAO group n = 4/group.
17-DMAG reduced infarction, improved neurological deficits and inhibited BBB disruption
MCAO was performed to dertermine the effect of HSP90 inhibition on cerebral ischemia. As shown in Figure 3A, cortical blood flow was successfully decreased by 80% after occlusion. HSP90 inhibition by 17-DMAG both reduced infarct volume (Figure 3B) and protected BBB integrity (Figure 3C). 17-DMAG also improved neurological deficits of MCAO mice at 24 h after reperfusion compared to vehicle (Figure 3D).
Figure 3.

17-DMAG reduced infarction, improved neurological deficits and inhibited BBB disruption. A. In the mouse MCAO model, laser -doppler flowmeter showing that cortical blood flow was reduced more than 80%. B. 17-DMAG significantly decreased infarction size in cerebral ischemia. C. 17-DMAG significantly attenuated MCAO-induced Evans blue extravasation. D. 17-DMAG significantly improved neurological function in MCAO mice. *p < 0.05, compared to MCAO group n = 10/group.
17-DMAG attenuated MCAO-induced MMP9 elevation and tight junction degradation
MMP9 activities in cortex of MCAO mice were investigated by zymography. Consistant with previous studies, MMP9 activity was strongly increased in MCAO mice, whereas 17-DMAG treatment significantly suppressed the induction of MMP9 activity in cortex (Figure 4A). Real-time PCR showed that 17-DMAG treatment significantly suppressed MMP9 gene expression induced by MCAO, similar to the results obtained with zymography (Figure 4B). Conversely, protein levels of Occludin and ZO-1, tight junction proteins involved in BBB integrity, were markedly decreased in MCAO group, which were robustly prevented by 17-DMAG treatment (Figure 4C and 4D).
Figure 4.
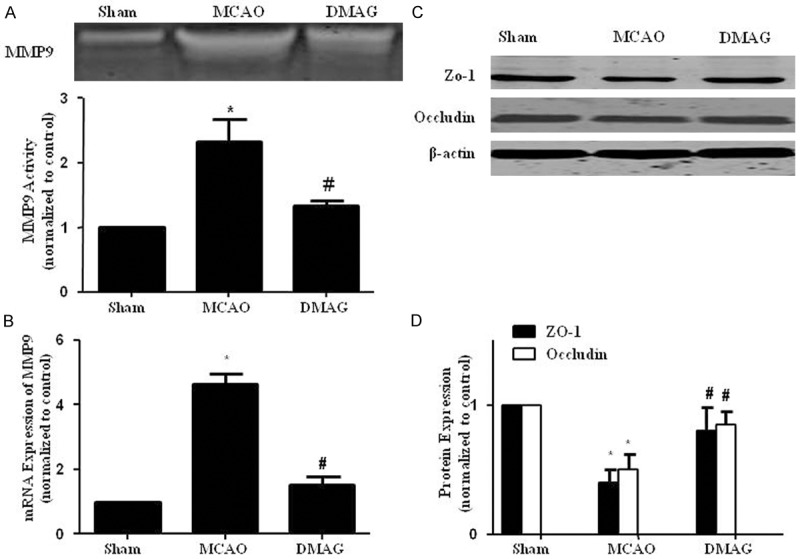
17-DMAG attenuated MCAO-induced MMP9 elevation and tight junction degradation. A. Zymography showed that 17-DMAG inhibitied the elevation of total MMP9 activities 24 hr after reperfusion. B. qPCR showed that 17-DMAG inhibited the increase of MMP9 mRNA expression. C, D. 17-DMAG restored Occludin and ZO-1 downregulation. *p < 0.05, compared with sham group, #p < 0.05, compared with MCAO group n = 8/group.
17-DMAG suppressed inflammatory responses in the ischemic brain
Local inflammation reponses contribute to BBB disruption induced by MCAO [23]. Microglia are stimulated within hours after ischemia onset, followed by upregulation of a variety of inflammatory factors [24]. We quantified the number of activated microglia (assessed by IBa1 staining) in the peri-infarct region within the ipsilateral ischemic cortex. Regions of interest were selected based on criteria published previously [25]. Figure 5A shows more intensely stained IBa1-positive cells in a brain section of a vehicle treated mice compared with 17-DMAG treated mice at 24 h after MCAO. Gene expression profiles of pro-inflammatory cytokines in the ischemic cortex were also examined. As expected, compared to sham group, MCAO significantly induced mRNA expression of IL-1β and MCP-1, which was remarkably attenuated by 17-DMAG (Figure 5C and 5D).
