

Abstract
Evolutionary Action analyses of The Cancer Gene Atlas data sets show that many specific p53 missense and gain-of-function mutations are selectively overrepresented and functional in high-grade serous ovarian cancer (HGSC). As homozygous alleles, p53 mutants are differentially associated with specific loss of heterozygosity (R273; chromosome 17); copy number variation (R175H; chromosome 9); and up-stream, cancer-related regulatory pathways. The expression of immune-related cytokines was selectively related to p53 status, showing for the first time that specific p53 mutants impact, and are related to, the immune subtype of ovarian cancer. Although the majority (31%) of HGSCs exhibit loss of heterozygosity, a significant number (24%) maintain a wild-type (WT) allele and represent another HGSC subtype that is not well defined. Using human and mouse cell lines, we show that specific p53 mutants differentially alter endogenous WT p53 activity; target gene expression; and responses to nutlin-3a, a small molecular that activates WT p53 leading to apoptosis, providing “proof of principle” that ovarian cancer cells expressing WT and mutant alleles represent a distinct ovarian cancer subtype. We also show that siRNA knock down of endogenous p53 in cells expressing homozygous mutant alleles causes apoptosis, whereas cells expressing WT p53 (or are heterozygous for WT and mutant p53 alleles) are highly resistant. Therefore, despite different gene regulatory pathways associated with specific p53 mutants, silencing mutant p53 might be a suitable, powerful, global strategy for blocking ovarian cancer growth in those tumors that rely on mutant p53 functions for survival. Knowing p53 mutational status in HGSC should permit new strategies tailored to control this disease.
Abbreviations: CASP3, caspase 3; CDKN1A, cyclin dependent protein kinase 1, P21; CNV, copy number variation; DNAJC3, DnaJ homolog subfamily C; GFP, green fluorescent protein; GOF, gain-of-function; HGSC, high-grade serous ovarian cancer; HOX, homeobox genes; LOH, loss of heterozygosity; MDM2, mouse double minute 2; TCGA, The Cancer Genome Atlas; TP53, tumor protein 53, p53; UPR, unfolded protein response; WT, wild type
Introduction
High-grade ovarian cancer in women is a complex and deadly disease characterized by tumor protein 53 (TP53; p53) mutations and genomic instability [1], [2], [3], [4]. Recent studies have sought to identify subtypes of ovarian cancer for more selective and successful therapeutic strategies. Based on transcriptional profiles provided in The Cancer Gene Atlas (TCGA), high-grade serous ovarian cancer has been divided into specific molecular subtypes [3]. Based on genomic instability, defects in homologous recombination, and sensitivity to platinum-based therapies, two subtypes have been identified that exhibit either high or low levels of loss of heterozygosity (LOH) or allelic imbalance [5]. The high-LOH group was further divided into two groups based on high or low sensitivity to platinum therapies. Remarkable in the high-LOH subtype was the consistent and specific loss of chromosome 17 that contains the TP53 locus. Not only is the loss of chromosome 17 an apparent defining feature in high-grade serous ovarian cancer [5], these tumors have the highest frequency of mutations in the TP53 gene than any other tumor in women [5]. Many of these mutations confer gain-of-function (GOF) activities that drive tumor growth independent of wild-type (WT) p53 [6]. Molecular taxonomy studies further verify that the molecular signature of ovarian cancer is distinct from that of other cancers but similar to the tissue of origin [7]. These unique characteristics of high-grade serous ovarian cancer indicate that targeting mutant p53 as well as WT p53 might provide new effective, “personalized” therapeutic strategies. In addition, recent analyses of the TCGA data sets, as described herein, indicate that a significant subset of human ovarian tumor samples expresses heterozygous, not homozygous, specific “hot-spot” TP53 mutants, raising additional questions about targeting therapies for these tumors. Yet the functional activities of different mutant p53 proteins in this heterozygous subtype (compared with WT or homozygous mutant) have not been analyzed in detail in ovarian cancer.
It is now clear from many studies and analyses of the TCGA data sets not only that are there many p53 mutants in most human cancers but also that not all mutants are structurally and functionally equivalent [8], [9], [10], [11]. For example, the R248Q mutant aggregates and is associated with metastasis, whereas the R248W mutant does not aggregate and is less metastatic [12]. R175H inhibits cell cycle arrest and apoptosis in response to DNA damage, whereas the R175P only blocks cell cycle arrest [8]. P53 mutants also exert different phenotypic outcomes whether they are expressed with a WT p53 allele or are expressed with a null allele. For example, the pioneering studies of Olive et al. [13] showed that mice expressing a WT p53 allele with an R175H, R270H, or null allele develop more carcinomas and fewer lymphomas, whereas mice expressing R175H/R175H homozygous alleles do not develop carcinomas but rather develop more sarcomas and lymphomas [8], [13]. Different p53 mutants also interact with different partners and regulate different pathways: R248Q and R273H (but not R175H) bind the MRE11 nuclease leading to increased genomic instability. R175H, R248, and R273 exhibit distinct gene expression patterns related to different metabolic states in colon cancer cells [11]; R273H appears to be highly related to steroid metabolism [10]. Using a mouse model of ovarian cancer, we have shown that the functional status of p53 in ovarian epithelial tumors impacts tumor growth, metastasis, and response to steroid hormones [14], [15]. Specifically, WT p53 promotes papillary tumor growth, whereas depletion of p53 impairs tumor growth [14]. These results are consistent with the role of WT P53 as a regulator of cell proliferation in normal and cancer cells [16], [17], [18], [19]. However, the p53 null cells are highly sensitive to the steroid hormone estradiol, undergo rampant metastases to the peritoneal cavity, and exhibit some features of high-grade ovarian cancer [15]. Introduction of the specific p53 mutant R172H into the mouse ovarian tumor cells in vivo changes ovarian tumor progression, metastasis, morphology, and response to steroids. Moreover, the tumor phenotype is markedly more aggressive, metastatic, and similar to high-grade ovarian cancer when homozygous R172H mutant alleles are expressed compared with when the mutant is expressed in the presence of a WT allele (heterozygous) (Ren et al., in revision), making these cells a valuable resource for understanding p53 functions in ovarian cancer.
Clearly, the outcomes of expressing homozygous mutant alleles that exhibit GOF are not equivalent to outcomes associated with loss of p53 alleles [10], [20], [21]. This may be dictated, in part, because they can bind to similar interacting partners but lead to diverse outcomes and greater genomic instability [9], [10]. These complexities in p53 function have made it challenging to target p53 for therapeutic purposes. However, several small molecules designed to maximally activate WT p53 are currently available and lead to cell cycle arrest and apoptosis [8], [10]. One such drug, currently in clinical trials, is nutlin-3a that disrupts the interactions of p53 with mouse double minute 2 (MDM2) leading to increased p53 stability and activity [14], [22], [23]. Drugs to reactivate WT p53 functions in mutant p53 protein, such as PRIMA-1, are available and are also being tested [22]. However, this approach is not straightforward [24]. Because p53 mutants acquire their own specific functions and alter tumor functions, disrupting mutant p53 is an alternative attractive approach [6], [10]. Recent studies have shown in breast cancer and colon cancer cells in culture that disrupting p53 expression using siRNA leads to catastrophic events and cell death in cells expressing mutant p53 without affecting cell viability in cells with WT p53 [25], [26].
Based on these considerations, the primary goals of these studies were 1) to use a novel computational approach, termed Evolutionary Action (EAp53), which has been developed to stratify p53 mutants as high and low survival risk [27], [28], to identify p53 mutants that are overrepresented in human high-grade ovarian cancer and thus most likely to be exerting functional regulation on tumor cell survival, metastatic activity, and responses to drug therapies and 2) to determine the functional characteristics of the heterozygous versus the homozygous tumor subtypes by expressing or silencing WTp53 or specific p53 mutants overrepresented in ovarian cancer. The small molecule nutlin-3a was used to maximally activate WT p53 in each context [14], [23]. In addition, p53 siRNA was used to knock down WT and mutant p53 in human and mouse cells to determine the degree of dependence of p53 status on cell survival in each context. Our results provide “proof of principle” that human and mouse ovarian cancer cells expressing one WT allele and one mutant allele are functionally distinct from cells homozygous for either WT or mutant alleles and clearly support previous studies [13], [21]. Moreover, we show that specific p53 mutants in the presence of a WT p53 allele differentially regulate specific target genes and the responses of cells to nutlin-3a. Thus, knowing the mutant status of TP53 can be used to define the responses of cells to specific drugs and when combined with analyses of LOH could be used to improve the design of new therapeutic approaches.
Materials and Methods
Cell Lines
Trp53(+) cells from Pten;KrasG12D;Ptenfl/fl;Amhr2-Cre mice and Trp53(−) cells from Trp53fl/fl;LSL-KrasG12D;Ptenfl/fl;Amhr2-Cre mice were isolated and cultured as previously described [29]. Mice carrying the germ line Trp53R172H mutation were generated by Dr. Guillermina Lozano (MD Anderson Cancer Center, Houston, TX) [21]. These mice were bred to Ptenf/f;KrasG12D;Amhr2-Cre (PK) mice described previously [30] to obtain PK mice expressing WT Trp53 (PKP53+/+) and either heterozygous (PKP53H/+) or homozygous mutant Trp53R172H (PKP53H/H). Several human high-grade serous ovarian carcinoma cell lines were used (TP53 mutation status is indicted in parentheses): OVCAR3 (R248Q), OVCA420 (R273H), OVCA433 (E258K), SKOV3 (H179R), and ALST (WT). The SKOV3 cell line is an established p53-mutant cell line which does not express p53 at the protein or mRNA level and was therefore used as a negative control [31]. The MDM2 inhibitor Nutlin-3a was synthesized by the MD Anderson Pharmaceutical Chemistry Facility, Houston, TX. Cell lines were incubated in a humidified atmosphere at 37°C with 5% CO2 and cultured in RPMI 1640 media supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. For mouse Western blot and reverse transcriptase polymerase chain reaction (RT-PCR) analysis, cells transfected with mutant TRP53 were treated with 10 μM nutlin-3a, a dose that was previously determined to be the IC50 for WT TRP53 in Trp53(+) cells [14].
P53 siRNA and Cell Transfections
In experiments using transfected vector DNA, cells were transfected with 0.20 μg of DNA with Attractene Transfection Reagent (Qiagen, Valencia, CA) according to the manufacturer’s protocol. P53 siRNA experiments were performed using ON-TARGETplus Human TP53 siRNA-SMARTpool according to manufacturer protocol (Dharmacon, Pittsburgh, PA). The optimized concentration and time points for each cell line are indicated in the figure legends.
Determination of Cell Viability and IC50 of Nutlin-3a in Human Ovarian Carcinoma Cell Lines
For cell viability and IC50 determinations, cells were plated at a density of 9 × 10 [3] cells per well in 96-well plates. After 24 hours, media samples were exchanged, and cells were treated with incremental concentrations of nutlin-3a (5 μM, 10 μM, 25 μM, 50 μM, and 75 μM). After 24, 48, and 72 hours of incubation, WST-1 (Roche, Pleasanton, CA) was added to each well according to manufacturer’s protocol. The IC50 was defined as the concentration at which a 50% reduction in cell viability occurred, as we reported previously and verified by Annexin V FACS [32] and as documented for these studies (Supplemental Figure 1)
Supplemental Figure 1.
