

Abstract
Oncogenic NRAS mutations are prevalent in human myeloid leukemia, especially in chronic myelomonocytic leukemia (CMML). NrasG12D mutation at its endogenous locus in murine hematopoietic stem cells (HSCs) leads to CMML and acute T-cell lymphoblastic lymphoma/leukemia in a dose-dependent manner. Hyper-activated MAPK and STAT5 pathways by oncogenic Nras contribute to the leukemogenesis in vivo. However, it is unclear whether these conclusions remain true in a more human relevant model. Here, we evaluated the effects of NRASG12D on human hematopoiesis and leukemogenesis in vitro and in vivo by ectopically expressing NRASG12D in human cord blood stem/progenitor cells (hSPCs). NRASG12D expressing hSPCs preferentially differentiated into myelomonocytic lineage cells, demonstrated by forming more colony forming unit-macrophages than control hSPCs in cultures. Transplantation of NRASG12D expressing hSPCs initiated myeloproliferative neoplasm in immune deficiency mice. All the recipient mice died of myeloid tumor burdens in spleens and bone marrows and none developed lymphoid leukemia. Phospho-flow analysis of CD34+ CD38- hSPCs confirmed that NRASG12D hyper-activated MAPK, AKT and STAT5 pathways. Our study provides the strong evidence that NRASG12D mutation mainly targets monocytic lineage cells and leads to myelomonocytic proliferation in vivo in a highly human relevant context.
Keywords: NRAS, hematopoietic stem/progenitor cells, immune deficiency mice, myelomonocytic proliferation
Introduction
NRAS, HRAS, and KRAS belong to the RAS family, whose mutations occur in all human cancer types [1]. Interestingly, NRAS mutations are prevalent in myeloid leukemia, especially in over 50% patients with myeloproliferative variant of chronic myelomonocytic leukemia (MP-CMML) [2]. Oncogenic RAS mutations lead to constitutive binding of GTP and thus hyperactivate the downstream effectors [3]. Knocking down of NRAS expression level in human cord blood CD34+ cells results in decreased colony forming unit-macrophage (CFU-M) colonies in a colony forming assay [4], while overexpression of oncogenic NRAS mutants enhances myelopoiesis and blocked erythropoiesis [5,6]. However, it remains elusive whether NRASG12D in human cord blood stem/progenitors (hSPCs) could initiate myeloid diseases in vivo.
Nras mutation in murine hematopoietic stem/progenitor cells (HSPCs) produces variable disease phenotypes, including acute myeloid leukemia (AML), CMML and acute T cell lymphoblastic leukemia/lymphoma (TALL), depending on its expression levels [7-10]. Using an advanced mouse model, we have systematically investigated the effects and mechanisms of endogenous NrasG12D mutation on hematopoiesis and leukemogenesis [8,10,11]. We first observed that a single copy of NrasG12D mutation confers growth advantage on hematopoietic stem cells (HSCs) and thus competitively depletes wild type HSCs counterparts. We also revealed that lower dose of NrasG12D signaling predominantly leads to CMML disease, while higher dose of NrasG12D efficiently initiates TALL in BMT models [10]. Our mechanistic studies provide the strong evidence that the magnitude of MAPK signaling balances the stemness of HSCs in mice. However, it is unclear whether these phenotypes and signaling mechanisms caused by NrasG12D mutation in mouse blood system are conserved in human counterpart.
It is unrealistic to obtain endogenous NRASG12D mutation in human HSCs by direct gene editing approach, due to the technical bottlenecks to induce human HSCs from pluripotent stem cells and the failure to maintain and propagate human HSCs in long-term cultures. Recently, several advanced immune deficiency mouse strains have been used to stably engraft human blood cells, including NOD/SCID and NOD/SCID/ IL2Rg-/- mice [12,13]. These strains provide a great opportunity to test oncogene functions in a more human relevant model.
Here, we report that ectopoic expression of oncogenic NRASG12D in human cord blood CD34+ stem/progenitor cells (hSPCs) preferentially enhances CFU-M formation and blocks erythroid colony formation in vitro. These hSPCs successfully induce a lethal myelomonocytic proliferation in immune deficiency mice in vivo. Single cell phospho-flow assay in CD34+ CD38- hSPCs indicates that NRASG12D constitutively hyper-activates MAPK, AKT and STAT5 signaling pathways, which are conserved between human and mouse species.
Materials and methods
Cord blood acquisition and CD34+ HSPCs enrichment
Healthy newborns’ fresh umbilical cord blood was obtained from Guangdong Cord Blood Bank. Mononuclear cells were then isolated using Ficoll-Hypaque (AXIS-SHIELD, 1114547). After lysis of the residual red cells cells were incubated with the human CD34 MicroBead Kit (Miltenyi Biotec, 130-046-702) and enriched for CD34+ cells using magnetic beads separator (Miltenyi Biotec, 130-042-401). The purity of CD34+ cells was assessed using flow cytometry with APC-conjugated anti- human CD34 antibody (eBioscience, 17-0349-42).