Figure 5.
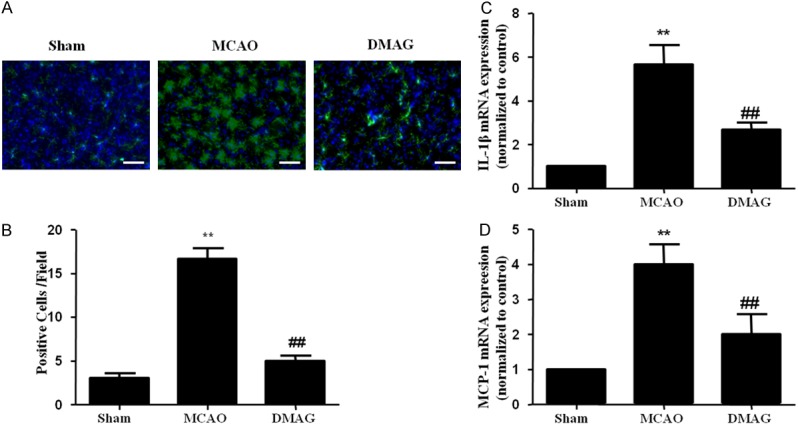
17-DMAG inhibited inflammatory responses in the ischemic cortex. A. Representative IBa1 staining within the ischemic cortex of mice following MCAO show less intense staining in 17-DMAG treated mice. B. Fewer IBa1 (n = 8/group) were detectable in the peri-infarct regions of 17-DMAG treated mice, compared with vehicle treated mice (n = 7/group). C, D. qPCR showed that 17-DMAG inhibited the increase of IL-1β and MCP-1 mRNA expression. (**p < 0.01, compared with sham group; ##p < 0.01, compared with MCAO group).
17-DMAG suppressed NF-κB activation in ischemic brain
NF-κB activation has been reported to up-regulate transcription of various inflammatory genes including MMP9 after cerebral ischemia [26]. Nuclear translocation of NF-κB p65 subunit is an indicator of NF-κB activation. Compared to sham group, elevated p65 in the nuclear fraction and reduced p65 levels in cytoplamic fraction were observed in ischemic cortex at 24 h after MCAO. 17-DMAG treatment inhibited MCAO-induced nuclear translocation of p65 in ischemic cortex (Figure 6A). Activation of NF-κB requires phosphorylation of IκBa by IKK and degradation by the proteasome, allowing NF-κB to transfer to the nucleus. Therefore, we also investigated the effects of HSP90 inhibitors on IKK expression. As expected, 17-DMAG inhibited IKK expreesion in vivo (Figure 6B).
Figure 6.
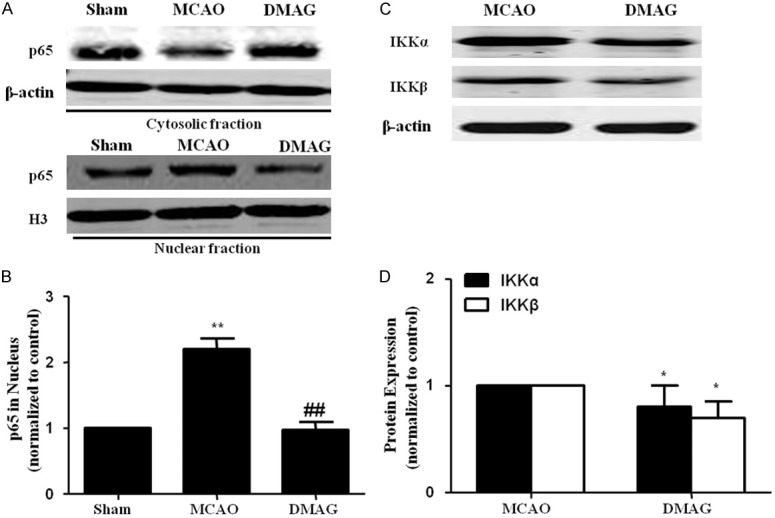
17-DMAG suppressed NF-κB activation in ischemic brain. A. Representative images of western-blot analysis of p65 in nuclear fractions extracted from the cortical tissue. B. Bar graph showed that p65 nuclear level in cortex were increased in vehicle treated group, which was restored by 17-DMAG. C, D. 17-DMAG inhibited IKKα and IKKβ expressions in cortex of MCAO mice.