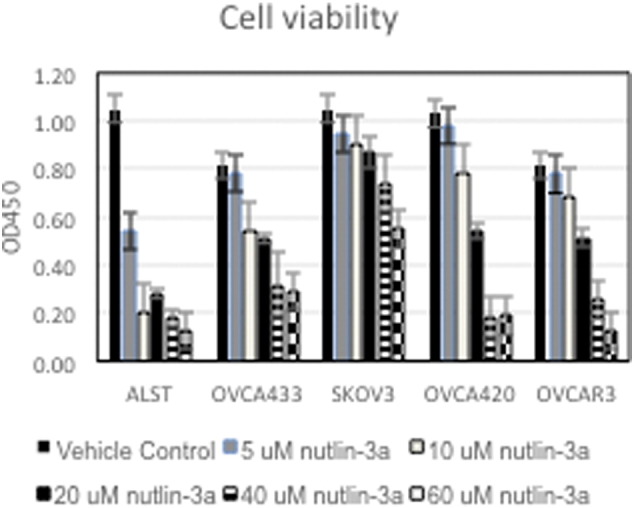
Human ovarian cancer cells were cultured with increasing doses (5, 10, 20, 40, 60 μM) of nutlin-3a for 24 hours. Cell viability was measured by the WST-1 assay as described in Methods. N = 3 replicates.
P53 Activity Measurement
P53 activity was measured in lysates of cells treated with DMSO or 10 μM nutlin-3a for 24 hours for the transfected Trp53(+) and Trp53(−) mouse tumor cell lines or for 24 hours of DMSO or nutlin-3a at the IC50 of each cell line (above) for the human cell lines. P53 activity was determined using the Cignal p53 Reporter (luciferase) Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions.
Western Blot Analysis
Cells were plated at a density of 20,000 cells per well in 24-well plates and transfected with 0.25 μg of plasmid DNA using Attractene Transfection Reagent (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Protein lysates were prepared in radioimmunoprecipitation assay buffer. Electrophoresis of lysates (10 μg for transfected and human cells and 25 μg for nontransfected cells to detect endogenous p53) was carried out on an 8% sodium dodecyl sulfate polyacrylamide gel and blotted with the following primary antibodies: P53 (1:1000 dilution, sc-6243; Santa Cruz Biotechnology, Inc, Santa Cruz, CA), CDKN1A/p21 (1:000, sc-397; Santa Cruz), and β-actin (1:1000 dilution, AAN01-A; Cytoskeleton, Inc, Denver, CO). Densitometry was performed using ImageJ v. 1.44 (Rasband, 1997 to 2012). β-Actin densitometry values were used to account for loading differences between lanes.
Sequencing for TP53 Mutations
DNA was extracted from three human cell lines according to manufacturer’s instructions using the Invitrogen Purelink Genomic DNA Mini Kit (Carlsbad, CA). DNA was amplified by PCR, and PCR products were then purified using the Invitrogen Purelink PCR Purification Kit. Exons 5 to 9 of TP53 were then sequenced for mutation analysis in all samples via Sanger Sequencing at the MD Anderson Sequencing and Microarray Facility using the BigDye Terminator v3.1 Cycle Sequencing Kits and the 3730xl DNA Analyzer (Applied Biosystems, Carlsbad, CA). Sequences were then analyzed using both Finch TV v1.3.1 and Lasergene SeqMan Pro. The negative control cell line SKOV3 did not possess any mutations in exons 5 to 9 but is known to contain a single nucleotide deletion in exon 4. The absence of p53 protein and transcript expression in Western blot and quantitative RT-PCR confirmed the absence of p53 activity; we therefore did not resequence SKOV3 exon 4.
Quantitative Real-Time RT-PCR
RT-PCR was performed as previously described [15]. Data are presented as the mean ± SEM of at least three experiments performed in triplicate. Differences among groups were analyzed by Student's t test. *P < .05 was considered statistically significant.
Evolutionary Impact Model
To estimate the functional impact of p53 mutations in human ovarian carcinoma tissue, we used the Evolutionary Action (EA) method [27]. The EA method models the genotype-to-phenotype relationship to derive an equation for the mutation impact, which equals to the product of the functional importance of the mutated residue and of the amino acid similarity of the substitution. These two terms are approximated by Evolutionary Trace ranks and substitution matrices, respectively. The EA scores were validated, among others, on a retrospective data set of TP53 mutants tested for transactivation activity in yeast, resulting in a nearly linear correlation. In addition, EA was found to estimate the functional impact of cystathionine-beta-synthase mutations and the clinical impact of CDKN2A (p16) mutations better than current state-of-the-art methods at the international contests organized by The Critical Assessment of Genome Interpretation in 2011 and in 2012 to 2013, respectively (https://genomeinterpretation.org/).
TP53 Genomic Status Analysis
As described previously [33], DNA copy number aberrations were determined using TCGA level 1 Affymetrix SNP6.0 data, which were processed by Nexus Copy Number version 7.5 software (BioDiscovery, Inc, Hawthorne, CA) with allele-specific copy number analysis of tumors algorithm [34]. The frequency of each segmented region with DNA copy number aberration for each sample group with the p53 mutation at the same amino acid (e.g. R175) was compared with all the samples by Mann-Whitney U test to identify significant regions that are associated with specific p53 mutant. To identify potential upstream regulators for gene expression changes in samples with similar p53 mutations, gene expression profile analysis using RNAseq data from the TCGA data set was performed. Genes that are differentially expressed between the samples with specific p53 mutation and all other samples with missense p53 mutations were selected by simple t test (with Reads Per Kilobase Per Million mapped reads [RPKM] > 0.1 and P value < .05). The list of genes was then uploaded into Ingenuity Pathway Analysis for upstream regulator analysis as described [35]. The TP53 status of the 316 high-grade serous TCGA samples has been described previously [33]. The TP53 status of additional ovarian cancer samples was retrieved using the Catalogue of Somatic Mutations in Cancer database [36].
Results
Identification TP53 Mutants Selectively Expressed in HGSC and Their Relation to LOH, Copy Number Variation (CNV) and Specific Regulatory Pathways
Recent analysis of TCGA data has revealed that the TP53 gene is mutated in the vast majority (83%) of high-grade ovarian cancers [3]. To better understand how these mutations influence p53 function in ovarian cancer, we applied the recently described EA method [27] for ranking the functional impact of missense mutations upon protein function [28]. We calculated the EA scores for 233 different missense mutations found in a set of 464 ovarian tumor samples from the TCGA and plotted their distribution (Figure 1A). Compared with the background distribution of missense mutations found in other genes, the mutations observed within p53 are clearly different (Kolmogorov-Smirnov one-tailed test P value = 10− 96). The strong bias of these mutations toward high action indicates a strong selection to impact p53 functions overall.
Figure 1.

P53 mutant expression plasmid vectors were created for mutants in serous ovarian carcinomas with high evolutionary impact scores. (A) Calculated biological impact data (EA score) for p53 missense mutations in ovarian tumor samples (data from TCGA). Out of 464 samples, 83% had p53 mutations (385). Of these, 233 were missense mutations (black and gray) and 99 are unique (black). (B) All p53 hot-spot mutations in ovarian tumor samples (TCGA) are predicted to have biologic function. One hundred ten ovarian tumors have 1 of the 14 hot-spot mutations defined as mutations that appear in more than 1% of the ovarian tumors in TCGA. (C) All of the hot-spot mutations are located in the DNA-binding domain.
Next, we focused our analysis on the frequently occurring p53 mutations (observed in more than 1% of the ovarian tumor samples). Compared with all ovarian tumor p53 mutants, this subset has a statistically significant shift in its distribution (Kolmogorov-Smirnov one-tailed test = .018) away from total loss of function, suggesting that these frequently occurring substitutions maintain some p53 function (Figure 1B). The frequency of these hot-spot mutations within TP53 was higher in ovarian tumors than in nonovarian tumors for 10 of the 14 mutations and was distributed equally into conformation and DNA binding mutants (Table 1A) (Figure 1C).
Table 1A.
Frequency of Hot-Spot Mutations in Ovarian Tumors and H179R and C277F Mutations Which Occurred Less Frequently
| Mutation | Ovarian Cancer Frequency | Other Cancer Frequency | Ratio |
|---|---|---|---|
| R175H* | 3.90% | 3.65% | 1.07 |
| R273H* | 3.12% | 2.68% | 1.16 |
| Y220C* | 2.60% | 1.67% | 1.56 |
| R248Q | 2.60% | 3.26% | 0.80 |
| I195T* | 2.34% | 0.57% | 4.09 |
| R273C | 2.08% | 3.96% | 0.53 |
| V157F | 1.82% | 0.75% | 2.43 |
| R248W* | 1.82% | 2.02% | 0.90 |
| R282W | 1.56% | 2.33% | 0.67 |
| C176Y* | 1.56% | 0.48% | 3.22 |
| S241F* | 1.30% | 0.40% | 3.28 |
| Y163C | 1.30% | 0.66% | 1.97 |
| H193R* | 1.30% | 0.84% | 1.56 |
| G245D | 1.30% | 0.44% | 2.95 |
| H179R* | 0.78% | 1.01% | 0.77 |
| C277F* | 0.26% | 0.18% | 1.48 |
Additional analyses of the TCGA data sets revealed that although some high-grade ovarian cancers exhibit LOH (~ 31.3%; Table 1B), this percentage is less than anticipated or assumed. In fact, there are a significant number of tumors that are diploid and express one WT and one mutant allele (~ 24%). Among the mutants overrepresented in ovarian cancer, the R273H(9)/R273C(6)/R273L(2)/R273P(1) mutation uniquely has a strong association with heterozygosity (6 out of 18 tumors; P < .0158) compared with other mutants, including the most frequently occurring R175H mutation (2 out of 5; P < .193). In addition, ovarian cancer is associated with extensive CNV [2]. When CNV of tumors was assessed in relation to specific p53 mutations, the analyses revealed that tumors with the TP53p.R175H mutation (8 of 8; 100%) uniquely exhibited a high frequency of copy number loss on chromosome 9 (9q22.1-9q31.1) compared with all other p53 mutants (Figure 2). This region contains many DNA repair and tumor suppressor genes, miRNAs, and RNA binding proteins, indicating that their loss somehow confers a selective survival advantage in the R175H mutant tumor cells (Suppl. Table 1). These results also indicate that the tumors with the R175H mutant are different from others [11].
Table 1B.
Frequency of TP53 Allele Heterozygosity (TCGA, N = 316)
| Copy Loss/LOH |
No Copy Loss/Diploid |
|
|---|---|---|
| Mutation status | N (%) | N (%) |
| WT | 28 (8.9%) | 17 (5.4%) |
| Missense | 99 (31.3%) | 76 (24%) |
| Other mutation | 53 (16.8%) | 37(11.7%) |
| 2 mutant alleles | NA | 6 (1.9%) |
Figure 2.

Analyses of p53 mutants associated CNV. TCGA tumors with the TP53p.R175 (N = 8) mutant had higher frequency of 9q22.1-q31.1 copy number loss than tumors with all other TP53 mutations (N = 171) and genes associated with this region (Suppl. Table 1).