Generation of NRAS mutation and preparation of lentivirus
Wildtype NRAS cDNA was amplified from total RNA extracted from U937 cell line (ATCC, CRL-3253TM) using specific forward and reverse primers: forward-5’ actctgcagaccatgactgagtacaaactg 3’; reverse-5’ actgaattctacatc accacacatggc 3’. Subsequently the nucleotide G at codon 12 was mutated into A using the synthetic long forward primer-5’ atgactgagtacaaactggtggtggttggagcagatggtgttgggaaaagcgca 3’. Then the NRASG12D cDNA was cloned into the MND-PGK-green fluorescent protein (GFP) lenti-viral vector [14], which facilitates ectopic expression of genes in human white blood cells.
Plasmids transfection of 293T cells (ATCC CRL-3216) was performed using calcium phosphate transfection method as described before [15]. Three packaging vectors (pLP1, pLP2, and pLP/VSVG) were purchased from Biovector Science Lab (01128569). 5.5×106 293T cells were plated into each 10 cm plate in DMEM (Hyclone) with 10% FBS (Natocor) one day before transfection. 10 ug of NRASG12D or GFP lenti-vectors and 15 ug pLP1, pLP2, and pLP/VSVG packaging vector plasmids (1:1:1) were mixed with calcium phosphate and added to each 293T dish. Twelve hours (hrs) after transfection, the cells were rinsed with PBS and changed with fresh medium. The NRASG12D and GFP virus supernatants were collected 48 hrs after transfection, and concentrated using ultracentrifugation at 50000 g for 2 h at 4°C. The virus titers were determined effectively using U937 cell line (ATCC® CRL-1593.2™).
Cell culture and viral transduction
Human cord blood CD34+ stem/progenitor cells (1×106/ml) were pre-stimulated for ~16 hrs in StemSpan (StemCell Technologies) culture medium containing cytokines (100 ng/ml of SCF and Flt3l, 20 ng/ml of IL-6). Virus transduction was performed as before [16]. Briefly, the stimulated cells were transferred into StemSpan (StemCell Technologies, 09600) culture medium with concentrated-viruses. The culture dish was pre-coated with 10 ug/cm2 human recombinant fibronectin (TaKaRa, T100A/B) and incubated with concentrated virus before transduction. The cells were infected for 24 hrs at 37°C, and then the medium was replaced with fresh StemSpan medium. The transduction efficiency was measured using flow cytometry, and the NRASG12D expression level was determined using qRT-PCR.
CFU assay
Two days after virus transduction, the virus-transduced cells were harvested, washed with PBS, and suspended in DAPI solution. GFP+ cells were sorted using a FACS ARIA II sorter (BD Biosciences). One thousand sorted GFP+ cells were plated into each 35 mm culture dish (StemCell Technologies, 27150) containing the methylcellulose H4434 medium (StemCell Technologies). After 13 days in culture, all types of colonies were counted, including CFU-G, CFU-M, CFU-GM, BFU-E and CFU-mix. Representative images were taken using a microscope (Zeiss, Axio Vert. A1).
Transplantation
Eight to ten-week-old NOD/SCID/IL2Rg-/- mice [13] were sublethally irradiated (1.5 Gy) using a Biological Research Irradiator (RS 2000, Rad Source). Four hours after irradiation, the mice were anesthetized with avertin (400 mg/kg) for transplantation process. 3×105 transduced NRASG12D CD34+ cells (~50% were GFP+) in 20-30 μl of PBS were injected intramedullary into the irradiated mice. The mice were treated with water containing neomycin for two weeks to prevent infection. They were housed in the SPF grade animal care facility at the Guangzhou Institutes of Biomedicine and health (GIBH). All experiments were conducted with the ethical approval of the Animal Ethics Committee of GIBH.
Engraftment analysis
Peripheral blood samples were collected and processed as before [15]. At a moribund stage, recipient mice were sacrificed for flow cytometric analysis and pathological evaluation. Human white white blood cells were detected using PE-cy7-conjugated antibody against human CD45 (eBioscience, 25-0459-42). Human myeloid cells were detected by PE-conjugated antibody against human CD33 (eBioscience, 12-0339-42), B-lymphoid cells were detected by APC-conjugated antibody against human CD19 (eBioscience, 17-0199-42). To detect T cells, primary biotin-conjugated antibodies against human CD3/CD4/CD8 (eBioscience, 12-0037-80/13-0048-80/13-0086-80) were used, followed by secondary Percp-cy5.5-conjugated straptavidin (eBioscience, 45-4317-82). All the samples were analyzed using a Fortessa instrument (BD Biosciences). FACS data were analyzed using Flowjo vx.0.7 software (FlowJo).
Histological analysis of spleen and liver
Spleens and livers were weighed using a scale. Half of the tissues were fixed in 4% paraformaldehyde, then processed for histologic hematoxylin and eosin-staining (Pathology Lab, GIBH). The remaining tissues were grinded into single cell suspension for subsequent FACS analysis.