HSP90 inhibition suppressed MMP9 secretion and gene expression in bEnd.3 cells via inhibition of NF-κB pathway
As NF-κB transcription factor is essentially involved in regulating MMP9 expression in bEnd.3 cells [27], we investigated whether 17-DMAG/17-AAG inhibits expression of MMP9 via inhibition of NF-κB transcriptional activity. 17-DMAG dose dependently inhibited MMP9 mRNA expression (Figure 7A). However, treatment of mouse bEnd.3 cells with OGD markedly increased the activity and mRNA levels MMP9, which were significantly inhibited by treatment of cells with 17-DMAG/17-AAG (Figure 7B and 7C).
Figure 7.

HSP90 inhibition inhibited NF-κB dependent MMP9 expression in endothelial cells after oxygen-glucose deprivation/reoxygenation. A. bEnd.3 cells were treated with vehicle or 17-DMAG (dose as indicated) for 30 min. RT-PCR for MMP9 was then examined. B, C. bEnd.3 cells were pretreated with vehicle, 17-DMAG or 17-AAG (10 nM) for 30 min before OGD. Zymography for MMP9 activity and RT-PCR for MMP9 mRNA expression were examined at 1 h reoxygenation after oxygen–glucose deprivation (OGD) for 6 h. D. The effects of 17-DMAG on the binding of p65 to MMP-9 gene promoters were detected by ChIP analysis. E. Representative immunoblot images showing HSP90 was successfully inhibited by siHSP90. F. Genetic inhibition of HSP90 decreased basal MMP9 mRNA expression.
Because the promoter region of MMP9 has a proximal NF-κB binding site that is functionally critical for the augmented production of MMP9. We then investigated the effect of 17-DMAG/17AAG on NF-κB binding to the MMP9 promoters in bEnd.3 cells by ChIP assays. In microglial cells, OGD treatment markedly increased the binding of NF-κB to MMP9 promoters, as shown in OGD-treated cells, after immunoprecipitation with primary antibody to p65, but not with control immunoglobulin G (IgG). Importantly, 17-DMAG/17AAG markedly attenuated the NF-κB binding to MMP9 promoters (Figure 7D). Taken together, these results indicate that HSP90 inhibitors inhibit OGD-induced expression of MMP9, at least in part, via inhibition of NF-κB transcriptional activity (Figure 7E).
To study the specific effect of HSP90 inhibition, we measured basal MMP9 production under genetic silencing of HSP90. We found that genetic inhibition of HSP90 decreased MMP9 mRNA expression (Figure 7F).
Discussion
It is well established that the heat shock protein 90 (HSP90) is over expressed and significantly correlated with MMP9 in many malignancies [14]. Studies on brain tissue and serum of patients suffering ischemic stroke demonstrated that MMP9 may play an important role in BBB disruption during ischemic stroke under clinical condition [28-30]. Previous study has demonstrated that HSP90 is over expressed in animal model of cerebral ischemia [12]. In this study, we observed HSP90 expressed higher and correlated with MMP9 significantly in serum of ischemic stroke patients (Figure 1A and 1B), which suggested that HSP90 may be a key molecule for increased MMP9 expression and subsequent BBB disruption in ischemic stroke.
Although the inhibitors of HSP90 are of therapeutic interest primarily in cancer [31], evidence is emerging for the potential beneficial role of HSP90 inhibitors in the treatment of other inflammatory diseases including ischemic stroke [19]. Indeed, one major mechanism undelying ischemic stroke-induced BBB disruption is enhanced inflammatory responses [23]. Particularly, pro-inflammatory cytokine induction of MMP9 expression and activity is a major cascade in destructive opening of BBB after stroke [32]. Importantly, inhibition of MMPs by the broad-spectrum MMP inhibitor minocyline has been shown to inhibit BBB disruption in ischemic stroke [33]. In agreement with previous studies, our data demonstrated that HSP90 inhibitor 17-DMAG significantly decreased infarc volume and inflammatory reponses in MCAO mice (Figures 3 and 5). Consistent with these observations, 17-DMAG reduced the expression and activity MMP9 both in vivo and in bEND cells (Figures 4 and 7). It is well known that tight junction proteins such as occludin and ZO-1 are essential components of BBB and known to be substrates of MMPs [22]. Our data show that occludin and ZO-1 were rapidly degraded following MCAO and 17-DMAG significantly inhibited degradation of these proteins (Figure 4C and 4D). Taken together, these results suggest that 17-DMAG effectively prevents BBB disruption, in part by inhibiting degradation of tight junction proteins via inhibition of inflammatory responses and subsequent MMP9 expression after ischemic stroke.