High-grade serous ovarian cancer is also uniquely associated with specific upstream signaling pathways [7], and thus we hypothesized that these pathways might also be related to specific p53 mutations. Further analyses revealed that several unique and commonly activated pathways are strongly associated with p53 mutation status (Table 2, A and B; Suppl. Table 2). Of note, R175 has more unique pathways associated with it than R273, perhaps reflecting the more complex genomic mutational variance in the R175 mutants compared with the R273 mutants that are more likely to be heterozygous for the WT allele (see above). The unique presence of the KMT2A methyltransferase in the R175 tumors indicates that specific epigenetic signatures likely occur in these tumor cells [37] that may involve the expression of specific HOX genes [38], [39], [40], [41]. In addition, the R175 mutant is associated with immune-related genes and HOX genes, indicating that it is related to both the immune and mesenchymal signature subtypes [42]. The H179 mutant is linked to TGFβ1 signaling and many inflammatory/immune-related genes, indicating that it is perhaps specifically associated with the immune ovarian cancer subtype [42]. The R248 mutant is highly enriched for stem-like factors SOX2, TBX2, and TGFB2 [43], [44] as well as chemokines, cytokines, and MMP signaling. Intriguingly, R179, R248, R175, and S241 all share high activation scores for SMARCA4, a member of the SWI/SNF chromatin-remodeling complex [45], [46], and TBX2 [43], [47]. Several mutants appear linked to retinoic acid signaling that is known to impact the expression of HOX family genes [48], which can mediate many tumor cell behaviors including metastasis [40], [41]. The I195T mutant is strongly associated with estradiol actions as well as with the transcription factors GATA4 and TBX5 [43]. Distinctively, tumors expressing the I195, Y220, and S241 mutants do not show immune-associated factors and are different from each other and the R179, R174, and R248 tumors. Thus, P53 mutant status is clearly related to distinct regulatory pathways, the most notable of which appear to be associated with the immune, TGF, and HOX related genes and tumor subtypes [39], [40], [41], [42].
Table 2A.
Specific p53 mutants that are frequently present in ovarian cancer are associated with specific regulatory molecules and pathways
| Mutation | Upstream Regulator | Molecule Type | Activation Z-Score |
|---|---|---|---|
| R175 | SMARCA4 | transcription regulator | 3.317 |
| P38 MAPK | group | 3.13 | |
| dexamethasone | chemical drug | 3.035 | |
| OSM | cytokine | 2.699 | |
| D-glucose | chemical - endogenous mammalian | 2.527 | |
| tretinoin | chemical - endogenous mammalian | 2.461 | |
| HNF1A | transcription regulator | 2.449 | |
| CREBBP | transcription regulator | 2.425 | |
| CTNNB1 | transcription regulator | 2.425 | |
| butyric acid | chemical - endogenous mammalian | 2.425 | |
| KMT2A | transcription regulator | 2.386 | |
| PPIF | enzyme | 2.236 | |
| CREB1 | transcription regulator | 2.234 | |
| INHBA | growth factor | 2.219 | |
| isoproterenol | chemical drug | 2.207 | |
| TO-901317 | chemical reagent | 2.187 | |
| alitretinoin | chemical drug | 2.186 | |
| estrogen | chemical drug | 2.183 | |
| Pka | complex | 2.166 | |
| CXCL12 | cytokine | 2.138 | |
| CEBPB | transcription regulator | 2.101 | |
| IL1A | cytokine | 2.083 | |
| tributyrin | chemical drug | 2 | |
| PTF1A | transcription regulator | 2 | |
| mycophenolic acid | chemical drug | 2 | |
| R273 | tretinoin | chemical - endogenous mammalian | 2.252 |
| decitabine | chemical drug | 2.415 | |
| Y220 | dexamethasone | chemical drug | 2.611 |
| tretinoin | chemical - endogenous mammalian | 2.247 | |
| R248 | tretinoin | chemical - endogenous mammalian | 3.726 |
| JUN | transcription regulator | 3.252 | |
| Cg | complex | 3.11 | |
| lipopolysaccharide | chemical drug | 3.036 | |
| IL1 | group | 2.914 | |
| SMARCA4 | transcription regulator | 2.789 | |
| HGF | growth factor | 2.772 | |
| OSM | cytokine | 2.626 | |
| EP300 | transcription regulator | 2.621 | |
| Vegf | group | 2.592 | |
| F2 | peptidase | 2.588 | |
| I195 | beta-estradiol | chemical - endogenous mammalian | 4.137 |
| Cg | complex | 3.256 | |
| GATA4 | transcription regulator | 3.237 | |
| CTNNB1 | transcription regulator | 3.119 | |
| MEF2C | transcription regulator | 2.931 | |
| TBX5 | transcription regulator | 2.923 | |
| estrogen | chemical drug | 2.905 | |
| SIM1 | transcription regulator | 2.828 | |
| ARNT2 | transcription regulator | 2.828 | |
| trichostatin A | chemical drug | 2.76 | |
| cyclic AMP1 | chemical - endogenous mammalian | 2.749 | |
| H179 | TGFB1 | Growth factor | 8.05 |
| Lipopolysaccharide | Chemical drug | 7.019 | |
| EGF | Growth factor | 6.497 | |
| Vegf | Group | 6.44 | |
| CSF2 | Cytokine | 6.392 | |
| SMARCA4 | Transcription regulator | 6.229 | |
| ERK1/2 | Group | 6.008 | |
| Phorbol myristate acetate | Chemical drug | 5.872 | |
| ERK | Group | 5.852 | |
| F2 | peptidase | 5.828 | |
| S241F | SMARCA4 | transcription regulator | 2.778 |
| pirinixic acid | chemical toxicant | 2.369 | |
| benzo(a)pyrene | chemical toxicant | 2.158 | |
| B | |
|---|---|
| P53 mutant | Genes uniquely associated with upstream regulatory pathways related to each p53 mutant |
| Immune-related | |
| R175: | CCL24, CXCL3, CD70, CYP24A1, MMP13, PPARG, TNFSF11 |
| H179: | CCL17, CCL18, CCL2, CCL25, CCL3, CD14, IL10, IL16, IL1B, MMP13 |
| R248: | CXCL1, CXCL13, CXCL5, CXCL6 HOXA5, HOXA9, MMP13, SOX2, TBX2, TGFB2 |
| Others | |
| R273: | HOXA3, HOXA5, HOXA9, MAGEA1, MAGEA11, MAGEC2 |
| Y220: | COL11A2, FOXD1, GCK, MIA, MMP13, NAT8B, NPY, SLC5A5, TRIM31 |
| I195: | GATA4, GDF10, INHA, OXTR, PDYN, PGR, SCARB1, STAR, TIMP4, WNT4 |
| S241: | CYP1A1, CYP1A2, CYP4A11, CYP8B1, HNF1B, HPX, LCN2, POPDC3, SLC23A1 |
To assess the expression of selected upstream pathway genes, five human cell lines in which p53 status has been determined were analyzed: 1) ALST cells express WT TP53, 2) OVCA433 cells express heterozygous alleles (E258K/WT), 3) OVCA420 cells are homozygous for the R273H mutant allele (R273H/R273H), 4) OVCAR3 cells are homozygous for the R248Q allele (R248Q/R248Q), and 5) SKOV3 cells are TP53 null cells with a homozygous deletion (C267) leading to a frameshift mutation (Suppl. Table 3) [31]. Analyses of these cell lines confirmed that expression levels of TBX2, FOXA2, HOXA5, HOXA9, PPARG, and SMARCA4 were distinct for each cell line and p53 mutant status (Figure 3). Expression of HOXA5 and HOXA9 but not PPARG confirms gene expression patterns noted in R248 tumors but not for R273 tumors (Table 2B; Suppl. Table 2). Collectively, these analyses show 1) that specific TP53 mutants in ovarian cancer are associated with distinct functional characteristics including changes in CNV, LOH, and specific upstream regulatory pathways, the most notable of which are immune and HOX gene related, and 2) that some mutants are preferentially present with a WT p53 allele, representing a new significant subtype of ovarian cancer in which the functional status of the WT allele and the mutant allele needs to be analyzed to determine if therapeutic drugs that target p53 by either deletion of the mutant p53 or activation of WT p53 are effective in this heterozygous tumor type.
Supplemental Table 3.
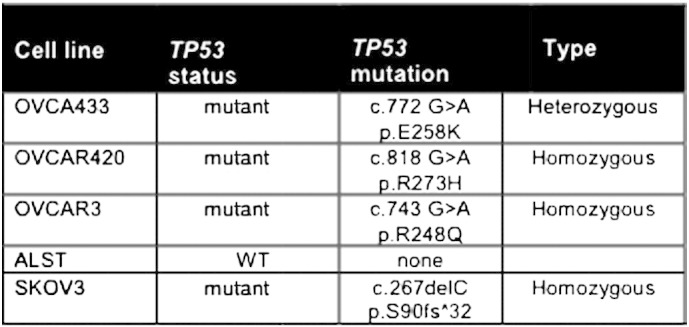
This table shows the TP53 mutations that are expressed in each human ovarian cancer cell line and that have been sequenced and verified for these studies.
Figure 3.

Expression levels of genes selectively associated with specific p53 mutations in human ovarian cancer cell lines. The expression of selected genes associated with specific p53 mutant up-stream regulatory pathways (Suppl. Table 2) in human ovarian cancer (TCGA data sets) was analyzed in human ovarian cancer cell lines with known p53 status (WT, heterozygous, null, or homozygous mutant). Specific genes were selectively expressed in each cell line (*P < .05 compared with ALST cells, N = 3).
Expression levels of genes selectively associated with specific p53 mutations in human ovarian cancer cell lines. The expression of selected genes associated with specific p53 mutant up-stream regulatory pathways (Suppl. Table 2) in human ovarian cancer (TCGA data sets) was analyzed in human ovarian cancer cell lines with known p53 status (WT, heterozygous, null, or homozygous mutant). Specific genes were selectively expressed in each cell line (*P < .05 compared with ALST cells, N = 3).
Sensitivity of Human Ovarian Carcinoma Cells to Activation and/or Overexpression of WT TP53 or Knockdown of Endogenous WT or Mutant TP53
To determine if p53 status (WT, heterozygous, null, or mutant) in human ovarian cancer cell lines determines their expression levels and responses to nutlin-3a, cells were exposed to nutlin-3a alone without (Figure 4A) or with exogenous WT p53 (Figure 4C). All cell lines, with the exception of the SKOV3 cells, expressed detectable basal levels of p53 protein in the absence of nutlin-3a. However, the basal levels of TP53 protein were higher in the two homozygous mutant cell lines where increased stability is known to occur [8], [10] (Figure 4, A and C). Nutlin-3a increased endogenous WT TP53 protein in ALST and OVCA433 cells but not in null or homozygous mutant cells (Figure 4A). Nultin-3a increased p53 promoter luciferase reporter activity in the OVCA433 cells, whereas the elevated TP53 activity in the ALST cells was not further increased by nutlin-3a (Figure 4B). Cells lacking TP53 or containing only mutant TP53 were nutlin-3a resistant as expected and exhibited either no TP53 protein or no further increase in protein, respectively, and no increase in p53 activity (Figure 4, A and B) [14].
Figure 4.