Phos-flow assay
Forty-eight hours after the initiation of viral transduction, NRASG12D and GFP control transduced CD34+ cells were harvested and washed in PBS. Phos-flow analysis was performed as described before [17]. Briefly, virus transduced cells were immediately fixed with paraformaldehyde at a final concentration of 2% (Electron Microscopy Sciences) for 10 minutes at 37°C. hSPCs were detected with PE-conjugated antibody against human CD34 and PE-cy7-conjugated antibody against human CD38. Phospho-ERK1/2 was detected by a primary antibody against p-ERK (Thr202/Tyr204; Cell Signaling Technology), followed by APC-conjugated donkey anti-rabbit F(ab’)2 fragment (JacksonImmuno Research). p-STAT5 signaling pathway was detected by Alexa 647 conjugated primary antibody against phospho-STAT5 (Tyr 694; BD Biosciences). p-AKT was detected by a primary antibody against p-AKT (Ser473; Cell Signaling Technology), followed by APC-conjugated donkey anti-rabbit F(ab’)2 fragment. All the samples were analyzed using a Fortessa (BD Biosciences) instrument. All the data were analyzed using the Flowjo software.
Results
Ectopic expression of NRASG12D in human cord blood CD34+ hSPCs via lentiviral transduction
In mice, expression of endogenous oncogenic Nras (NrasG12D/+) in hematopoietic cells leads to expansion of myeloid progenitors, and CMML-like phenotypes [8]. To study whether the oncogenic NRASG12D has the same effects on hSPCs, we overexpressed NRASG12D in human cord blood CD34+ hSPCs using a lentiviral transduction approach (Figure 1A). We inserted NRASG12D into a lentiviral vector, which is optimized for overexpressing genes in human CD34+ cord blood cells [14] (Figure 1B). Further sequencing of the viral construct confirmed the mutation of nucleotide G to A at the codon 12 of NRAS cDNA (Figure 1C). To ensure the viral titers for experimental reproducibility, the MOI of each batch of packaged viruses was measured using U937 cell line. To control the viability of human cord blood CD34+ cells, only fresh cord blood samples were used. Two days after the initiation of NRASG12D and GFP viral transduction with an MOI of 20, flow cytometric analysis showed that the transduction efficiency was nearly 50% (Figure 1D), which is an ideal efficiency to achieve ~50% GFP+ cells and ~50% GFP- cells as an internal control. To confirm the overexpression of NRASG12D, total RNA was extracted from two samples transduced with NRASG12D and GFP control viruses. Quantitative RT-PCR assay using primers flanking the NRASG12D cDNA verified the efficient expression of NRASG12D in the cord blood CD34+ progenitors (Figure 1E). Thus, we successfully overexpressed NRASG12D in hSPCs using a lentiviral system.
Figure 1.
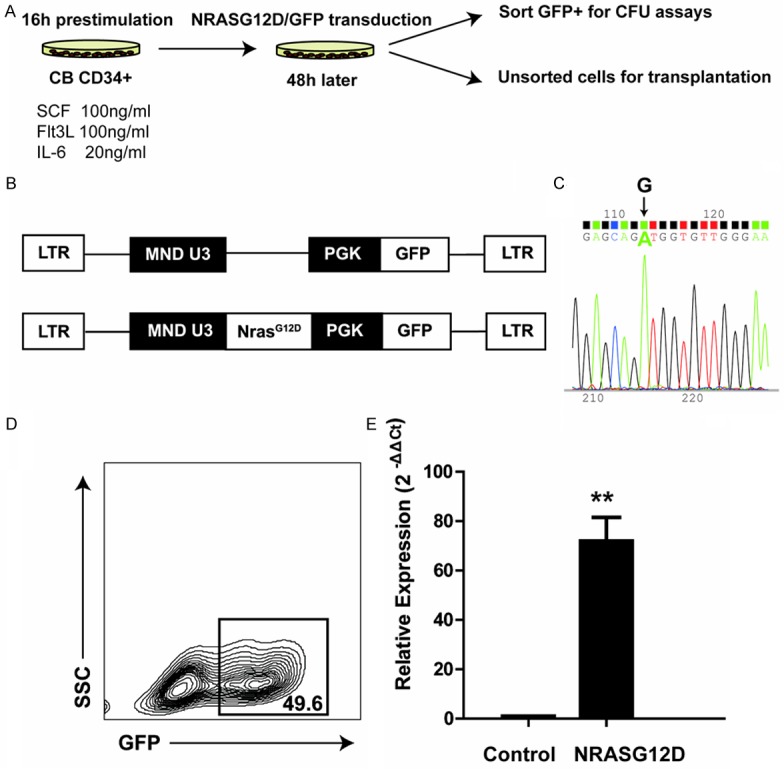
Ectopic expression of NRASG12D in CD34+ human cord blood SPCs. A. Schematic diagram of experimental strategy. We overexpressed NRASG12D in the cord blood CD34+ progenitors using lentiviral transduction, and evaluated its effects in vitro (CFU assay) and in vivo (transplantation). B. Schematic picture of lentiviral constructs. The promoter region is shown in black. C. Sequencing of NRASG12D mutation using forward primer. Arrow indicates the mutation of nucleotide G to A at the codon 12. D. Transduction efficiency of cord blood CD34+ cells after 48 hours incubation with viruses. E. Confirmation of ectopic expression f NrasG12D at the mRNA level. Total RNA was extracted from CD34+ hSPCs transduced with NRASG12D or GFP control viruses. Data are shown as mean + SD (**, P<0.001). Triplicates from one representative experiment are shown.