Among the different client proteins of HSP90 involved in inflammatory diseases, NF-κB is one of the most representative examples. NF-κB activation has been reported to up-regulate transcription of various inflammatory genes including MMP9 after cerebral ischemia [26]. Indeed, both genetic and pharmacological inhibition of NF-κB pathway has been reported to protect against ischemic stroke [34]. Since IKK exists in complexes with HSP90, disruption of these complexes by HSP90 inhibitors may block IKK function and subsequent NF-κB activation. In accordance with this hypothesis, we found that inhibition of HSP90 by 17-DMAG markedly attenuated the IKK expression and MMP9 secretion in MCAO mice. Likewise, we further demonstrated that 17-DMAG potently inhibited the binding of P65 to the promoter regions of MMP9 in bEnd.3 cells.
The protective effects of HSP90 inhibitors in inflammatory diseases could be due to up-regulation of antiinflammatory HSP expression (especially HSP70) [35]. Moreover, mice overexpressing HSP70 showed decreased number of activated macrophages and inhibition of NF-κB in ischemic stroke [36]. Therefore, further studies are needed to clarify whether the anti- inflammatory effects observed with HSP90 inhibitors is due to HSP70 up-regulation, inhibition of HSP90 client proteins, or both.
In conclusion, HSP90α was overexpressed and correlated with MMP9 level in patients with ischemic stroke. Inhibition of HSP90 by 17-DMAG markedly attenuated BBB disruption following cerebral ischemia in mice through inhibiting degradation of tight junction proteins via inhibition of inflammatory responses and subsequent MMP9 expression. Currently, more than 10 HSP90 inhibitors in various oncological indications are undergoing clinical trials [37]. Therefore, our findings may have direct translational implications for ischemic stroke, especially hemorrhagic transformation after stroke.
Acknowledgements
This study was supported by funds from Shanghai Municipal Commission of Health and Family Planning (201540301), and Natural Science Foundation of Shanghai (15ZR1414600).
Disclosure of conflict of interest
None.
References
- 1.Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- 2.Del Zoppo GJ, Saver JL, Jauch EC, Adams HP Jr American Heart Association Stroke Council. Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke. 2009;40:2945–2948. doi: 10.1161/STROKEAHA.109.192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Lee SR, Arai K, Lee SR, Tsuji K, Rebeck GW, Lo EH. Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- 4.Date I, Takagi N, Takagi K, Tanonaka K, Funakoshi H, Matsumoto K, Nakamura T, Takeo S. Hepatocyte growth factor attenuates cerebral ischemia-induced increase in permeability of the blood-brain barrier and decreases in expression of tight junctional proteins in cerebral vessels. Neurosci Lett. 2006;407:141–145. doi: 10.1016/j.neulet.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 5.Lee CZ, Xue Z, Zhu Y, Yang GY, Young WL. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke. 2007;38:2563–2568. doi: 10.1161/STROKEAHA.106.481515. [DOI] [PubMed] [Google Scholar]
- 6.McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucivero V, Prontera M, Mezzapesa DM, Petruzzellis M, Sancilio M, Tinelli A, Di Noia D, Ruggieri M, Federico F. Different roles of matrix metalloproteinases-2 and -9 after human ischaemic stroke. Neurol Sci. 2007;28:165–170. doi: 10.1007/s10072-007-0814-0. [DOI] [PubMed] [Google Scholar]
- 8.Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. 2001;86:324–333. [PubMed] [Google Scholar]
- 10.Harada K, Suzuki Y, Yamakawa K, Kawakami J, Umemura K. Combination of reactive oxygen species and tissue-type plasminogen activator enhances the induction of gelatinase B in brain endothelial cells. Int J Neurosci. 2012;122:53–59. doi: 10.3109/00207454.2011.623808. [DOI] [PubMed] [Google Scholar]
- 11.Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- 12.Kawagoe J, Abe K, Aoki M, Kogure K. Induction of HSP90 alpha heat shock mRNA after transient global ischemia in gerbil hippocampus. Brain Res. 1993;621:121–125. doi: 10.1016/0006-8993(93)90306-8. [DOI] [PubMed] [Google Scholar]
