

Abstract
Breast cancer is the second leading cause of cancer induced death in women. Tamoxifen is an endocrine therapy which is administered to 70% of all breast cancer patients with estrogen receptor alpha (ERα) expression. Despite the initial response, most patients eventually acquire resistance to the drug. MicroRNAs (miRNAs) are a class of small non-coding RNAs which have the ability to post-transcriptionally regulate gene expression. Although the role of a few miRNAs has been described in tamoxifen resistance, little is known about how concerted actions of miRNAs targeting biological networks contribute to its resistance. In this study, we identified that miR-155 is frequently up-regulated in breast cancer with tamoxifen resistance. Ectopic expression of miR-155 induces cell survival and resistance to TAM, whereas inhibition of miR-155 causes cells to apoptosis and enhances TAM sensitivity. Further, we identified SOCS6 as a new direct target of miR-155. Sustained overexpression of miR-155 resulted in repression of SOCS6 protein and mRNA levels, and knockdown of miR-155 increased SOCS6 expression. Introduction of SOCS6 cDNA lacking the 3’-UTR abrogated miR-155-induced cell survival and chemoresistance. Finally, it was verified that SOCS6 or inhibition of STAT3 could inhibit miR-155 STAT3 activation and cell proliferation. In conclusion, our study reveals a molecular link between miR-155 and SOCS6-STAT3 and presents an evidence that miR-155 is a critical therapeutic target in breast cancer.
Keywords: miR-155, breast cancer, SOCS6, STAT3, tamoxifen resistance
Introduction
Most of breast cancer patients carring the tumors expressing the estrogen receptor (ERα) and are candidates for endocrine therapy. The selective ERα modulator tamoxifen, is the most commonly prescribed endocrine therapy, but 30-40% of patients fail adjuvant tamoxifen therapy and nearly all patients with metastatic disease develop tamoxifen resistance [1-3]. Unfortunately, de novo and acquired tumor resistance to tamoxifen therapy remains a poorly understood and serious clinical problem [4-7].
MicroRNAs (miRNAs) are about 22 nucleotide RNAs that negatively regulate gene expression in eukaryotes [8]. miRNAs are one of the most abundant classes of regulatory molecule in mammals, and mounting evidence indicates that miRNAs are key regulators of animal development and involve in human diseases such as cancer [9]. OncomiRs are upregulated in tumors functioning in tumor development mainly via repressing the expression of tumor suppressor or tumor suppressor-like genes [9,10].
miR-155 is located within a region known as B cell integration cluster, which was originally thought to be a proto-oncogene associated with lymphoma [11]. miR-155 was first implicated in the oncogenesis of hematopoietic malignancies based on the finding that BIC/mir-155 expression is upregulated in B-cell lymphomas and chronic lymphocytic leukemia [12]. Consistent with this observation, transgenic expression of miR-155 in B cells causes acute lymphoblastic leukemia or high-grade lymphoma [13]. mir-155 is also overexpressed in various solid tumors including breast, lung, colon, pancreatic, and thyroid cancers [14]. Moreover, high expression levels of miR-155 have been found to correlate with poor prognoses of lung cancer and pancreatic tumor. All of reports are consistent with the notion that miR-155 functions as an oncogenic miRNA (oncomiR) in human cancers [15-17].
Although miR-155 has been found to be upregulated in breast cancer, its role in TAM resistance in breast cancer [18] has not been clear. In the present study, we found that miR-155 expression in TAM resistant breast cancer tissues or cell lines were up-regulated. Further research identified suppressor of cytokine signaling 6 (SOCS6) was a novel target of miR-155 in breast cancer cells. SOCS6 is a tumor suppressor that normally functions as a negative feedback regulator of Janusactivated kinase (JAK)/signal transducer and activator of transcription (STAT) signaling. Furthermore, the results show that overexpression of miR-155 promoted cell proliferation, colony formation, xenograft tumor growth and induced TAM resistance in breast cancer cells through the repression of SOCS6.
Materials and methods
Patients and tumor tissues
Human breast cancer tissues with tamoxifen resistance and their controls were obtained during the surgery at Shandong Provincial Hospital (China) between January 2006 and December 2011. The diagnosis was based on pathological evidence and the specimens were immediately snap-frozen and stored at -80°C. None of the patients received chemotherapy or radiotherapy before the surgical excision. All patients provided written informed consent for the use of their tissues and the study protocol was approved by the Ethics Committee of the Hospital.
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was isolated from breast cancer tissues or cells using Trizol reagent (Invitrogen, Carlsbad, USA). miR-155 and U6 were polyadenylated using poly-A polymerase based First-Strand Synthesis kit (TaKaRa Bio, Japan) following the manufacturer’s protocol. To quantify the SOCS6 and GAPDH mRNA levels, 1 ug of total RNA was subjected to first-strand cDNA synthesis for 15 min at 37°C and 5 s at 85°C using a PrimeScript RT Reagent kit (TaKaRa). qPCR was performed using SYBR Green PCR master mix (TaKaRa) on the ABI 7500HT System. U6 or GAPDH were used as an endogenous control. The relative fold expressions were calculated with the 2-ΔΔCT method. All the qRT-PCR reactions were run in triplicate. All the primers were ordered from Invitrogen.