NRASG12D overexpression increases CFU-M colony formation and blocks BFU-E colony formation
We first investigated the effect of NRASG12D mutation on hSPCs using a colony-forming assay. One thousand sorted GFP+ hSPCs from either NRASG12D or GFP control virus transduced cells were plated in methylcellulose H4434 medium. After two weeks incubation, all types of colonies were counted under microscope (Figure 2A). As expected, CD34+ blood cells transduced with GFP control viruses generated all types of normal colonies, including burst forming units-erythroid (BFU-Es) and colony forming unit-mix (CFU-Mix) (Figure 2C). Interestingly, cells transduced with NRASG12D viruses formed significantly more colony forming unit-macrophage (CFU-M) colonies than cells transduced with GFP control (P<0.05). Moreover, the size of the CFU-Ms was much larger (Figure 2B), indicating that the CD34+ hSPCs expressing NRASG12D tend to differentiate into myelomonocytic lineage cells. Meanwhile, the BFU-E colonies disappeared in the NRASG12D culture, demonstrating that enforced expression of NRASG12D in CD34+ hSPCs inhibited erythropoiesis.
Figure 2.

Overexpression of NRASG12D in CD34+ hSPCs enhances CFU-M formation and inhibits BFU-E formation. CD34+ hSPCs were infected with NRASG12D and GFP viruses. Two days after viral transduction, 1000 GFP+ cells were sorted and plated in semisolid medium. (A) Colonies were numerated after 13 days in culture (n=3). *P<0.05, **P<0.01 and ***P<0.001. (B, C) Representative images of different CFU subtypes from cells transduced with NRASG12D (B) or GFP control viruses (C).
NRASG12D expression enhances myelopoiesis and initiates a lethal myelomonocytic proliferation in immune deficiency mice
To further investigate the physiological effects of oncogenic NRASG12D in vivo, we performed a bone marrow transplantation (BMT) assay. We transplanted 3×105 hSPCs either transduced with NRASG12D viruses or GFP controls into sublethally irradiated immunodeficient NSI mice via intrafemoral injection [13]. We bled the mice from the retro-obital vein and analyzed human hematopoietic lineages in peripheral blood six weeks post transplantation using flow cytometric analysis. As expected, we indeed observed that hSPCs expressing NRASG12D generated higher percentage of myeloid lineage cells (CD33+) in peripheral blood compared with control (P<0.05) (Figure 3A, 3C), which was consistent with our results from in vitro colony assay. Since 7 weeks after transplantation, recipient mice with hSPCs expressing NRASG12D began to die (Figure 4C). In order to find out the cause of death, we sacrificed some moribund mice and performed autopsy, flow cytometric analysis and hematoxylin and eosin (H&E) staining of tissue sections. We found that the spleens of the moribund mice were enlarged (Figure 4A). On average, the spleen weights from moribund mice were 2-fold heavier than those from control mice (P<0.01) (Figure 4A). H&E staining of spleen and liver sections indicated that both splenic and the hepatic parenchyma were infiltrated with myelomonocytic cells (Figure 4B). Further flow analysis confirmed the overburden of human myeloid cells (CD33+) in bone marrows and spleens of the moribund mice (Figure 3B, 3D). Taken together, oncogenic NRASG12D expression in human CD34+ blood cells promotes a lethal myelomonocytic proliferation in vivo.
Figure 3.