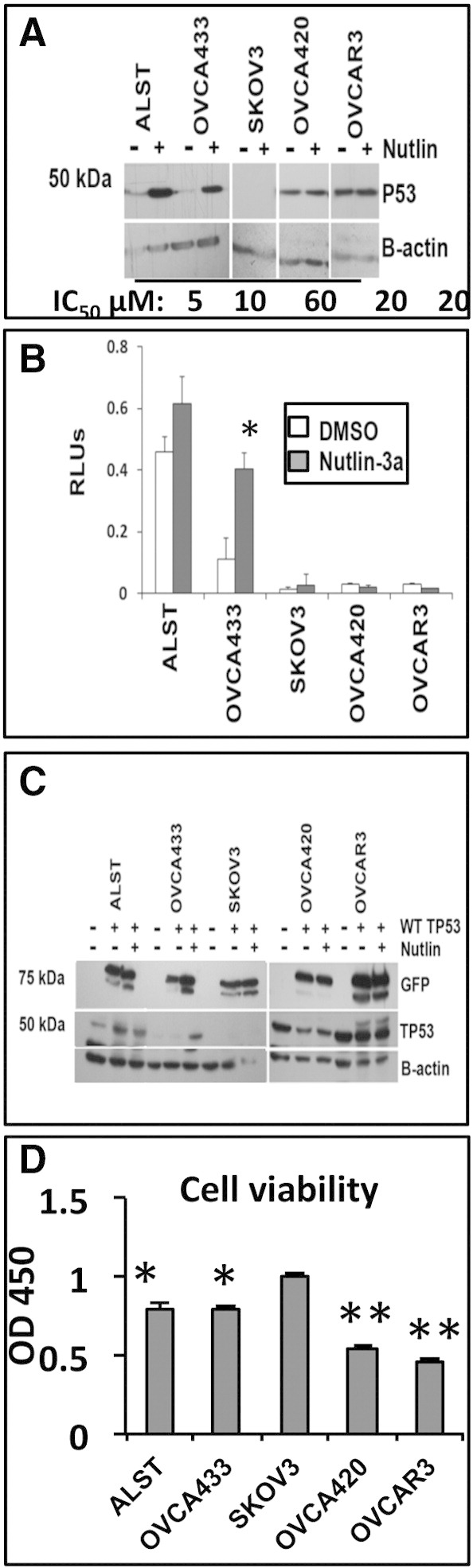
Sensitivity of human ovarian carcinoma cells to nutlin-3a is altered by the presence of endogenous and exogenous WT TP53. (A) Western blot of lysates from human cell lines treated with nutlin-3a at the IC50 for each cell line. (B) TP53 activity (luciferase assay) was measured in lysates of cells treated with DMSO or nutlin-3a at their predetermined IC50. (C) Western blots of GFP-tagged transfected WT TP53 and endogenous TP53 in cells treated with DMSO or nutlin-3a at the IC50 for each cell line. (D) Viability of cells transfected with empty vector or WT TP53. All assays were run at least three times; Western blots are representative of three experiments.
Consistent with the known role of highly activated p53 in mediating apoptosis, the ALST and OVCA433 cells that express WT TP53 were more sensitive (IC50 = 5 and 10 μM, respectively) to low doses of nutlin-3a in a cell viability assay than cells with no p53 (IC50 = 60 μM) or mutant p53 (IC50 = 20 μM) (Figure 4, A; Supplemental Figure 1). Collectively, these results show that basal activity of the WT p53 allele is reduced in the presence of the mutant allele but that the WT allele can be activated by nutlin-3a in this context, leading to apoptosis. Cells that are null for TP53 or that express mutant TP53 are more resistant to nutlin-3a. Although these data support our hypothesis that WT TP53 can be activated in the presence of a mutant allele, results from one human TP53 heterozygous cell line are not sufficient to make a general conclusion.
Therefore, to test our hypothesis further, we next introduced WT TP53 into the human cell lines using a vector expressing GFP-tagged WT TP53 (Figure 4C). GFP-WT TP53 protein was highly expressed in all cell lines and did not have a significant effect on endogenous TP53 protein levels in the cell lines analyzed. Western blot analyses showed that WT p53 increased the levels of CDNK1 (P21) protein, a known target of p53, in all cell lines including those expressing homozygous mutant p53, indicating that the WT p53 is active in the presence of the mutant alleles. Nutlin-3a reduced p21 protein levels, indicating that either higher levels of p53 activity are inhibitory or nutlin-3a exerts unknown effects on p21 protein stability (Supplemental Figure 3A). Cell viability assays showed that exogenous WT TP53 increased cell death in each cell line except the SKOV3 p53 null cells (Figure 4D). Moreover, we noted that the cell lines OVCA420 and OVCAR3 with homozygous mutant alleles exhibited significantly more rapid and extensive cell death (P < .001) compared with those (ALST and OVCA533) with WT alleles. Thus, WT TP53 maintains p53 activity in the presence of mutant protein, even in the absence of nutlin-3a; adding nutlin-3a had no further effect on p53 activity and even reduced p21 protein levels (data not shown). These results support and confirm observations in mouse models [49].
Supplemental Figure 3.

WT TP53 and p53 depletion with siRNA alters endogenous WT and mutant p53 protein levels and p21 protein levels in human and mouse ovarian cancer cell lines. (A) Human cancer cell lines were transfected with WT p53 and treated without or with nutlin-3a for 24 hours. Cell lysates were prepared, and Western blots were done to analyze the levels of p21 protein. Note that P21 is increased by WT p53 in all cell lines and is reduced by nutlin-3a by mechanisms not yet understood. (B) Human cells were treated with 20 nM siRNA for 24 hours. Lysates were prepared, and Western blots were done to determine the relative reduction of cellular levels of p53 and p21 in each sample. Note that SKOV3 cells lacking p53 do not express p21 and that OVCA420 and OVCAR3 cells expressing homozygous mutant alleles express low levels of p21 protein. (C–D) Mouse ovarian cancer cells expressing WT p53, heterozygous alleles (WT/R172H), or homozygous alleles (R172H) were treated with p53 siRNA at 0, 20, and 50 nM or 50 nM, respectively, for 72 hours. Note that the mouse cell lines are less sensitive to p53 siRNA knockdown of p53 and p21. Representative of 3 separate experiments.
WT p53 Activity by Mutant p53 in Mouse Ovarian Cancer Cell Lines
Because only two human cell lines that we know of are heterozygous for a WT and mutant allele and because the mutant allele in the cell line available to us is not one highly overrepresented in ovarian cancer, we next sought to determine the impact of specific p53 mutants on tumor cell behaviors in the presence and absence of WT p53. Expression vectors containing GFP-tagged WT p53 and 10 mutants were created to study the impact of specific p53 mutants (asterisk) that are present in the DNA-binding domain (Figure 1C) that lead to either conformation changes or lack of binding. Mutants were selected based on 1) their high frequency of expression in ovarian cancer (R175H, R273H, Y220C) (Figure 1) [9], 2) their high frequency of expression as heterozygous alleles in ovarian cancer (R175H, R273H, R248W) (Table 1A and B), 3) their overrepresentation in ovarian cancer (I195T, S241F, C176Y, Y220C, H193R, and C277F) (Figure 1), and 4) those occurring in human ovarian cancer cell lines OVCAR3 (R248Q) and OVCA420 (R273H) (Suppl. Table 3) that are used routinely for studying ovarian cancer. These vectors expressing WT TP53 and specific TP53 mutants were transfected into our mouse epithelial ovarian cancer cell lines that express WT p53 (Trp53(+) or are null for Trp53) [14]. Endogenous WT TRP53 protein is expressed at low levels in the Trp53(+) cells (Suppl Figure 1A) as reported previously [14]. The expression levels of GFP-WT TP53 and most GFP-mutant TP53 proteins were elevated above those in control cells and relatively similar in Trp53(+) and Trp53 null cells (Suppl Figure 1B) as determined by Western blotting with GFP and p53 specific antibodies.
To determine the relative functional activity of WT p53 alone and in the presence of TP53 mutants, we cotransfected a p53 consensus promoter-luciferase reporter construct with WT or mutant TP53 expression vectors. Luciferase activity was measured (Figure 5A) and normalized to protein levels (Supplemental Figure 2, A–C). The activity of transfected WT TP53 in the Trp53 null cells was lower than that of the C227F mutant. However, basal p53 activity was significantly lower than that of WT in the presence of other mutants (Figure 5A). Importantly and as predicted from our computational (Figure 1), gene expression (Table 2&B; Figure 3), and copy number analysis (Figure 2; Suppl. Table 1), p53 activity was significantly different in the presence of specific mutants compared with each other. Notably, p53 activity was higher in the presence of R175H, C176Y, I195T, and S241F, the latter two of which are uniquely expressed in ovarian cancer cells. When p53 activity was tested in cells expressing endogenous WT p53, basal activity was highest in the presence of exogenous WT p53 and the C277F mutant but was reduced in the presence of mutant p53. However, two mutants overrepresented in ovarian cancer (Y220C and S241F) and one frequently expressed as a heterozygous allele (R248W) showed higher activity than the other mutants, indicating that these mutants exerted less dominant negative effects on endogenous WT p53 than the other mutants. These data show that specific TP53 mutants overrepresented in ovarian cancer differentially exhibit some WT p53 activity in the p53 null cells and suppress endogenous WT p53 activity in the p53 + mouse cell lines and support the observation that basal p53 activity was lower in the heterozygous OVCA433 human cell (Figure 4B).
Figure 5.
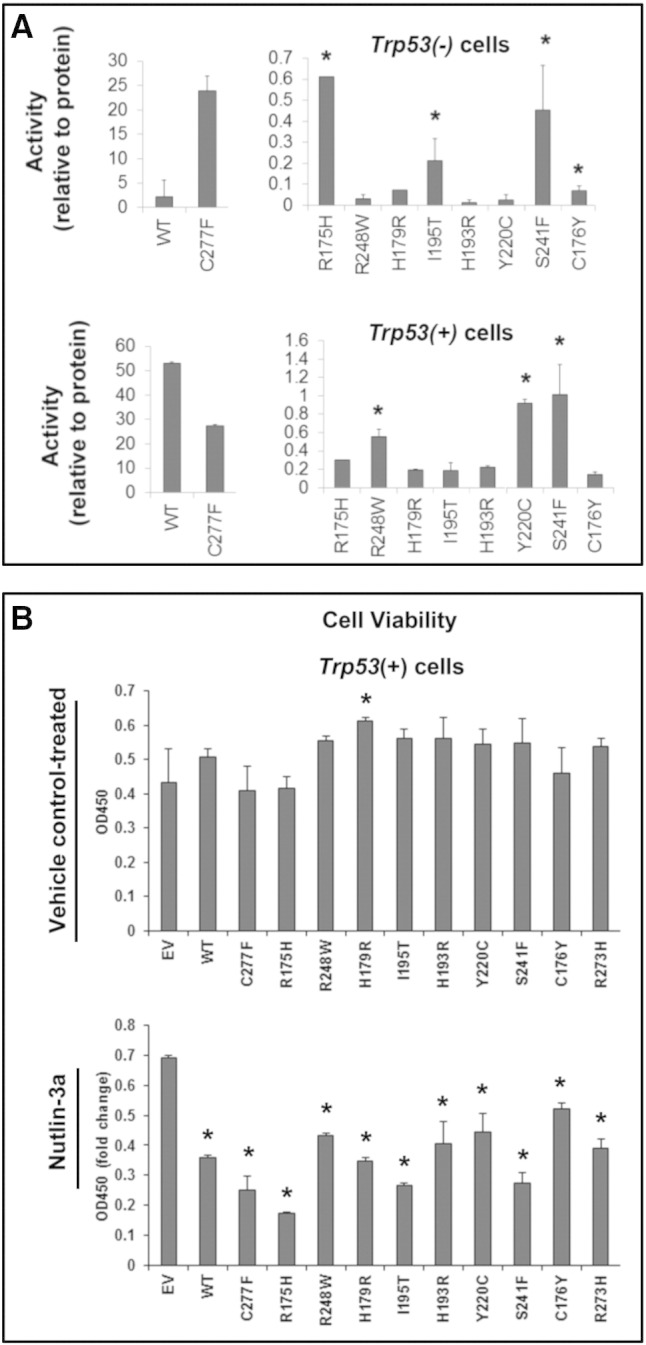
Mutant TP53 activity and nutlin-3a–induced activation of TP53 is gene and cell context specific. (A) P53 (luciferase) activity (left and right panels) was measured in Trp53(−) and Trp53(+) cells following transfection with vectors expressing different TP53 mutant proteins (*P < .05, N = 3). Luciferase activity was expressed relative to protein levels for each mutant as determined by Western blot and densitometry analyses (Suppl. Figure 2, A–C). (B) Cell viability was measured in Trp53(−) and Trp53(+) cells following transfection with vectors expressing different TP53 mutant proteins and treatment without or with 10 μM nutlin-3a (*P < .05, N = 3).