Cell lines and culture
Human breast cancer MCF7 and SKBR3 cell lines were primary obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and were maintained in RPMI-1640 medium supplemented with. The cells were grown in a humidified incubator at 37°C with 5% CO2.
Transfection
The microRNAs were designed and synthesized by RiboBio (Ribobio Co., Guangzhou, China). The microRNA transfection was performed using Lipofectamine 2000 (Invitrogen). The breast cancer cells were seeded in 12-well plates and were grown up to 40% confluence before the transfection. The RNA and proteins were extracted at 48 h after the transfection. The final concentration of miR-155 mimic and anti-miR-155 was 50 nM. Lentiviral miR-155 (LV-miR-155) and empty lentiviral vector (LV-NC) were constructed by Genechem Company (Shanghai, China) and were transfected into the breast cancer cells according to the manufacturer’s instruction.
Cell proliferation assay
Cell proliferation was determined by MTT assay. Briefly, the breast cancer cells (5×103 per well) were plated in 96-well plates in RPMI 1640 and 10% FBS. After 24 h of being in the culture, the cells were transfected with 50 nM miR-155 mimics, anti-miR-155 and their respective control using Lipofectamine 2000 (Invitrogen). The cells were then cultured in the medium for another 48 h and were assessed by a colorimetric assay using MTT solution (5 mg/mL) at 570 nm. All the experiments were performed three times with five replicates.
Colony formation assay
Cells were tranfected with miR-155, or plasmids were digested after 24 h. Then, these cells were added to 6-well plates (200 cells/well) followed by incubation at 37°C in an environment with saturated humidity and 5% CO2 for 24 h. Non-adherent cells were removed. Cell were cultured for 10-14 days and colonies were present to count.
Cell apoptosis assay
Cell apoptosis was detected by an Annexin V-FITC/PI Apoptosis Detection kit I (BD Biosciences) in accordance with the manufacturer’s protocols. Cells were seeded into 6-well plate at the density of 1×105/well. After incubation for 24 h at 37°C, Cochicine was added to the 6-well plate and incubated for another 24 h. Cells were collected and washed twice with PBS and then resuspended, and aliquots of 1×105 cells were transferred into new 5 mL culture tubes in 100 uL of 1 × binding buffer. Then, 5 uL of Annexin V-FITC and 5 uL of propidium iodide were added to the resuspended cells. After incubation at room temperature for 15 min in the dark, 400 uL of binding buffer were added to the resuspended cells. Flow cytometry (Becton Dikinson, USA) was used to assess the apoptotic cells. The quantitation of apoptotic cells was calculated by CellQuest software.
Hoechst staining
In order to visualize nuclear morphology and the induction of apoptosis, Hoechst 33342 dye (Molecular Probes, Eugene, OR, USA) was used to stain the nuclei. Following treatment with either Green 1 or NSC 51046, cells were incubated with 10 µM of the Hoechst 33342 dye for 10 minutes at 37°C. Images were obtained using a Leica DM IRB inverted fluorescence microscope (Wetzlar, Germany) at 400X magnification.
Western blot analysis
Breast cancer cells were collected after 48 h treatment with 50 nM miR-155 mimic or anti-miR-155 and corresponding controls. Protein extraction, SDS-PAGE gel electrophoresis and blotting were performed as we previously described. Several different primary antibodies were used including: SOCS6, STAT3 (Cell Signaling Technology, Danvers, USA) and GAPDH (Santa Cruz Biotechnology, Santa Cruz, USA). The secondary antibody incubations were performed for 2 h at room temperature and protein bands were visualized on the X-ray film using an enhanced chemiluminescence ECL substrate.
Dual luciferase assay
Co-transfection experiments were performed in 96-well plates. A total of 1×104 cells were seeded per well in 200 µl medium. A total of 100 ng wild type (WT) or mutant (MUT) reporter constructs were co-transfected with Lipofectamine 2000 transfection reagent into the breast cancer cells with 50 nM miR-155 or miR-control according to the manufacturer’s instruction. After 48 h, luciferase activity was measured with the Dual-Luciferase reporter assay system (Promega). Firefly luciferase activity was then normalized to the corresponding Renilla luciferase activity.
Breast cancer xenograft model
Four-week-old female nude mice (BALB/c-nude) were used to examine the tumorigenicity. A total of 100 uL cell solutions (containing 1.5×107 cells) were subcutaneously injected into the right flank of the mice. The tumor size was measured with a vernier caliper every four days and the tumor volumes were calculated using the formula: length×width 2×0.5. The use of nude mice complied with the Guide for the Care and Use of Laboratory Animals and the study was approved by Animal Care and Use Committee of Shengjing Hospital.
Statistical analysis
The miR-155 expression was compared in breast cancer tissues and cells by the unpaired Student’s t test. The relationships between miR-155 and clinicopathologic parameters were evaluated by χ2 test. The survival rates for miR-155 expression were estimated by using the Kaplan-Meier method and the difference in survival curves were tested by log-rank test. All statistical analyses were carried out by SPSS13.0 software and the data were presented as means ± standard deviation (SD). A p lower than 0.05 was considered significant.