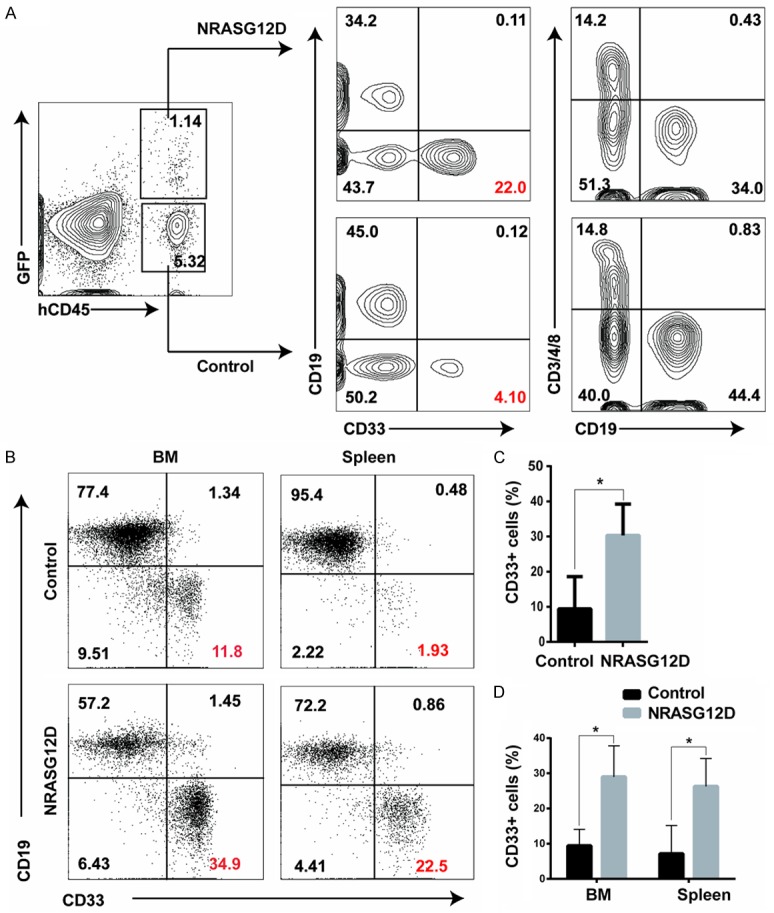
NRASG12D-expressing CD34+ hSPCs show bias of myelopoiesis in vivo. Sublethally irradiated (1.5 Gy) immunodeficient mice were transplanted with 3×105 total CD34+ cord blood cells infected with NRASG12D or GFP control viruses. The transduction efficiencies were adjusted to ~50%. Samples were collected from NRASG12D moribund recipients. Debris and dead cells (DAPI positive) were excluded from analysis. A. Engraftment analysis of hematopoietic lineages in peripheral blood of recipients six weeks after transplantation. B. Flow cytometric analysis of human white blood cells in bone marrow and spleens from a representative moribund mouse. Markers used: B-lymphoid cells, CD19+; myeloid cells, CD33+; and T-lymphoid cells, CD3/4/8+. C. Statistical analysis of GFP+ and GFP- human myeloid (CD33+) cells in peripheral blood of transplanted recipeints. D. Statistical analysis of GFP+ and GFP- human myeloid (CD33+) cells in BM and spleens of transplanted NSI mice (n=3, *, P<0.05).
Figure 4.
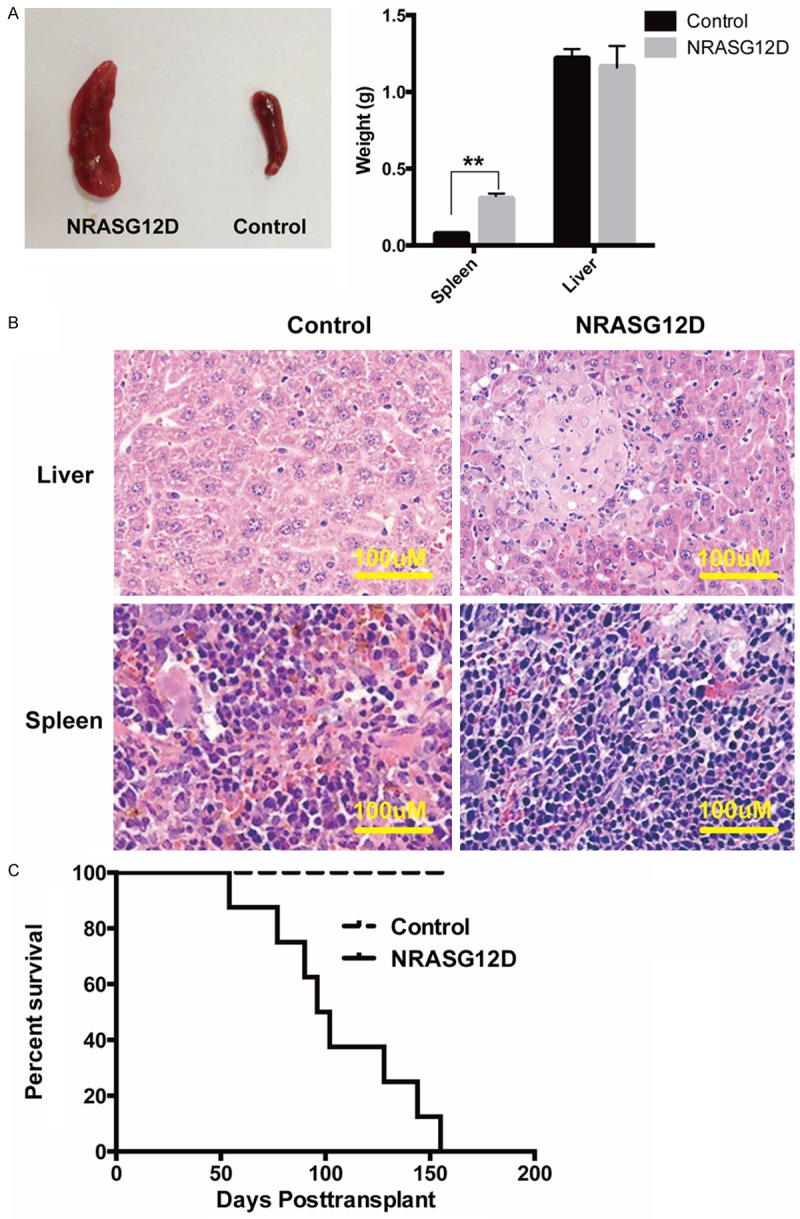
NRASG12D initiates myelomonocytic proliferation in NSI mice. Sublethally irradiated (1.5 Gy) immunodeficient mice were transplanted with 3×105 total CD34+ cord blood cells infected with NRASG12D or GFP control viruses. The transduction efficiencies were adjusted to ~50%. Samples were collected from NRASG12D moribund recipients. A. Splenomegaly from a representative recipient transplanted with NRASG12D or GFP control transduced CD34+ cord blood cells (left). Results are presented as averages of spleen weights or liver weights + SD (Right). (n=3, **P<0.01) B. Representative histologic hematoxylin and eosin stained sections from spleen and liver. C. Kaplan-Meier comparative survival curves of transplanted mice. Cumulative survival was plotted against days after transplantation. NRASG12D group, n=8; control group, n=4.