Mutant TP53 activity and nutlin-3a–induced activation of TP53 is gene and cell context specific. (A) P53 (luciferase) activity (left and right panels) was measured in Trp53(−) and Trp53(+) cells following transfection with vectors expressing different TP53 mutant proteins (*P < .05, N = 3). Luciferase activity was expressed relative to protein levels for each mutant as determined by Western blot and densitometry analyses (Suppl. Figure 2, A–C). (B) Cell viability was measured in Trp53(−) and Trp53(+) cells following transfection with vectors expressing different TP53 mutant proteins and treatment without or with 10 μM nutlin-3a (*P < .05, N = 3).
Supplemental Figure 2.

Mutant p53 stability and activation depend on TP53 mutation status in Trp53(+) and Trp53(−) cells. (A) Western blot of endogenous TRP53 protein in lysates of Trp53(+) and Trp53(−) cells. Twenty-five micrograms of cell lysate was analyzed to detect low-level endogenous TRP53 protein. (B) Western blot analysis of GFP and TP53 protein expression in 10 μg of lysate from Trp53(+) and Trp53(−) cells transfected with GFP-tagged mutant TP53 expression plasmid vectors (~ 77 kDa). (C) Average densitometry analysis of TRP53 protein in lysates of cells treated with DMSO or nutlin-3a for 24 hours (N = 3) as analyzed by Western blot. Some mutant TP53 protein was more stable compared with WT (*P < .05), and the stability of some mutants was increased or decreased by nutlin-3a compared with untreated (**P < .05).
To determine the effects of p53 mutants on cell viability, additional cultures with mouse cells expressing endogenous WT p53 were transfected with WT or specific mutant p53 and treated with or without nutlin-3a (Figure 5B). In cells expressing WT p53, the introduction of WT or the p53 mutants did not lead to a major difference in cell viability, with the exception of R179H in which cell viability was significantly higher that WT. However, when the cells were exposed to nutlin-3a that maximally activates WT p53, cell viability was reduced significantly when exogenous WT p53 was present. Specific mutants (C277F, R175H, I195T, and S241F) caused even greater decreases in cell viability in the presence of nutlin-3a, indicating that they may potentiate the effects of endogenous p53 activity or have apoptotic functions of their own in cells where WT p53 is activated. These novel data support the enhanced apoptosis mediated by WT p53 when introduced into human cell lines expressing homozygous mutant alleles (Figure 4D).
Diverse WT p53 Regulated Gene Expression in Response to P53 Mutants
Human cells
These results demonstrating differential effects of mutant TP53 on WT p53 activity and cell viability led us to analyze the expression of specific p53-target genes related to cell cycle arrest and apoptosis in the human cell lines (Figure 6) [14]. CDKN1A mRNA is expressed in ALST cells that have high levels of endogenous WT TP53 and is induced significantly by nutlin-3a. Expression of exogenous WT TP53 and treatment with nutlin-3a further increased the CDKN1A mRNA in the ALST cells. Nutlin-3a also increased CDKN1A mRNA in the OVCA433 cells. Although exogenous WT p53 and nutlin-3a increased CDNK1A mRNA expression, the induction by nutlin-3a was less than that observed in the absence of exogenous WT p53. CDKN1A was also increased following expression of WT TP53 in cells that are TP53 null or contain homozygous TP53 mutations. However, the levels of CDKN1A mRNA were low, and nutlin-3a did not further increase in CDKN1A expression relative to that induced by WT TP53 alone. The expression pattern of MDM2 in these cells was similar to that of CDKN1A. MDM2 was induced by nutlin-3 in the ALST cells, but expression of WT TP53 did not increase this response further. MDM2 mRNA was also increased by the introduction of WT TP53 in the TP53 mutant cell lines, but the levels were low and the response of these cells to nutlin-3a was blunted or absent. These results show that expression of CDNK1A and MDM2 are elevated in cells expressing one or two copies of the endogenous WT allele and that cells null for p53 or that express GOF mutants fail to express either mRNA. Because MDM2 is low in homozygous GOF mutants, it is surprising that exogenous WT p53 did not induce higher expression of these genes, indicating that mechanisms to restrict their activity have been imposed. However, because MDM2 is low, it is not surprising that nutlin-3a had no effect.
Figure 6.

The sensitivity of human ovarian carcinoma cells to WT TP53 reintroduction correlates with p53 status. RT-PCR was performed on lysates from human cell lines expressing different p53 proteins (ALST:WT, OVCA433:WT/E248K, SKOV3:null, OVCA420:R273H, and OVCAR3:R248Q) following transfection with WT TP53 for 24 hours (*P < .05, N = 3).
Because WT p53 dramatically reduced cell viability in the OVCA420 and OVCAR3 cells compared with the ALST, OVCA433, and SKOV3 cells and because cell viability was not strictly related to the levels of p21 mRNA (Figure 6) or protein (Suppl. Figure 3A), we sought to determine what mechanisms other than, or in addition to, cell cycle arrest might mediate these effects. Therefore, we analyzed the expression of genes that mediate apoptosis (PUMA) and caspase 3 (CASP3) as well as a factor (DNAJC3) involved in the unfolding protein response (UPR) that has been shown to increase in response to specific drugs [50]. As shown (Figure 6), PUMA was induced markedly in the null and mutant cells lines but not in the cell lines expressing WT p53, indicating that WT p53 can induce this apoptotic related factor in the presence of GOF mutants. The response to nutlin-3a may be nonspecific because MDM2 levels are low. Surprisingly, CASP3 is not induced in the mutant cells but is increased in the heterozygous cell line. The marked increase in DNAJC3 suggests that the exogenous WT p53 impacts the UPR in the presence of the homozygous mutants.
Mouse cells
To determine if WT TP53 had a similar effect on p53 activity in mouse ovarian cancer cells, WT p53 was introduced into our mouse ovarian cancer cell lines that are either homozygous or heterozygous for the p53 R172H mutation [15], which is equivalent to the human R175H mutation. As shown, Cdkn1a and Mdm2 expression levels were low and unresponsive to nutlin-3a in the absence of exogenous WT p53 but increased significantly following WT TP53 expression in both the p53 R172H heterozygous and homozygous cells and rendered these cells responsive to nutlin-3a (Figure 7). These results indicate that the mouse tumor cells are less sensitive to nutlin-3a but support those in the human cell lines, indicating that WT TP53 is active at low levels in the presence of mutant p53 in these mouse ovarian cancer cells (Figure 6).
Figure 7.
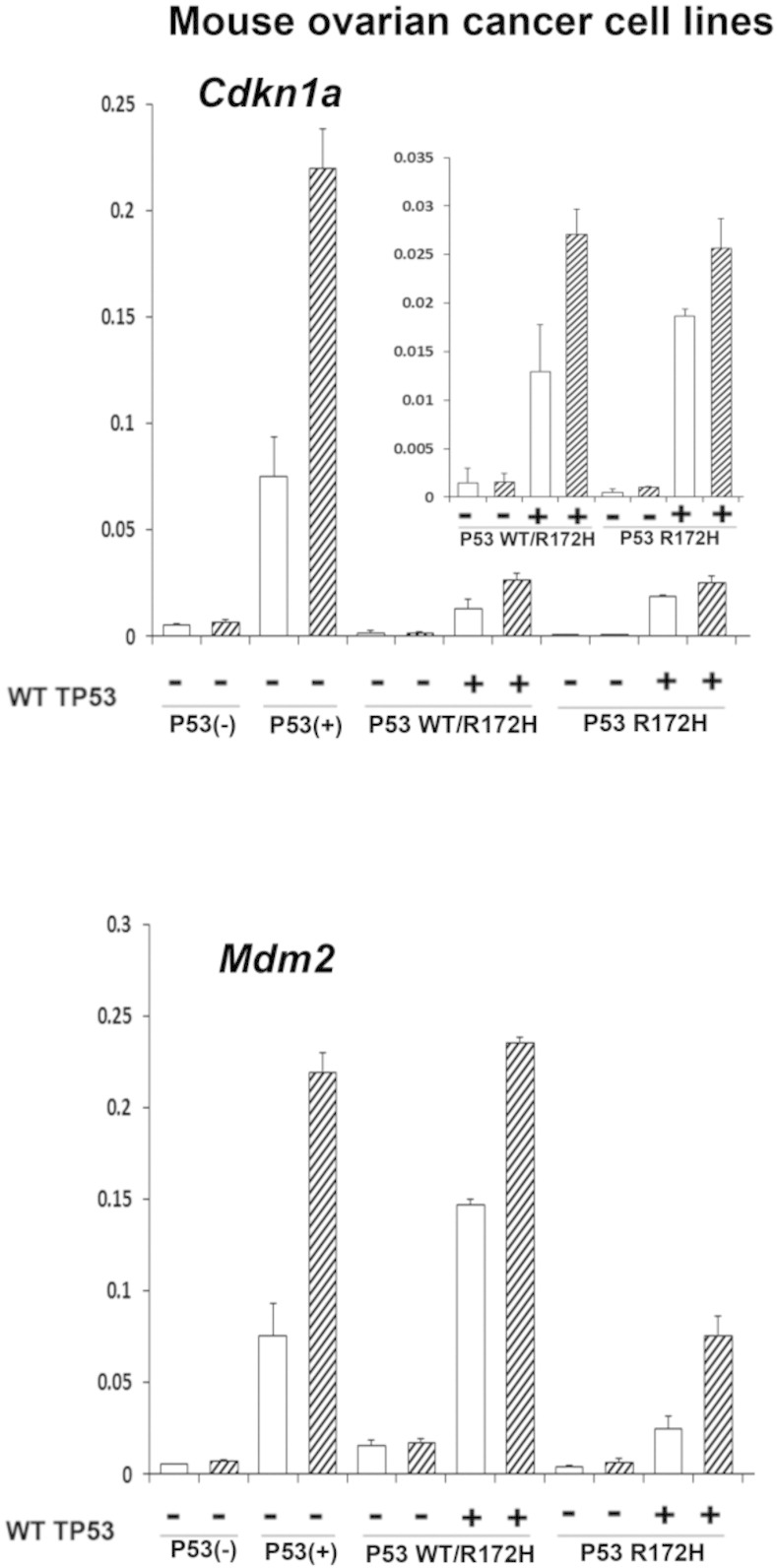
Sensitivity of mouse ovarian cancer cells to WT TP53 reintroduction correlates with p53 status. RT-PCR was performed on lysates from mouse cell lines (p53 null, p53 WT, P53 WT/R172H, P53 R712H/R172H) transfected with WT TP53 for 24 hours (*P < .05, N = 3).