- 13.Xu XH, Hua YN, Zhang HL, Wu JC, Miao YZ, Han R, Gu ZL, Qin ZH. Greater stress protein expression enhanced by combined prostaglandin A1 and lithium in a rat model of focal ischemia. Acta Pharmacol Sin. 2007;28:1097–1104. doi: 10.1111/j.1745-7254.2007.00624.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Cui S, Zhang X, Wu Y, Tang H. High expression of heat shock protein 90 is associated with tumor aggressiveness and poor prognosis in patients with advanced gastric cancer. PLoS One. 2013;8:e62876. doi: 10.1371/journal.pone.0062876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musial K, Zwolinska D. Matrix metalloproteinases (MMP-2,9) and their tissue inhibitors (TIMP-1,2) as novel markers of stress response and atherogenesis in children with chronic kidney disease (CKD) on conservative treatment. Cell Stress Chaperones. 2011;16:97–103. doi: 10.1007/s12192-010-0214-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon HM, Kim Y, Yang SI, Kim YJ, Lee SH, Yoon BW. Geldanamycin protects rat brain through overexpression of HSP70 and reducing brain edema after cerebral focal ischemia. Neurol Res. 2008;30:740–745. doi: 10.1179/174313208X289615. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, Wang G, Chen Z. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321–1333. doi: 10.4161/auto.25132. [DOI] [PubMed] [Google Scholar]
- 18.Bradley E, Zhao X, Wang R, Brann D, Bieberich E, Wang G. Low dose Hsp90 inhibitor 17AAG protects neural progenitor cells from ischemia induced death. J Cell Commun Signal. 2014;8:353–62. doi: 10.1007/s12079-014-0247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi J, Han X, Liu HT, Chen T, Zhang JL, Yang P, Bo SH, Lu XT, Zhang J. 17-Dimethylaminoethylamino-17-demethoxygeldanamycin Attenuates Inflammatory Responses in Experimental Stroke. Biol Pharm Bull. 2014;37:1713–1718. doi: 10.1248/bpb.b14-00208. [DOI] [PubMed] [Google Scholar]
- 20.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X, Zhang Z, Wang Y, Yang GY. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflammation. 2014;11:177. doi: 10.1186/s12974-014-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke. 2013;44:190–197. doi: 10.1161/STROKEAHA.112.670299. [DOI] [PubMed] [Google Scholar]
- 24.Morioka T, Kalehua AN, Streit WJ. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J Comp Neurol. 1993;327:123–132. doi: 10.1002/cne.903270110. [DOI] [PubMed] [Google Scholar]
- 25.Wang GJ, Deng HY, Maier CM, Sun GH, Yenari MA. Mild hypothermia reduces ICAM-1 expression, neutrophil infiltration and microglia/monocyte accumulation following experimental stroke. Neuroscience. 2002;114:1081–1090. doi: 10.1016/s0306-4522(02)00350-0. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu H, Saito S, Higashiyama Y, Nishijima F, Niwa T. CREB, NF-kappaB, and NADPH oxidase coordinately upregulate indoxyl sulfateinduced angiotensinogen expression in proximal tubular cells. Am J Physiol Cell Physiol. 2013;304:C685–692. doi: 10.1152/ajpcell.00236.2012. [DOI] [PubMed] [Google Scholar]
- 27.Lee EJ, Kim HS. Inhibitory mechanism of MMP-9 gene expression by ethyl pyruvate in lipopolysaccharide-stimulated BV2 microglial cells. Neurosci Lett. 2011;493:38–43. doi: 10.1016/j.neulet.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 28.Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. doi: 10.1016/s0304-3940(97)00859-8. [DOI] [PubMed] [Google Scholar]
- 29.Switzer JA, Hess DC, Ergul A, Waller JL, Machado LS, Portik-Dobos V, Pettigrew LC, Clark WM, Fagan SC. Matrix metalloproteinase-9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke. 2011;42:2633–2635. doi: 10.1161/STROKEAHA.111.618215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- 31.Soti C, Nagy E, Giricz Z, Vigh L, Csermely P, Ferdinandy P. Heat shock proteins as emerging therapeutic targets. Br J Pharmacol. 2005;146:769–780. doi: 10.1038/sj.bjp.0706396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to bloodbrain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 34.Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Kohrmann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- 35.Madrigal-Matute J, Lopez-Franco O, Blanco-Colio LM, Munoz-Garcia B, Ramos-Mozo P, Ortega L, Egido J, Martin-Ventura JL. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc Res. 2010;86:330–337. doi: 10.1093/cvr/cvq046. [DOI] [PubMed] [Google Scholar]
- 36.Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 2008;28:53–63. doi: 10.1038/sj.jcbfm.9600502. [DOI] [PubMed] [Google Scholar]
- 37.Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem. 2009;9:1479–1492. doi: 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]