NRASG12D hyper-activates AKT, MAPK and STAT5 pathways in CD34+ hSPCs
To investigate the signaling mechanisms underlying NRASG12D overexpression induced aberrant myelopoiesis, we performed single cell phospho-flow analysis of the canonical MAPK, AKT and STAT5 signaling pathways in hSPCs, which are the effector pathways of Ras signaling in murine cells [3]. We overexpressed NRASG12D in CD34+ hSPCs, and detected these signaling pathways two days after viral transduction. To prevent dephosphorylation, we immediately fixed the cells without starvation. We specifically analyzed the RAS signaling pathways in CD34+ CD38- hSPCs expressing NRASG12D (Figure 5A). Compared with GFP control, the levels of phosphorylated ERK1/2, AKT and STAT5 were much higher in NRASG12D expressing hSPCs (Figure 5B). Statistical analysis showed that the median intensities of p-AKT (P<0.01), p-ERK1/2 (P<0.05) and p-STAT5 (P<0.01) were significantly higher in NRASG12D transduced hSPCs than those of GFP control (Figure 5C), indicating that the MAPK, AKT and STAT5 pathways were hyper-activated by NRASG12D overexpression.
Figure 5.
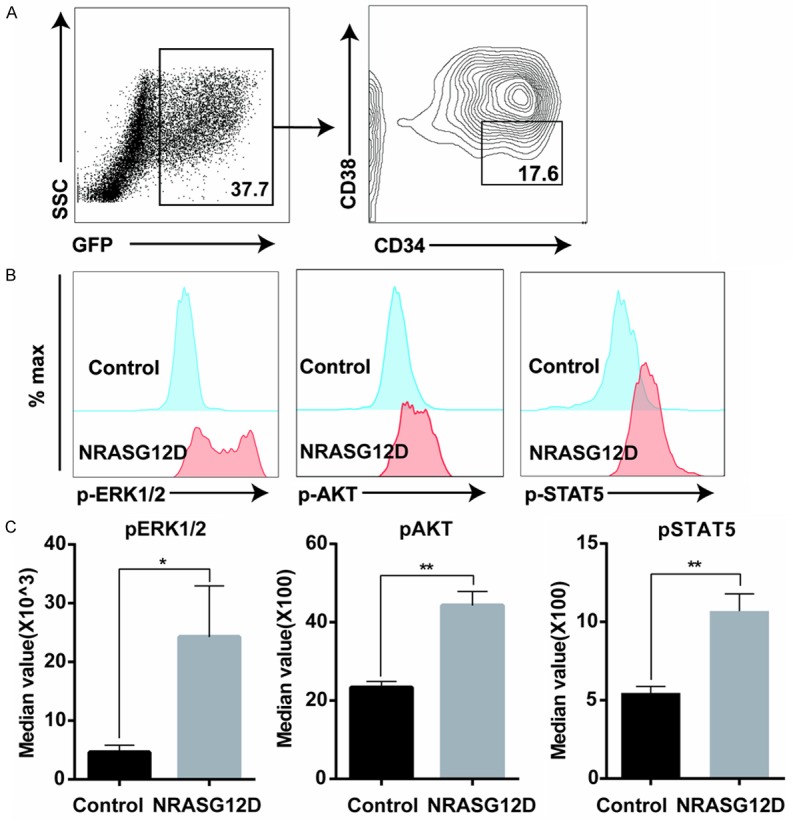
Hyper-activation of AKT, MAPK and STAT5 pathways in NRASG12D-overexpressing CD34+ hSPCs. CD34+ enriched cord blood cells were transduced with GFP and NRASG12D viruses. Two days after viral transduction, cells were immediately fixed, permeablized and analyzed using flow cytometry. A. NRASG12D expressing hSPCs were gated as GFP+ CD34+ CD38- cells. B. Representative plots of phosphorylated AKT, ERK1/2 and STAT5. Levels of phosphorylated AKT, ERK1/2 and STAT5 were shown from cells expressing GFP control and NRASG12D. C. Statistical analysis of the levels of phosphorylated AKT, ERK1/2 and STAT5. Median intensities of p-AKT, p-ERK1/2 and p-STAT5 were analyzed from individual samples. For each experiment, CD34+ hSPCs samples were isolated from the same donor to reduce variability. Data from one representative experiment with three replicates were shown as mean + SD. *P<0.05, and **P<0.01.