Results
miR-155 expression is up-regulated in tamoxifen resistant breast cancer cells
Firstly, we determined if upregulation of miR-155 expression was a common feature in tamoxifen resistant breast cancer cells. MCF-7 and SKBR3 cells (both expresssing ERα) were continuously exposed to 5 mM tamoxifen for about 6 months until cells had acquired resistance to tamoxifen. miR-155 expression was examined by real time RT-PCR in multiple tamoxifen (TAM) sensitive and resistant MCF-7 and SKBR3 cells. Interestingly, tamoxifen sensitive cells expressed low levels of miR-155 whereas the tamoxifen resistant cell lines exhibited dramatically increased levels of miR-155 (Figure 1A and 1B). To confirm this result, we used breast cancer tissues from tamoxifen non-sensitive and sensitive to compare miR-155 expression. TAM resistance or sensitive was determined from the clinic investigation, patients with ER positive were treated with TAM and good therapeutic effects were classcified as sensitive, others as non-sensitive. it was found that miR-155 increased in tamoxifen resistant breast cancer cells (Figure 1C).
Figure 1.
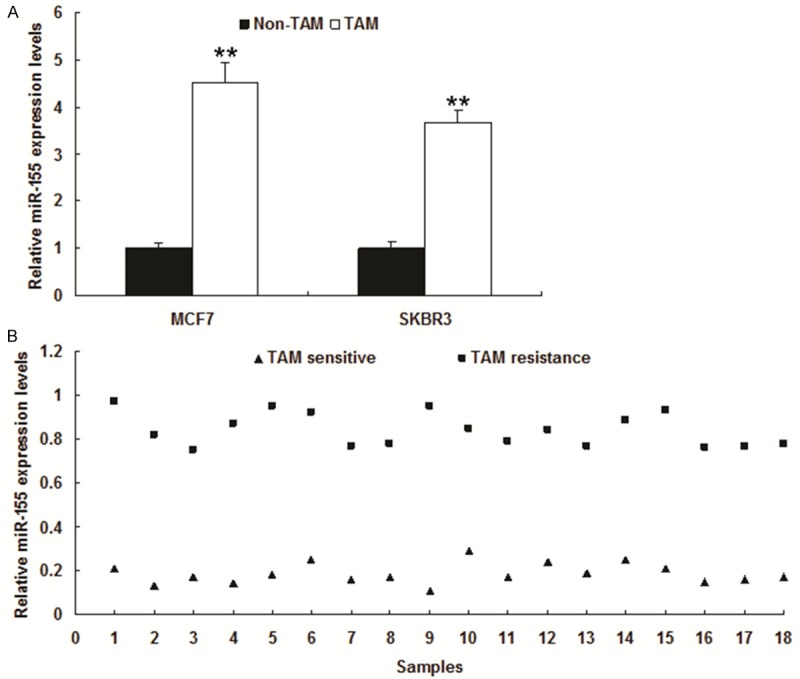
Expression of miR-155 is increased in tamoxifen resistant breast cancer cells. A. Total RNA was isolated from tamoxifen sensitive breast cancer cells (MCF-7 or SKBR3) and tamoxifen resistant cells (MCF-7/TAM and SKBR3/TAM). RNA was analyzed by real time RT-PCR for miR-155 levels. B. Total RNA was isolated from breast cancer samples from the patients with tamoxifen sensitive and resistance. RNA was analyzed by real time RT-PCR. Data represents the mean ± SE of triplicate samples. **P<0.01.
Suppression of miR-155 increases breast cancer cells sensitive to tamoxifen
To determine if modulation of miR-155 expression impacts tamoxifen response, we suppressed miR-155 expression in the tamoxifen resistant cell lines. miR-155 expression was down-regulated in MCF7 and SKBR3 transfected with anti-miR155 by real time RT-PCR (Figure 2A). MCF7 and SKBR3 cells became more sensitive to tamoxifen (Figure 2B and 2C). Transient transfection of cells with a miR-155 inhibitor induced apoptosis (Figure 2D and 2E). These results suggested that expression of miR-155 could induce resistance of breast cancer cells to tamoxifen treatment and tamoxifen resistance can be acquired through miR-155.
Figure 2.