To further investigate the impact of specific p53 mutants on endogenous WT p53 transcriptional activities, we analyzed the expression of selected p53 target genes (Cdkna1 and Hmgb2) in Trp53 + mouse tumor cells expressing either WT p53 or specific p53 mutants (Figure 8). Cdkn1a (p21) mRNA was elevated in the Trp53 + cells as reported previously [14]; expression of exogenous WT TP53 did not further increase in levels of p21 mRNA. Furthermore, expression R175H, R273H, and C176Y did not alter p21 mRNA levels in the Trp53 + cells, indicating that they did not impact the functional activity of endogenous WT p53 on this gene. However, R248W, H179R, and S241F mutants significantly decreased the expression of p21 mRNA, indicating that they exert dominant negative effects on endogenous p53 activity in these cells. Hmgb2 is a p53-suppressed gene encoding a chromatin-associated high mobility–group protein that plays a role in DNA unwinding [51]. WT, R175H, and S241F significantly repressed the expression of Hmgb2 mRNA, suggesting that these mutants retain some p53 activity on this gene in this context or possibly enhance the functions of endogenous p53 [8]. None of the mutants altered Mdm2 or Brca1 expression levels, indicating that the effects of mutants are gene specific (data not shown).
Figure 8.

P53 mutants differentially impact p53 target gene expression and responses to nutliin-3a in mouse Trp53(+) tumor cells. P53-responsive genes were analyzed by RT-PCR in untreated Trp53 + cells transfected with vectors expressing different TP53 mutant proteins (left panel). Nutlin-3a induction of p53-responsive genes was analyzed by RT-PCR following transfection of mutant TP53 vectors into Trp53(+) cells (right panel). Results in the nutlin-3a–treated cells are presented as the fold change of induction with 10 μM nutlin-3a for 24 hours compared with cells transfected with control empty vector alone (EV, lane 1; *P < .05, N = 3).
Nutlin-3a increased the levels of p21 mRNA three-fold in the Trp53 + cells (Figure 8; empty vector); this induction was not increased further by exogenous WT TP53 or R175H. However, nutlin-3a–mediated induction of p21 mRNA was enhanced significantly by several mutants (R248W, H179R, R273H, S241F, and C176Y), suggesting that they enhance endogenous WT p53 activation and/or stabilization in this context, perhaps by their high affinity for MDM2 and protecting WT p53 from degradation. Except for S241F, these mutants also enhanced the suppression of Hmgb2 in the presence of nutlin-3a. These data indicate that endogenous p53-mediated induction of p21 mRNA and suppression of Hmgb2 are maintained or even enhanced by specific p53 mutants. These results further support our hypothesis that, in the heterozygous ovarian cancer subtype, WT TP53 can be activated and its activation by nutlin-3a can even be enhanced, not suppressed, by specific mutants. These results support those observed in the heterozygous human cell lines and highlight the usefulness of nutlin-3a as a therapeutic agent for patients with heterozygous ovarian tumors.
Tumor Cell Functions Following Depletion of Mutant p53
Although nutlin-3a appears to be a useful and effective drug to activate WT TP53 and promote apoptosis of ovarian tumor cells (Figure 4) [14], [32], it does not impact ovarian tumor cells expressing homozygous mutants (Figure 4) which appear to have acquired specific mechanisms for maintaining cell survival [6], [8]. Therefore, we next determined if knocking down mutant TP53 using siRNA (targeting both WT and mutant p53) in the human and mouse cells lines would alter their cell survival–promoting functions. Following transfection of p53 siRNA, successful depletion of TP53 protein and its target gene p21 was verified in each cell line by Western blot analyses (Suppl Figure 3, B–D), and changes in cell viability were analyzed (Figure 9A). Specifically, human and mouse cells with WT p53 or those heterozygous for WT and mutant alleles or those that were p53 null were less sensitive to p53 siRNA treatment than were the homozygous TP53 mutant cell lines (Figure 9A). Moreover, the mouse cell lines were less sensitive to p53 siRNA knockdown of p53 and expression of p21 (Suppl. Figure 3, C and D). OVCAR3 cells expressing the R248Q mutant p53 were more sensitive to siRNA than all other cell lines, and almost all cells were dead by 48 hours. Human breast cancer cells with R175H (SK-BR-3) and L194F (T47D) mutant p53 were more sensitive than cells with WT p53 as previously described [26]. However, the breast cancer cells were more resistant than the ovarian cancer cells to siRNA depletion even at 50 nM p53 siRNA. These results indicate that the presence of homozygous R248Q, R273H, or R172H alleles confers a survival advantage and dependency on the mutant p53 protein compared with the other cell lines. To determine the impact of p53 knock down on gene expression in the human cells, RT-PCR was performed to measure p53 target genes, and genes involved in apoptosis and cell stress responses (Figure 9B). CDKN1A was decreased in ALST cells following silencing of WT p53 expression, whereas there was no change in cells with heterozygous mutant p53. By contrast, CDKN1A expression was increased by p53 siRNA in OVCA420 cells but to a lesser degree in OVCAR3 cells [52]. CASP3, a mediator of apoptosis, and DNAJC3, a component of the UPR stress pathway, were highly induced in response to p53 siRNA in OVCA420 (10- and 179-fold, respectively) and OVCAR3 (640- and 250-fold, respectively) but not in cells expressing WT p53. PUMA was increased in the OVCA420 cells but was low in the OVCAR3 cells, indicating that the cells homozygous for the R273H mutation differ from those with the R248Q mutation in this context. Furthermore, p53 siRNA reduced expression of the upstream regulatory factor SMARCA4 in cells expressing WTp53, had no effect in the p53 null cells, and may increase expression in the R273H homozygous mutant cells. When mouse cells homozygous for the R172H allele were exposed to p53 siRNA, Casp3 but not Cdkn1a expression was increased and is associated with decreased cell viability. Expression of Puma and Dnajc3 was low and not altered, indicating that other pathways appear to be activated in these cells, perhaps relating to the fact that the mouse cells exhibit less genomic instability and are not as susceptible to the UPR. As in the human cells, p53 siRNA reduced expression of Smarca4 in mouse cells expressing WT p53 but did not alter its expression in the R172H homozygous mutant cells (Figure 9B), indicating that Smarca4 may be a p53-regulated gene.
Figure 9.

Human and mouse ovarian cancer cells with WT or mutant p53 exhibit distinct responses to depletion of p53. Human and mouse ovarian cell lines were treated with siRNA at doses and times determined to reduce cell viability. Human and mouse ovarian cancer cell lines were transfected with 20 nm p53 siRNA. Human breast cancer cell lines that were resistant to 20 nm were transfected with 50 nM P53 siRNA. Cell viability and gene expression patterns were analyzed by methods described above. (*P < .05, N = 3).
Collectively, these data indicate that cells heterozygous for mutant and WT p53 respond to nutlin-3a and exhibit increased p53 activity, target gene expression, and apoptosis. Conversely, ovarian cancer cells expressing WT p53 can withstand acute removal of p53 and avoid rapid cell death. However, cells dependent on mutant p53 are highly sensitive to either the 1) the removal of the mutant p53 protein leading to a stress-induced cell death or 2) an increase in WT p53 and consequent activation of p53 mediated apoptosis, including increased p21 protein levels.
Discussion
High-grade epithelial ovarian cancer, unlike other epithelial cancers, is unique because of genomic instability and the vast number of TP53 mutations that occur in this disease. Hence, it has become imperative to determine how these mutants impact ovarian tumor progression and response to therapeutic drugs. Using evolutionary computer analyses, selected human ovarian cancer cell lines that are homozygous or heterozygous for specific mutants, and tumor cell lines derived from our mouse models expressing WT or mutant alleles, we document herein that 1) specific missense and GOF mutations that are predicted to have biologic function are overrepresented in ovarian cancer; 2) a subset of the most highly represented TP53 mutations in ovarian tumors is heterozygous for a WT and mutant TP53 allele and, hence, represent a specific ovarian cancer subtype not previously analyzed in detail; 3) the R273H mutant in particular is more strongly linked to heterozygosity than other mutants; 4) the R175H mutant is uniquely associated with CNV and allelic LOH on chromosome 9; 5) upstream pathways associated with specific TP53 mutants show distinct gene expression differences; 6) p53 activity (luciferase activity, target gene expression, and cell viability in response to nutlin-3a) in tumor cells expressing WT and mutant heterozygous alleles differs from that in cells homozygous for WT or mutant alleles; and 7) cells with homozygous mutant alleles are more sensitive to p53 siRNA than are cells with WT TP53 and exhibit specific apoptotic and UPR-related genes but also show distinct differences in the expression of specific genes associated with specific regulatory pathways.
Specifically, by interrogating the ovarian cancer TGCA data sets in the context of specific p53 mutations, we have revealed several novel findings relevant to TP53 mutations, their prevalence in ovarian cancer, and their links to CNV and heterozygosity. The data sets also show that upstream pathways [7] linked to specific TP53 mutations exhibit striking differences as well as a few similarities. For example, the R273H mutation has few upstream pathways that are unique, whereas the I195T mutations show high association with estrogen receptor signaling and the R175H mutation is linked to multiple pathways, including retinoic acid signaling. In addition, we show that several TP53 mutations, including R175H, R273H, Y220C, I195T, and S241F, are overrepresented in ovarian cancer and that each exhibits specific functional differences when expressed endogenously or exogenously in the tumor cells. In particular, human tumors with the R175H mutation are associated with a high frequency of CNV in a specific region of chromosome 9. That this occurs in eight of eight tumors analyzed suggests that LOH in this region of chromosome 9 somehow confers early survival advantages to these R175H-expressing tumors. Many of the 124 genes identified in the 9q22.1-33.1 region control DNA repair, cell cycle progression, or tumor suppressor functions; many others are microRNAs that can have global effects in cells.
The R273H mutation that is strongly linked to heterozygosity (four of nine; P < .016) in ovarian tumors also exhibits fewer unique upstream regulatory pathways compared with tumors expressing R175H, R179H, R248Q, and I195T. The presence of both a WT and R273H allele in the tumor samples most likely renders them similar to cells expressing only WT TP53 alleles; R273H enhanced nutlin-3a–mediated induction of Cdnk1a and suppression of Hmgb2 in mouse cells expressing WT p53. Evidence presented herein in human and mouse ovarian cancer cells supports previous studies of other tumor types [13], [21] and our hypothesis that ovarian tumor cells expressing heterozygous p53 alleles belong to a specific ovarian cancer subtype with specific functional characteristics that may also be dependent on the p53 mutant that is expressed. For example, although the human ovarian cell line that expresses the R273H mutation is homozygous for the mutant allele, data from the OVCA433 cell line expressing one WT allele and one mutant allele show that the presence of one WT allele impacts tumor cell behavior such that the cells respond more like cells with WT p53. Specifically, the human OVCA433 cells exhibit increased p53 (luciferase) activity and Cdnk1a and Mdm2 expression and reduced cell viability in response to nutlin-3a, all targets of WT p53. Moreover, the responses of the OVCA433 cells to nutlin-3a were highly similar to those of the ALST cells expressing only WT TP53. Conversely, the human OVCA420 and OVCAR3 cells that are homozygous for R273H and R248Q alleles, respectively, do not respond or show reduced responses to nutlin-3a.