Discussion
Using an advanced murine model, we previously investigated the physiological phenotypes and leukemogenic mechanisms induced by endogenous oncogenic NrasG12D mutation. We reported that endogenous NrasG12D mutation initiates CMML and TALL in a dose-dependent manner [8,10,11]. However, it remains elusive whether these observations are conserved in human blood system. Currently, technical issues make it impossible to introduce NRASG12D mutation at the endogenous locus in human HSCs, as these cells failed to be derived from human pluripotent stem cells. Isolation of primary hematopoietic progenitors from patients with NRASG12D mutations is an alternative cell source to study the effects of oncogenic NRASG12D. However, patients were usually diagnosed at advanced disease stages when multiple abnormalities already accumulated (such as secondary mutations, epigenetic modifications, as well as disruptions of bone marrow niche). Therefore, the heterogeneity and complexity of blood samples from patients with NRASG12D mutations make it difficult to address the specific effects of NRASG12D on hematopoiesis and leukemogenesis in a more human relevant context. Ectopic expression of oncogenic Nras in murine HSPCs successfully produced CMML- and AML-like diseases in a BMT model [7]. Thus we turned to choose ectopic expression of NRASG12D in cord blood CD34+ hSPCs, and evaluated the phenotypes in in vitro CFU and in vivo hematopoietic reconstitution assays using immunodeficient mice. We indeed observed that overexpressing NRASG12D in CD34+ hSPCs promoted myelopoiesis and blocked erythropoiesis in vitro. When transplanted into immune deficiency mice, NRASG12D expressing cells predominantly produced myeloid lineage cells, and caused the death of all recipients with myeloid diseases in bone marrows and spleens. Signaling analysis indicated that NRASG12D expression hyper-activated MAPK, AKT and STAT5 pathways in CD34+ CD38- hSPCs, which was consistent with the observations in murine models [8,10]. Thus, targeting these conserved pathways is a rational approach for intervention therapy of leukemia patients with NRASG12D mutations.
NRASG12D specifically promoted CFU-M formation in cultures, demonstrated by higher counts and enlarged sizes of CFU-M colonies. Our result is consistent with the previous report that ectopic expression of oncogenic NrasG12D in mouse bone marrow preferentially led to monocytic leukemia in a BMT model [7]. The potential artificial effects due to the variable virus titers and viral integration sites were excluded by multiple independent experiments using several cord blood samples from different donors. Our finding that NRASG12D caused pathologic effects mainly in monocytic lineage cells might explain why NRAS mutations are prevalent in CMML patients [18]. We also observed that overexpressing NRASG12D disturbed formation of BFU-Es, which was consistent with earlier studies using human CD34+ blood cells [6]. However, our previous studies using mouse models demonstrated that endogenous NrasG12D mutation alone fails to inhibit erythropoiesis [8,10,11]. This is likely to be caused by drastic differences of expression levels of oncogenic Nras. This possibility is supported by a previous support that overexpression of oncogenic Nras in mouse fetal liver erythroid progenitors blocks their terminal differentiation [19]. Nevertheless, CMML-like mice do develop monocytosis and anemia simultaneously, which highly correlate to CMML patients.
We transplanted NRASG12D viruses transduced CD34+ cells into sublethally irradiated immune deficiency mice [13]. All the mice died of myelomonocytic proliferation with a short latency. In our previous research in murine BMT models, endogenous oncogenic NrasG12D initiates CMML disease with a very long latency, while biallelic mutations of endogenous NrasG12D initiated T cell leukemia with a short latency [10]. In our transplantation model, we never observed T cell leukemia, which was consistent with the clinical observation that NRAS mutations are rare in human T cell leukemia [20]. It is possible that the murine thymic niche fails to mimic human counterpart and thus suppressed the development of lymphoid malignancy. According to our previous reports, the doses of oncogenic Nras mutation play an important role in developing different types of leukemia [10,21]. Since human NRAS mutations occur at its endogenous locus in human patients, ectopic expression using exogenous promoters might not mimic the true effects of endogenous mutations of NRAS. Once human HSCs could be successfully derived from PSCs, or large-scale expansion of human HSCs is made possible, human HSCs with endogenous NRASG12D mutation will be available by gene editing. Transplanting these HSCs into immunodeficient mice will better mimic the physiological effects of oncogenic NRASG12D.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. 31471117, 81470281), the Science and Technology Planning Project of Guangdong Province, China (2014B030301058), and the Major Scientific and Technological Project of Guangdong Province (2014B020225005), the Natural Science Foundation of Guangdong (No.2014A030313138) to ZXZ, and by R01 grants R01CA152108 and R01HL113066, and a Scholar Award from the Leukemia & Lymphoma Society to J.Z.