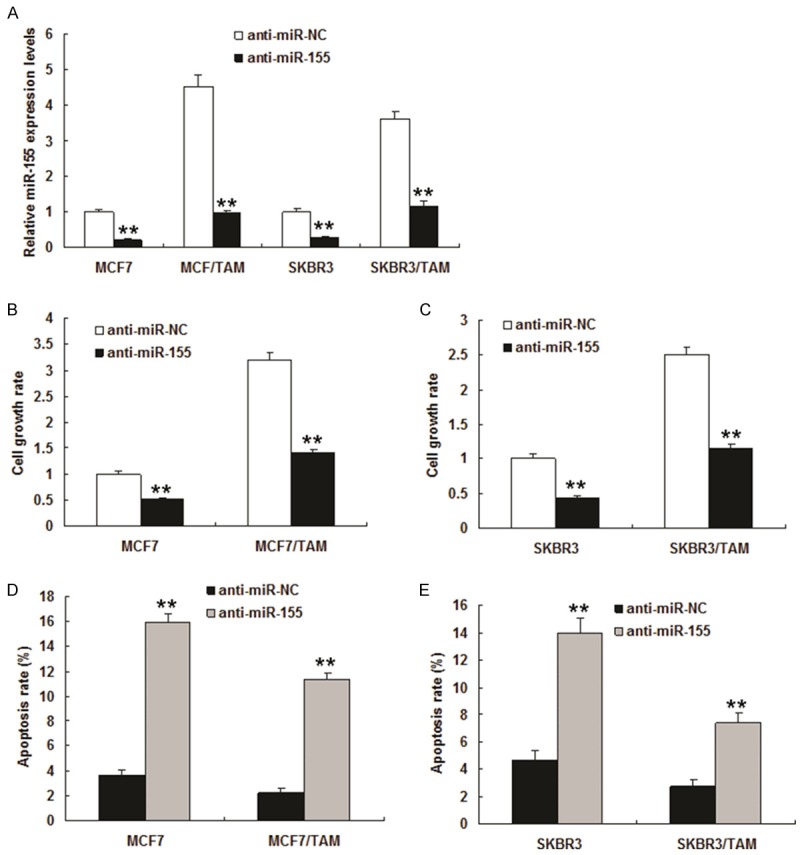
Suppression of miR-155 promotes TAM resistant cells to be sensitive to tamoxifen. The tamoxifen resistant cell lines were transfected with anti-miR-155 or anti-miR-control (anti-miR-NC). A. miR-155 expression in MCF7 and SKBR3 cells. MCF7 and SKBR3 cells were transfected with anti-miR-155 or its control and total RNA was extracted for quantative RT-PCR. B and C. Cell growth was examined in the breast cancer cells. MCF7 and SKBR3 cells were transfected with anti-miR-155 or its control and at the day 3, cell proliferation was assayed MTT method. D and E. Apoptosis was quantitated using flow cytometry. Data is represented as mean ± SE of three independent experiments. **P<0.01.
SOCS6 is a target of miR-155 in breast cancer cells
To explore the potential target through which the miR-155 induces tamoxifen resistance of breast cancer cells, we predicted the putative miR-155 targets by using TargetScan and miRanda. Our analysis identified SOCS6, a key tumor suppressor gene, as one of the potential targets for miR-155 (Figure 3A). Furthermore, the target sequences of SOCS6 3’UTR (wild type, WT) or the mutant sequence (mutant type, MUT) were cloned into the luciferase reporter vector, respectively. Our results showed that miR-155 significantly decreased the firefly luciferase activity in the reporter with wild type 3’UTR, but the activity of mutant 3’UTR vector remained unaffected in MCF7 and SKBR3 cells (Figure 3B and 3C). The MCF-7 and SKBR3 cells were further transfected with miR-155 and were examined for SOCS6 expression by qRT-PCR and Western blot. As shown in Figure 3C-E, miR-155 transfection led to an obvious decrease in SOCS6 mRNA and protein expression. On the contrary, transfection of anti-miR-155 resulted in an increase of SOCS6 protein levels (Figure 3E).
Figure 3.
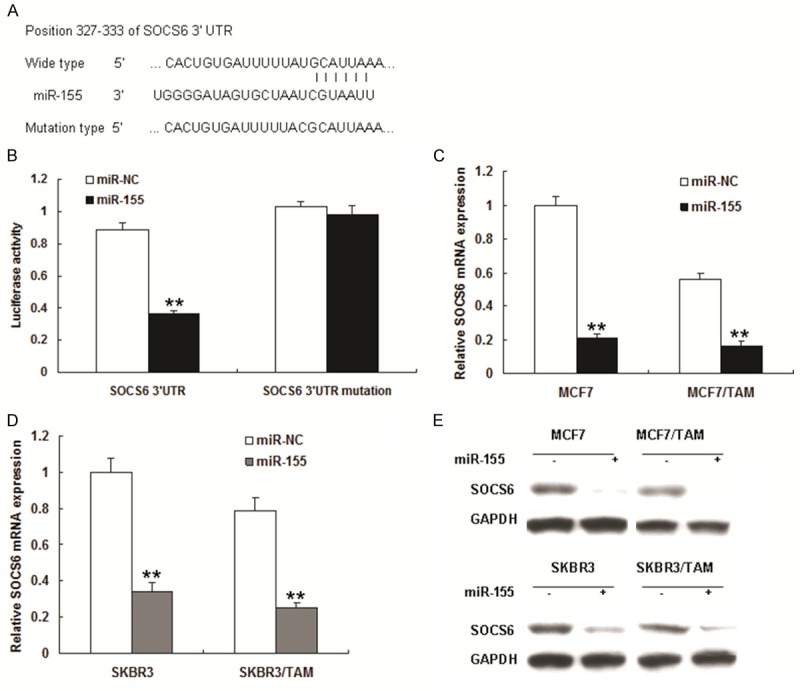
SOCS6 is a target of miR-155 in breast cancer cells. A. The miR-155 and the miR-155-binding site in the 3’UTR of SOCS6. B. Luciferase reporter assay with cotransfection of wild-type or mutant 3’UTR (100 ng) and miR-155 or miR-control in MCF7 cells. Firefly luciferase activity of each sample was normalized against Renilla luciferase activity. C. The effects of miR-155 on the expression of endogenous SOCS6 and SOCS6 mRNA in MCF7 and SKBR3 cells was tested by real time RT-PCR. D. The effects of anti-miR-155 on the expression of endogenous SOCS6 in MCF7 and SKBR3 cells. miR-155 was tested by real time RT-PCR. E. Western blot was used for monitoring the SOCS6 expression in MCF-7 and SKBR3 cells after the transfection for 48 h with miR-155 or anti-miR-155. All data from three separate experiments are presented as mean ± SD. **P<0.01 vs the control.