As a control, we tested whether or not introducing WT TP53 into the human cell lines would alter their responses without or with nutlin-3a. As expected, WT p53 increased p53 target gene expression (Cdnk1a, Mdm2) in each cell line. Although the levels of mRNA were much lower in the OVCA420 and OVCAR3 cells, the fold induction was highly significant, and these cells exhibited more dramatic cell apoptosis than the ALST or OVCA433 cells in response to the exogenous p53. This may be related, in part, to the elevated levels of p21 protein observed in the OVCA420 and OVCAR3 cells. That the elevated levels of p21protein in the SKOV3 cells are not related to enhanced apoptosis further reinforces the distinct phenotype of p53 null cells. That induction of CASP3 was far greater in the ALST or OVCA433 cells than in the homozygous mutant cells indicates that mechanisms other than, or in addition to, increased expression of CASP3 are operative in these mutant cells. The increase in PUMA suggests that WT p53 selectively increases this apoptotic-related gene in the homozygous mutant cells. It is also possible that the WT p53 blocks specific survival pathways in the mutant cells. To analyze this, we treated each cell line with p53 siRNA to deplete endogenous mutant p53. Whereas the cells expressing WT TP53 showed some increase in cell death, the homozygous mutant cells exhibit more dramatic increases in apoptosis. In the homozygous mutant cells but not in the cells expressing WT TP53, expression of DNAJC3, a gene involved in the unfolded protein stress response, was dramatically induced; CASP3 also increased, but the TP53 target PUMA was not induced. Collectively, these results indicate that WT p53 is functional in the mutant cells and appears to induce a specific set of genes that, combined with the loss of mutant p53, leads to distinct cellular responses and cell death in the homozygous mutant cells.
Data obtained in parallel from our mouse ovarian cancer cell lines extend and provide further support for the distinct functional activities of tumor cells expressing heterozygous versus homozygous 53 alleles and thus emphasize the importance of understanding the functional characteristics of the heterozygous tumor phenotype. Specifically, the data obtained in the mouse ovarian cancer cells document that although all of the p53 mutants tested suppressed basal p53 activity as measured by luciferase assays with a “consensus” p53 promoter element, some were more potent than others. In addition, the mutants exerted their own specific effects on p53 target gene expression in the Trp53 + cells. For example, we document that one p53 GOF and DNA binding mutant, R248W, which is overrepresented in ovarian cancer, suppressed endogenous WT p53-mediated luciferase activity and Cdkn1a mRNA expression but had no effect on Hmgb2 expression and unexpectedly enhanced nutlin-3a activation of Cdkn1a mRNA expression and suppressed Hmgb2 expression. In addition, the H179R mutant exerted similar effects to those of the R248W mutant on WT p53 activity and gene expression, indicating that these distinctly different mutants can impact WT p53 activity in a similar manner in this context, perhaps involving similar interacting partners [9], [10]. The S241F mutant provides another interesting example. It is highly overrepresented in ovarian tumors, its functional activities have not been analyzed, and no human ovarian cancer cell lines express this mutant. However, the S241F mutant 1) suppresses WT p53 activity in luciferase assays but not as much as other mutants, 2) suppresses Cdkn1a mRNA expression, and 3) more potently suppresses Hmgb2 mRNA than WT p53 in the Trp53 + cells. Moreover, S241F enhances nutlin-3a activation of Cdkn1a expression but does not alter nutlin-3a effects on Hmgb2 expression. Thus, the S241F mutant exerts both p53 activity and p53 antagonistic activity depending on context. Maybe it binds MDM2, protecting WT p53 from degradation. Collectively, these data show that specific p53 mutants can modulate WT p53 functional activities and thus tumor cell behavior in response to nutlin-3a, thereby confirming and extending results of others showing that different p53 mutants are not functionally equivalent [51], [53]. The ability of some mutants to retain WT p53 activity likely relates to their ability to bind similar p53 response elements in the target genes, whereas the ability of mutants to block p53 activity likely prevents WT p53 from targeting p53 consensus sites on target genes.
Furthermore, we have recently generated mice expressing the R172H (R175H in humans) allele. Tumors expressing either heterozygous or homozygous R172H alleles develop rapidly in these mice and exhibit markedly different phenotypes (Ren et al., in revision). Herein we show that cells derived from the heterozygous tumors or from tumors homozygous for the R172H allele exhibit increased p53 activity, as measured by Cdkn1a and Mdm2 mRNA expression, in response to exogenous WT p53 expression and nutlin-3a treatment (Figure 5). Moreover, the mouse data show clearly that cells homozygous for the R172H/R172H mutant protein do not totally suppress exogenous WT p53 activity. Furthermore, when the R175H mutant was expressed in the Trp53 + cells (Figure 2, Figure 3), it did not suppress basal or nutlin-3a Cdnk1a induction. Like the human cells, the homozygous R172H mutant mouse cells are sensitive to p53 siRNA, exhibit increased Casp3, and undergo cell death. That cell death is not as dramatic in the mouse cells as in the human cells may be related to the absence of an unfolded protein response and lack of induction of Dnajc3 or to the different genetic backgrounds of these cells. Like the human cells, the mouse cells expressing WTp53 are relatively insensitive to p53 siRNA.
Conclusions
These studies document that specific p53 missense and GOF mutations are selectively overrepresented in high-grade ovarian cancer. As homozygous alleles, specific mutants are differentially associated with specific LOH (chromosome 17) [5], CNV (R175H; chromosome 9), and specific upstream (SMARCA4, estrogen, retinoic acid, TGFβ) and cancer-related (TBX2/5, HOXA5, HOXA9, and PPARG) regulatory pathways. Of particular relevance, expression of immune-related cytokines was selectively related to p53 status, showing for the first time that p53 mutant status impacts, and is related to, the immune subtype [42], [54], [55] of ovarian cancer. Specific mutants differentially linked to retinoic acid signaling and specific HOX gene expression might provide potential targets for novel therapeutic strategies [40], [48]. Each mutant also differentially alters endogenous WT p53 activity, target gene expression, and responses to nutlin-3a, providing “proof of principle” that ovarian cancer cells expressing both a WT and mutant allele represent a distinct ovarian cancer subtype. Using human ovarian cancer cell lines with known p53 status, we show that 1) nutlin-3a activates WT p53 in the presence of mutant p53; 2) reintroduction of WT p53 into human cell lines that are homozygous for specific p53 mutants leads to target gene expression, elevated levels of p21 protein, and apoptosis even without nutlin-3a; and 3) knock down of endogenous p53 in cells expressing homozygous mutant alleles causes a stress-induced UPR and apoptosis. Despite different gene regulatory pathways associated with specific mutants, silencing mutant p53 might be a suitable and powerful strategy for blocking tumor growth in ovarian cancers that rely on mutant p53 functions for survival [6], [25], [26]. Thus, knowing the p53 status of human ovarian cancer cells and using drugs that either activate WT p53 or inactivate mutant p53 may provide alternative strategies for managing tumor growth.
The following are the supplementary data related to this article.
This table summarizes the genes that are deleted in the copy number loss of chromosome 9 (9q22.1-9q31.1) in all HGSCs expressing the R175H mutations.
This table summarizes p53 mutations and upstream signaling pathways that are uniquely associated with each p53 mutant analyzed.
Acknowledgements
Financial support: J. S. R. (NIH-CA-181808; NIH-HD-16229; U54-HD07945, Specialized Cooperative Centers Program in Reproduction and Infertility Research). K.-K. W. was supported in part by grants from the National Institutes of Health, including The University of Texas MD Anderson Cancer Center Specialized Program of Research Excellence in Ovarian Cancer (P50 CA08369), grant CA133057, and the Blanton-Davis Ovarian Cancer Research Program. O. L. (National Science Foundation [NSF] Computing and Communications Foundations [CCF]-0905536; NSF Division of Biological Infrastructure [DBI] 1062455; National Institutes of Health GM079656; National Institutes of Health GM066099). Nutlin-3a was synthesized by and was a generous gift of the MD Anderson Pharmaceutical Chemistry Facility.
Footnotes
Financial support: J. S. R. (NIH-CA-181808; NIH-HD-16229; U54-HD07945, Specialized Cooperative Centers Program in Reproduction and Infertility Research). K.-K. W. was supported in part by grants from the National Institutes of Health, including The University of Texas MD Anderson Cancer Center Specialized Program of Research Excellence in Ovarian Cancer (P50 CA08369), grant CA133057, and the Blanton-Davis Ovarian Cancer Research Program. O. L. (NSFCCF-0905536; NSFDBI 1062455; National Institutes of HealthGM079656; National Institutes of HealthGM066099).