Disclosure of conflict of interest
None.
References
- 1.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 2.Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000;96:1655–1669. [PubMed] [Google Scholar]
- 3.Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 4.Skorski T, Szczylik C, Ratajczak MZ, Malaguarnera L, Gewirtz AM, Calabretta B. Growth factor-dependent inhibition of normal hematopoiesis by N-ras antisense oligodeoxynucleotides. J Exp Med. 1992;175:743–750. doi: 10.1084/jem.175.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen SW, Dolnikov A, Passioura T, Millington M, Wotherspoon S, Rice A, MacKenzie KL, Symonds G. Mutant N-ras preferentially drives human CD34+ hematopoietic progenitor cells into myeloid differentiation and proliferation both in vitro and in the NOD/SCID mouse. Exp Hematol. 2004;32:852–860. doi: 10.1016/j.exphem.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Darley RL, Hoy TG, Baines P, Padua RA, Burnett AK. Mutant N-RAS induces erythroid lineage dysplasia in human CD34+ cells. J Exp Med. 1997;185:1337–1347. doi: 10.1084/jem.185.7.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006;108:2349–2357. doi: 10.1182/blood-2004-08-009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Liu Y, Li Z, Du J, Ryu MJ, Taylor PR, Fleming MD, Young KH, Pitot H, Zhang J. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116:5991–6002. doi: 10.1182/blood-2010-04-281527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haupt Y, Harris AW, Adams JM. Retroviral infection accelerates T lymphomagenesis in E mu-N-ras transgenic mice by activating c-myc or N-myc. Oncogene. 1992;7:981–986. [PubMed] [Google Scholar]
- 10.Wang J, Liu Y, Li Z, Wang Z, Tan LX, Ryu MJ, Meline B, Du J, Young KH, Ranheim E, Chang Q, Zhang J. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood. 2011;118:368–379. doi: 10.1182/blood-2010-12-326058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Kong G, Liu Y, Du J, Chang YI, Tey SR, Zhang X, Ranheim EA, Saba-El-Leil MK, Meloche S, Damnernsawad A, Zhang J, Zhang J. Nras(G12D/+) promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood. 2013;121:5203–5207. doi: 10.1182/blood-2012-12-475863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 13.Xiao Y, Jiang Z, Li Y, Ye W, Jia B, Zhang M, Xu Y, Wu D, Lai L, Chen Y, Chang Y, Huang X, Liu H, Qing G, Liu P, Li Y, Xu B, Zhong M, Yao Y, Pei D, Li P. ANGPTL7 regulates the expansion and repopulation of human hematopoietic stem and progenitor cells. Haematologica. 2015;100:585–594. doi: 10.3324/haematol.2014.118612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logan AC, Nightingale SJ, Haas DL, Cho GJ, Pepper KA, Kohn DB. Factors influencing the titer and infectivity of lentiviral vectors. Hum Gene Ther. 2004;15:976–988. doi: 10.1089/hum.2004.15.976. [DOI] [PubMed] [Google Scholar]
- 15.Yang D, Zhang X, Dong Y, Liu X, Wang T, Wang X, Geng Y, Fang S, Zheng Y, Chen X, Chen J, Pan G, Wang J. Enforced expression of Hoxa5 in haematopoietic stem cells leads to aberrant erythropoiesis in vivo. Cell Cycle. 2015;14:612–620. doi: 10.4161/15384101.2014.992191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohta H, Sekulovic S, Bakovic S, Eaves CJ, Pineault N, Gasparetto M, Smith C, Sauvageau G, Humphries RK. Near-maximal expansions of hematopoietic stem cells in culture using NUP98-HOX fusions. Exp Hematol. 2007;35:817–830. doi: 10.1016/j.exphem.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du J, Wang J, Kong G, Jiang J, Zhang J, Liu Y, Tong W, Zhang J. Signaling profiling at the single-cell level identifies a distinct signaling signature in murine hematopoietic stem cells. Stem Cells. 2012;30:1447–1454. doi: 10.1002/stem.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Lodish HF. Endogenous K-ras signaling in erythroid differentiation. Cell Cycle. 2007;6:1970–1973. doi: 10.4161/cc.6.16.4577. [DOI] [PubMed] [Google Scholar]
- 20.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Liu Y, Tan LX, Lo JC, Du J, Ryu MJ, Ranheim EA, Zhang J. Distinct requirements of hematopoietic stem cell activity and Nras G12D signaling in different cell types during leukemogenesis. Cell Cycle. 2011;10:2836–2839. doi: 10.4161/cc.10.17.17195. [DOI] [PMC free article] [PubMed] [Google Scholar]