SOCS6 makes breast cancer cells become sensitive to tamoxifen
MCF7, MCF7/TAM, SKBR3 and SKBR3/TAM were transfected with LV-SOCS6 or its control (LV-control), and SOCS6 protein was over-expressed (Figure 4A). To answer whether the role of SOCS6 in tamoxifen responses, MCF7/TAM and SKBR3/TAM cells were exposed to tamoxifen, and cell survival was examined. The results showed that over-expression of SOCS6 could make MCF7/TAM cells more sensitive to tamoxifen (Figure 4B), so did SKBR3/TAM cells (Figure 4C). SOCS6 induced apoptosis when the MCF-7 and SKBR3 cells were treated with tamoxifen (Figure 4D). Whether SOCS6 has an effect on miR-155 expression? The data indicated that the cells with SOCS6 knocking down was not companied with changes of miR-155 expression, which suggested that SOCS6 expression didn’t influence miR-155 expression (Figure 4E).
Figure 4.
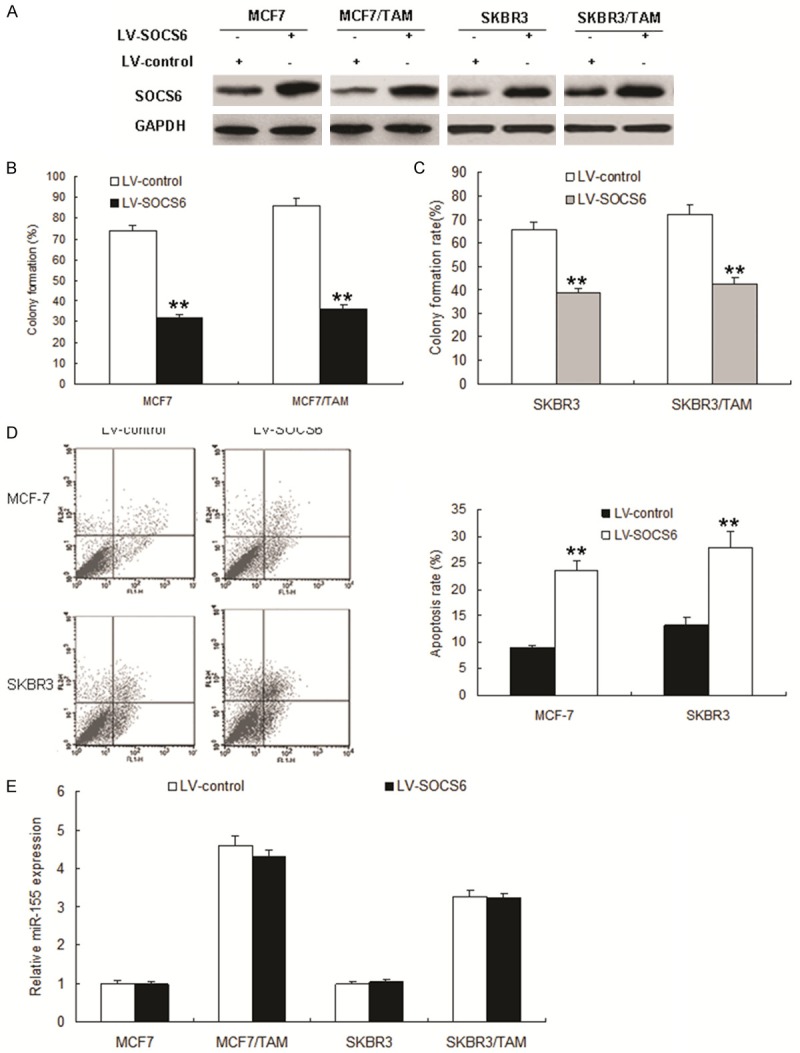
SOCS6 makes breast cancer cells become sensitive to tamoxifen. A. MCF-7, MCF-7/TAM cells, SKBR3 and SKBR3/TAM cells were infected with LV-SOCS6 or its control and total protein was extracted for immunoblotting. B and C. MCF-7, MCF-7/TAM cells, SKBR3 and SKBR3/TAM cells were infected with LV-SOCS6 using colony formation assay to test. D. MCF-7/TAM cells were infected with LV-SOCS6 and treated with TAM, and apoptosis was quantitated using flow cytometry (left panel). The right panel was quanlification of apoptosis rate from flow cytometry. E. MCF-7, MCF7/TAM, SKBR3 and SKBR3/TAM cells were infected with LV-SOCS6 or its control and total RNA was extracted for miR-155 expression by real time RT-PCR. Data is represented as mean ± SE of three independent experiments. **P<0.01.