References
- 1.Bast R.C., Hennessy B., Mills G.B. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [PMID: 19461667. PMCID: PMC12814299] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romero I.N., Bast R.C., Jr. Human ovarian cancer: biology, current management and paths to personalizing therapy. Endocrinology. 2012;153:1593–1602. doi: 10.1210/en.2011-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [PMID: 18842102. PMID: 21720365] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho K.R., Shih I.-M. Ovarian Cancer. Annu Rev Pathol. 2009;4:287–313. doi: 10.1146/annurev.pathol.4.110807.092246. [PMCID: PMC12679364] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z.C., Birkbak N.J., Culhane A.C., Drapkin R., Fatima A., Tian R., Schwede M., Alsop K., Daniels K.E., Piao H. Profiles of Genomic Instability in High-Grade Serous Ovarian Cancer Predict Treatment Outcome. Clin Cancer Res. 2012;18:5806–5815. doi: 10.1158/1078-0432.CCR-12-0857. [PMID: 22912389] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller Patricia A.J., Vousden Karen H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell. 2014;25:304–317. doi: 10.1016/j.ccr.2014.01.021. [PMID:24651012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S., Leiserson M.D.M., Niu B., McLellan M.D., Uzunangelov V. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [PMID: 25109877] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goh A.M., Coffill C.R., Lane D.P. The role of mutant p53 in human cancer. J Pathol. 2011;223:116–126. doi: 10.1002/path.2784. [PMID: 21125670] [DOI] [PubMed] [Google Scholar]
- 9.Brachova P., Thiel K.W., Leslie K.K. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int J Mol Sci. 2013;14:19257–19275. doi: 10.3390/ijms140919257. [PMID: 24065105] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freed-Pastor W.A., Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [PMID:22713868] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J., Wang J., Hu Y., Qian J., Xu B., Chen H., Zou W., Fang J.Y. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 2014;5 doi: 10.1038/cddis.2014.75. [PMID: 24603336] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanel W., Marchenko N., Xu S., Xiaofeng Yu S., Weng W., Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20:898–909. doi: 10.1038/cdd.2013.17. [PMID:23538418] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olive K.P., Tuveson D.A., Ruhe Z.C., Yin B., Willis N.A., Bronson R.T., Crowley D., Jacks T. Mutant p53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Mullany L.K., Liu Z., King E.R., Wong K.-K., Richards J.S. Wild-Type Tumor Repressor Protein 53 (TRP53) Promotes Ovarian Cancer Cell Survival. Endocrinology. 2012;153:1638–1648. doi: 10.1210/en.2011-2131. [PMID:22396451; PMCID:PMC23320246] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullany L.K., Liu Z., Wong K.K., Deneke V., Ren Y.A., Herron A., Richards J.S. Tumor Repressor Protein 53 and Steroid Hormones Provide a New Paradigm for Ovarian Cancer Metastases. Mol Endocrinol. 2014;28:127–137. doi: 10.1210/me.2013-1308. [PMID:24264574; PMCID: PMC23874458] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norquist B.M., Garcia R.L., Allison K.H., Jokinen C.H., Kernochan L.E., Pizzi C.C., Barrow B.J., Goff B.A., Swisher E.M. The Molecular Pathogenesis of Hereditary Ovarian Carcinoma: Alterations in the Tubal Epithelium of Women with BRCA1 and BRCA2 Mutations. Cancer. 2010;116:5261–5271. doi: 10.1002/cncr.25439. [PMID: 20665887] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdelalim E.M., Tooyama I. Knockdown of p53 suppresses Nanog expression in embryonic stem cells. Biochem Biophys Res Commun. 2014;443:652–657. doi: 10.1016/j.bbrc.2013.12.030. [PMID:24333425] [DOI] [PubMed] [Google Scholar]
- 18.Maddocks O.D.K., Berkers C.R., Mason S.M., Zheng L., Blyth K., Gottlieb E., Vousden K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [PMID:23242140] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen N., Satija Yatendra K., Das S. PGC-1α, a Key Modulator of p53, Promotes Cell Survival upon Metabolic Stress. Mol Cell. 2011;44:621–634. doi: 10.1016/j.molcel.2011.08.044. [PMID:22099309] [DOI] [PubMed] [Google Scholar]
- 20.Jackson J.G., Post S.M., Lozano G. Regulation of tissue- and stimulus- specific cell fate decisions by p53 in vivo. J Pathol. 2011;223:127–136. doi: 10.1002/path.2783. [PMID: 20957626] [DOI] [PubMed] [Google Scholar]
- 21.Lang G.A., Iwakuma T., Suh Y.-A., Liu G., Rao V.A., Parant J.M., Valentin-Vega Y.A., Terzian T., Caldwell L.C., Strong L.C. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [PMID: 15607981] [DOI] [PubMed] [Google Scholar]
- 22.Brown C.J., Lain S., Verma C.S., Fersht A.R., Lane D.P. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [PMID:19935675] [DOI] [PubMed] [Google Scholar]
- 23.Tovar C., Rosinski J., Filipovic Z., Higgins B., Kolinsky K., Hilton H., Zhao X., Vu B.T., Qing W., Packman K. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [PMID: 16443686 PMCID: PMC11413632] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vousden K.H., Privies C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [PMID: 19410540] [DOI] [PubMed] [Google Scholar]
- 25.Lim L.Y., Vidnovic N., Ellisen L.W., Leong C.O. Mutant p53 mediates survival of breast cancer cells. Br J Cancer. 2009;101:1606–1612. doi: 10.1038/sj.bjc.6605335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braicu C., Pileczki V., Irimie A., Berindan-Neagoe I. p53siRNA therapy reduces cell proliferation, migration and induces apoptosis in triple negative breast cancer cells. Mol Cell Biochem. 2013;381:61–68. doi: 10.1007/s11010-013-1688-5. [DOI] [PubMed] [Google Scholar]
- 27.Katsonis P., Lichtarge O. A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Res. 2014;24:2050–2058. doi: 10.1101/gr.176214.114. [PMID:25217195] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neskey D.M., Osman A.A., Ow T.J., Katsonis P., McDonald T., Hicks S.C., Hsu T.-K., Pickering C.R., Ward A., Patel A. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res. 2015;75:1527–1536. doi: 10.1158/0008-5472.CAN-14-2735. [PMID:25634208] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullany L.K., Fan H.Y., Liu Z., White L.D., Marshall A., Gunaratne P., Anderson M., Creighton C.J., Deavers M., Wong K.K. Molecular characterization of ovarian surface epithelial cells transformed by oncogenes. Oncogene. 2011;30:3522–3536. doi: 10.1038/onc.2011.70. [PMCID: PMC23139785] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan H.Y., Liu Z., Paquet M., Wang J., Lydon J.P., DeMayo F.J., Richards J.S. Cell type specific targeted mutation of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult in ovarian surface epithelial cells. Cancer Res. 2009;69:6463–6472. doi: 10.1158/0008-5472.CAN-08-3363. [PMID: 19679546. PMCID: PMC12741085] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaginuma Y., Westphal H. Abnormal Structure and Expression of the p53 Gene in Human Ovarian Carcinoma Cell Lines. Cancer Res. 1992;52:4196–4199. [PMID:1638534] [PubMed] [Google Scholar]
- 32.Crane E.K., Kwan S.-Y., Izaguirre D.I., Tsang Y.T.M., Mullany L.K., Zu Z., Richards J.S., Gershenson D.M., Wong K.-K. Nutlin-3a: A Potential Therapeutic Opportunity for TP53 Wild-Type Ovarian Carcinomas. PLoS One. 2015;10:e0135101. doi: 10.1371/journal.pone.0135101. [PMID:26248031] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong K.-K., Izaguirre D.I., Kwan S., King E.R., Deavers M.T., Sood A.K., Mok S.C., Gershenson D.M. Poor survival with wild-type TP53 ovarian cancer? Gynecol Oncol. 2013;130:565–569. doi: 10.1016/j.ygyno.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loo P., Nilsen G., Nordgard S., Vollan H., Børresen-Dale A.-L., Kristensen V., Lingjærde O. Analyzing Cancer Samples with SNP Arrays. In: Wang J., Tan A.C., Tian T., editors. Next Generation Microarray Bioinformatics. Humana Press; 2012. pp. 57–72. [PMID:22130873] [Google Scholar]
- 35.Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [PMID:24336805] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forbes S.A., Tang G., Bindal N., Bamford S., Dawson E., Cole C., Kok C.Y., Jia M., Ewing R., Menzies A. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–D657. doi: 10.1093/nar/gkp995. [PMID:19906727] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao R.C., Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334–346. doi: 10.1038/nrc3929. [PMID:25998713] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhatlekar S., Fields J., Boman B. HOX genes and their role in the development of human cancers. J Mol Med. 2014;92:811–823. doi: 10.1007/s00109-014-1181-y. [PMID:24996520] [DOI] [PubMed] [Google Scholar]
- 39.Ko S.Y., Lengyel E., Naora H. The Mullerian HOXA10 gene promotes growth of ovarian surface epithelial cells by stimulating epithelial-stroma interactions. Mol Cell Endocrinol. 2010;317:112–119. doi: 10.1016/j.mce.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ko S.Y., Naora H. HOXA9 promotes homotypic and heterotypic cell interactions that facilitate ovarian cancer dissemination via its induction of P-cadherin. Mol Cancer. 2014;13 doi: 10.1186/1476-4598-13-170. [170-170, PMID:25023983] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ko S.Y., Naora H. Adaptation of ovarian cancer cells to the peritoneal environment: Multiple mechanisms of the developmental patterning gene HOXA9. Cancer Cell Microenviron. 2014;1 [PMID:26000332] [PMC free article] [PubMed] [Google Scholar]
- 42.Tothill R.W., Tinker A.V., George J., Brown R., Fox S.B., Lade S., Johnson D.S., Trivett M.K., Etemadmoghadam D., Locandro B. Novel Molecular Subtypes of Serous and Endometrioid Ovarian Cancer Linked to Clinical Outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [PMID:18698038] [DOI] [PubMed] [Google Scholar]
- 43.Wansleben S., Peres J., Hare S., Goding C.R., Prince S. T-box transcription factors in cancer biology. Biochim Biophys Acta. 2014;1846:380–391. doi: 10.1016/j.bbcan.2014.08.004. [PMID:25149433] [DOI] [PubMed] [Google Scholar]
- 44.Wang X., Ji X., Chen J., Yan D., Zhang Z., Wang Q., Xi X., Feng Y. SOX2 Enhances the Migration and Invasion of Ovarian Cancer Cells via Src Kinase. PLoS One. 2014;9 doi: 10.1371/journal.pone.0099594. [PMID: 24937695] [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Romero O.A., Sanchez-Cespedes M. The SWI/SNF genetic blockade: effects in cell differentiation, cancer and developmental diseases. Oncogene. 2014;33:2681–2689. doi: 10.1038/onc.2013.227. [PMID: 23752187] [DOI] [PubMed] [Google Scholar]
- 46.Biegel J.A., Busse T.M., Weissman B.E. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C: Semin Med Genet. 2014;166:350–366. doi: 10.1002/ajmg.c.31410. [PMID:25169151] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wansleben S., Davis E., Peres J., Prince S. A novel role for the anti-senescence factor TBX2 in DNA repair and cisplatin resistance. Cell Death Dis. 2013;4 doi: 10.1038/cddis.2013.365. [PMID:24113180] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langston A.W., Gudas L.J. Retinoic acid and homeobox gene regulation. Curr Opin Genet Dev. 1994;4:550–555. doi: 10.1016/0959-437x(94)90071-a. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y., Suh Y.-A., Fuler M.Y., Jackson J.G., Xiong S., Terzian T., Quintas-Cardama A., Bankson J.A., El-Naggar A.K., Lozano G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893–904. doi: 10.1172/JCI44504. [PMID: 21285512. PMCID: PMC23049366] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrova K., Oyadomari S., Hendershot L.M., Ron D. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 2008;27:2862–2872. doi: 10.1038/emboj.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin Y.-J., Kim M.-S., Kim M.-S., Lee J., Kang M., Jeong J.-H. High-mobility group box 2 (HMGB2) modulates radioresponse and is downregulated by p53 in colorectal cancer cell. Cancer Biol Ther. 2013;14:213–221. doi: 10.4161/cbt.23292. [PMID:23255232] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vikhanskaya F., Lee M.K., Mazzoletti M., Broggini M., Sabapathy K. Cancer-derived p53 mutants suppress p53-target gene expression—potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007;35:2093–2104. doi: 10.1093/nar/gkm099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu D-p, Song H., Xu Y. A common Gain of function of p53 cancer mutants in inducing genetic instability. Oncogene. 2010;29:949–956. doi: 10.1038/onc.2009.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson B.H. New insights into tumor immunity revealed by the unique genetic and genomic aspects of ovarian cancer. Curr Opin Immunol. 2015;33:93–100. doi: 10.1016/j.coi.2015.02.004. [PMID: 25710852] [DOI] [PubMed] [Google Scholar]
- 55.Zsiros E., Duttagupta P., Dangaj D., Li H., Frank R., Garrabrant T., Hagemann I.S., Levine B.L., June C.H., Zhang L. The Ovarian Cancer Chemokine Landscape Is Conducive to Homing of Vaccine-Primed and CD3/CD28–Costimulated T Cells Prepared for Adoptive Therapy. Clin Cancer Res. 2015;1–11 doi: 10.1158/1078-0432.CCR-14-2777. [PMID: 25712684] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table summarizes the genes that are deleted in the copy number loss of chromosome 9 (9q22.1-9q31.1) in all HGSCs expressing the R175H mutations.
This table summarizes p53 mutations and upstream signaling pathways that are uniquely associated with each p53 mutant analyzed.