miR-155 induces tamoxifen resistance in breast cancer cells in vitro and in vivo by targeting SOCS6
Above data showed that miR-155 was overexpressed in breast cancers and that it may act as an oncomiR and SOCS6 was a new target gene of miR-155. To futher investigate whether miR-155 plays an oncomiR through SOCS6. We then performed rescue experiments to further validate that SOCS6 targeting is involved in miR-155-mediated oncogenesis in MCF-7 and SKBR3 cells. MCF-7 or SKBR3 cells were infected with LV-miR-155 or combinded with LV-SOCS6 and cell proliferation was analysized by colony formation assay. The results indicated that miR-155 significantly promoted the proliferation of MCF-7 and SKBR3 cells (Figure 5A and 5B) and stimulated MCF-7 and SKBR3 cells to grow more and larger colonies. When the cells with SOCS6 overexpression, the colonies became less and smaller. These results showed that miR-155 promoted the proliferation and anchorage-independent growth of breast cancer cells in vitro. Results of cell apoptosis showed that SOCS6 could promote cell apoptosis rate in the MCF-7 and SKBR3 cells with miR-155 overexpression (Figure 5C and 5D). Western blot analyses confirmed that SOCS6 in these cells was modulated by miR-155, and the expression of the proliferation biomarker PCNA was enhanced by miR-155 (Figure 5E). Taken together, our results show that miR-155 acts as an oncomiR in breast cancer cells by targeting SOCS6.
Figure 5.
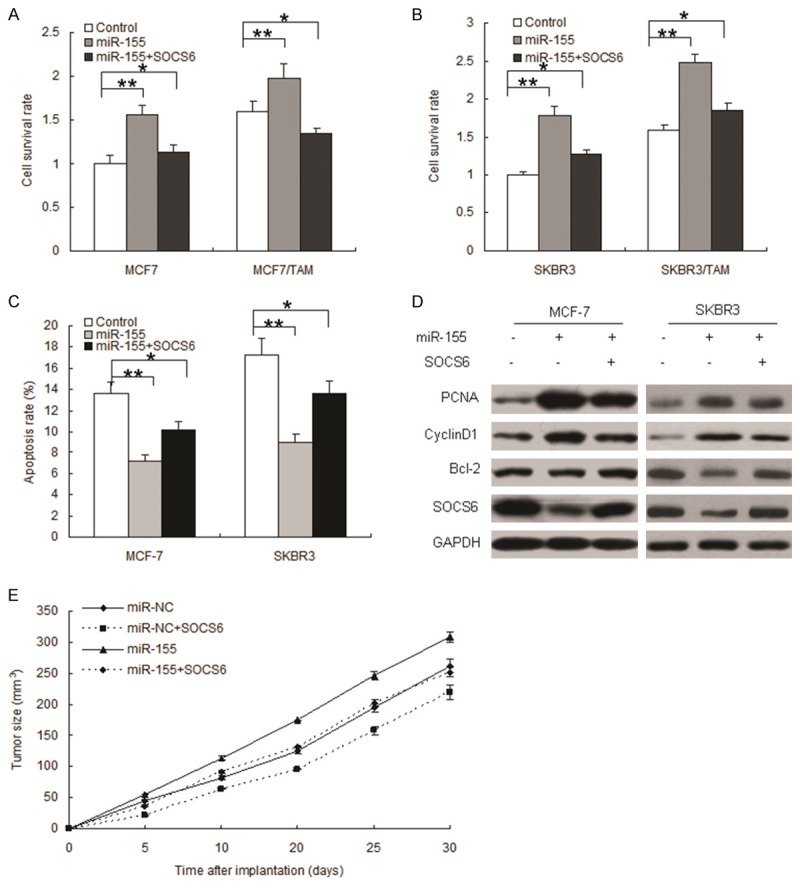
miR-155 acts as an oncomiR in breast cancer cells in vitro and in vivo. miR-155 promotes breast cancer cell proliferation. A. MCF-7 cells were transfected with miR-155, SOCS6, or control. MTT assay was performed 24 h after transfection. B. SKBR3 cells were transfected with miR-155, SOCS6, or control. MTT assay was performed 24 h after transfection. C. MCF-7 and SKBR3 cells were infected with miR-155, SOCS6 or control for 24 h and then treated with TAM for 24 h, straning FITC-Annexin V for flow cytometry. D. Western blotting was used to detect cell proliferation, and apoptosis associated proteins. E. miR-155 promoted breast cancer growth in vivo. Breast cancer models were set up using MCF-7 cells with miR-155 or combined with SOCS6 overexpression. Tumor growth curve was mapped to observe the tumor growth. Data is represented as mean ± SE of three independent experiments. **P<0.01.
miR-155 stimulates STAT3 signaling in breast canceMr cells by inhibiting SOCS6
Given that SOCS6 is repressor of JAK2/STAT3 signal pathway, we speculated that miR-155 overexpression in breast tumors may play a role in STAT3 signaling. In MCF-7/TAM cells, of which miR-155 was highly expressed and STAT3 was constitutively activated, we found that miR-155 expression resulted in decreased levels of SOCS6 protein and increased levels of phospho-JAK2 and phospho-STAT3 (Figure 6A-C). In contrast, the level of STAT3 protein was not affected by miR-155 in MCF-7/TAM and SKBR3/TAM cells (Figure 6A-C). Furthermore, we used JSI-124, a highly selective and potent inhibitor of STAT3, to validate miR-155-SOCS6-STAT3 axis functions. As shown in Figure 6D, miR-155 activated STAT3 in MCF7/TAM and JSI-124 reversed miR-155-promoting activation of STAT3 and cell growth, so did in SKBR3/TAM cells (Figure 6D). Results from MTT assay indicated that STAT3 inhibition led to cell proliferation becoming slower than that control (Figure 6E and 6F). The results suggested that miR-155 may function as an oncomiR in tamoxifen resistance by activating STAT3 signaling in breast cancer cells by suppressing SOCS6 expression.
Figure 6.

miR-155 stimulates STAT3 signaling in breast cancer cells by inhibiting SOCS6. A. miR-155 actived STAT3 signal pathway by targeting SOCS6. MCF7 and SKBR3 cells were tranfected with miR-155 combined with SOCS6 or its control and total protein was extracted from the cells for western blot analysis. Three independent experiments were performed. B. Qutification of protein bands from MCF-7/TAM cells (A). C. Qutification of protein bands from SKBR3/TAM cells. D. STAT3 inhibitor, JSI-124 suppressed miR-155 induced STAT3 activation in MCF-7/TAM and SKBR3/TAM cells. E. STAT3 inhibitor, JSI-124 suppressed STAT3 activation and inhibited miR-155 induced MCF-7 cell proliferation. F. JSI-124 suppressed STAT3 activation and inhibited miR-155 induced SKBR3 cell proliferation. Three independent experiments were performed. Data is represented as mean ± SE of three independent experiments. **p<0.01; *p<0.05.
Discussion
Endocrine resistance is still an complicated question in the breast cancer therapy [4-6]. Until recently, there are only a few useful tumor markers to guide the therapy for women with ERα positive breast cancers [2,3]. Here, we investigated the potential role of miRNAs in the regulation of tamoxifen response with the goal of identifying miRNAs that can be used to predict patient outcome during tamoxifen therapy. We found several miRNAs expression were altered in tamoxifen resistant breast tumor cells. It was further demonstrated that miR-155, an ERα associated miRNA, was dramatically up-regulated in two tamoxifen resistant breast tumor cell lines and in breast cancer patients who tamoxifen therapy failed. Importantly, inhibition of miR-155 could enhance breast cancer cells to tamoxifen therapy sensitivity, which suggests that miR-155 is an important regulator of tamoxifen response.
In this study, we profiled miRNA expression in tamoxifen-sensitive and resistant breast cancer cell lines and we identified several miRNAs differentially regulated in tamoxifen-resistant cells. It is shown that miR-155, which is up-regulated in tamoxifen resistant cells, is associated with decreased tamoxifen sensitivity of breast tumor cells. miR-155 plays important role in cancer progression [19]. Previously, miR-155 was a regulator of SOCS1 in breast cancer cells [20] and it also targeted other genes to function in the breat cancer cells like Fox3a [21].
A direct gene target of miR-155 remains to be experimentally confirmed and bioinformatics reveals many potential miR-155 target genes. As an approach to the identification of miR-155 targets we examined transcriptome changes in miR-155 overexpressing cells by microarray analysis. We observed significant alteration of 13 genes predicted by bioinformatics to be miR-155 targets. Interestingly, the majority of predicted target genes were down-regulated by miR-155 expression. Although miRNAs commonly suppress target gene mRNA levels, accumulating evidence suggests that miRNAs can target genes for up-regulation by at least two different mechanisms [22,23]. Although we could not identify an obvious association between the direct targets of miR-155 and tumor cell response to tamoxifen, Ingenuity Pathway Analysis of the entire set of genes significantly altered by miR-155 revealed a highly significant association of miR-155 regulated genes with cell apoptosis. This result is consistent with our observation that ectopic miR-155 expression reduced tamoxifen resistant cells to tamoxifen-induced apoptosis. Similarly, miR-155 expression in cancer cells results in tumor cell apoptosis [24-27]. Nevertheless, activity of miR-155 appears to be functionally different in colorectal and breast tumor cells. Our results indicate that miR-155 expression alone is not sufficient to induce cell growth, but anti-miR-155 sensitizes cells to apoptosis associated with estrogen-deprivation and tamoxifen exposure. In this context, the miR-155 indirect target thioredoxin-interacting protein (SOCS6) is the most dramatically down-regulated gene in response to tamoxifen.
Accordingly, SOCS6 expression is suppressed in tumor cells by various stresses including serum starvation, which mimics ERα inactivation in breast tumors, and enhanced SOCS6 expression has tumor-suppressor activity? Similar to our findings that inhibition of miR-155 sensitizes breast cancer cells to tamoxifen and SOCS6 sensitizes the cells to tamoxifen. Although these results are compelling, SOCS6 is an upregulated indirect target of miR-155 and deciphering the interplay between miR-155 and SOCS6 has proved difficult. Taken together our results provide a genomic basis to explore the role of miR-155 regulated genes in multiple tamoxifen actions including both apoptosis and cytostasis.
In summary, we have identified miR-155 as an important mediator of tamoxifen response in both breast tumor cell lines and breast cancer patients. With the potential of miRNA therapy, inhibition of miR-155 expression may constitute a novel therapeutic strategy to sensitize drug in breast cancer.
Disclosure of conflict of interest
None.
References
- 1.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG); Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 2.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Dowsett M. Molecular changes in tamoxifenresistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogenactivated protein kinase. J. Clin. Oncol. 2005;23:2469–2476. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 3.Cui Y, Zhang M, Pestell R, Curran EM, Welshons WV, Fuqua SA. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 2004;64:9199–9208. doi: 10.1158/0008-5472.CAN-04-2126. [DOI] [PubMed] [Google Scholar]
- 4.Herynk MH, Parra I, Cui Y, Beyer A, Wu MF, Hilsenbeck SG, Fuqua SA. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clin Cancer Res. 2007;13:3235–3243. doi: 10.1158/1078-0432.CCR-06-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 6.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 7.Osborne CK, Schiff R. Estrogen-receptor biology: continuing progress and therapeutic implications. J. Clin. Oncol. 2005;23:1616–1622. doi: 10.1200/JCO.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 8.Teng G, Papavasiliou FN. Shhh! Silencing by microRNA-155. Philos Trans R Soc Lond B Biol Sci. 2009;364:631–637. doi: 10.1098/rstb.2008.0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandhu SK, Volinia S, Costinean S, Galasso M, Neinast R, Santhanam R, Parthun MR, Perrotti D, Marcucci G, Garzon R, Croce CM. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc Nat Acad Sci U S A. 2012;109:20047–20052. doi: 10.1073/pnas.1213764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, Ferracin M, Delmas D, Latruffe N, Croce CM. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31:1561–1566. doi: 10.1093/carcin/bgq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donnem T, Eklo K, Berg T, Sorbye SW, Lonvik K, Al-Saad S, Al-Shibli K, Andersen S, Stenvold H, Bremnes RM, Busund LT. Prognostic impact of MiR-155 in non-small cell lung cancer evaluated by in situ hybridization. J Transl Med. 2011;9:6. doi: 10.1186/1479-5876-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 13.Calame K. MicroRNA-155 function in B Cells. Immunity. 2007;27:825–827. doi: 10.1016/j.immuni.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 15.Wu Q, Luo G, Yang Z, Zhu F, An Y, Shi Y, Fan D. miR-17-5p promotes proliferation by targeting SOCS6 in gastric cancer cells. FEBS Lett. 2014;588:2055–2062. doi: 10.1016/j.febslet.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 16.Lai RH, Hsiao YW, Wang MJ, Lin HY, Wu CW, Chi CW, Li AF, Jou YS, Chen JY. SOCS6, down-regulated in gastric cancer, inhibits cell proliferation and colony formation. Cancer Lett. 2010;288:75–85. doi: 10.1016/j.canlet.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 17.Hwang MN, Min CH, Kim HS, Lee H, Yoon KA, Park SY, Lee ES, Yoon S. The nuclear localization of SOCS6 requires the N-terminal region and negatively regulates Stat3 protein levels. Biochem Biophys Res Commun. 2007;360:333–338. doi: 10.1016/j.bbrc.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 18.Zhang CM, Zhao J, Deng HY. MiR-155 promotes proliferation of human breast cancer MCF-7 cells through targeting tumor protein 53-induced nuclear protein 1. J Biomed Sci. 2013;20:79. doi: 10.1186/1423-0127-20-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Research. 2010;70:3119–3127. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 20.Mattiske S, Suetani RJ, Neilsen PM, Callen DF. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:1236–1243. doi: 10.1158/1055-9965.EPI-12-0173. [DOI] [PubMed] [Google Scholar]
- 21.Kong W, He L, Coppola M, Guo J, Esposito NN, Coppola D, Cheng JQ. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J Biol Chem. 2010;285:17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Johansson J, Berg T, Kurzejamska E, Pang MF, Tabor V, Jansson M, Roswall P, Pietras K, Sund M, Religa P, Fuxe J. MiR-155-mediated loss of C/EBPβ shifts the TGF-β response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene. 2013;32:5614–5624. doi: 10.1038/onc.2013.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Czyzyk-Krzeska MF, Zhang X. MiR-155 at the heart of oncogenic pathways. Oncogene. 2014;33:677–678. doi: 10.1038/onc.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong W, He L, Richards EJ, Challa S, Xu CX, Permuth-Wey J, Lancaster J M, Coppola D, Sellers TA, Djeu JY, Cheng JQ. Upregulation of miRNA-155 promotestumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene. 2014;33:679–689. doi: 10.1038/onc.2012.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Z, Ye Y, Jiao D, Qiao J, Cui S, Liu Z. miR-155 and miR-31 are differentially expressed in breast cancer patients and are correlated with the estrogen receptor and progesterone receptor status. Oncol Lett. 2012;4:1027–1032. doi: 10.3892/ol.2012.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattiske S, Suetani RJ, Neilsen PM, Callen DF. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:1236–1243. doi: 10.1158/1055-9965.EPI-12-0173. [DOI] [PubMed] [Google Scholar]
- 27.Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]