

Abstract
AIM: To characterize phosphorylation of human glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and mobility of GAPDH in cancer cells treated with chemotherapeutic agents.
METHODS: We used proteomics analysis to detect and characterize phosphorylation sites within human GAPDH. Site-specific mutagenesis and alanine scanning was then performed to evaluate functional significance of phosphorylation sites in the GAPDH polypeptide chain. Enzymatic properties of mutated GAPDH variants were assessed using kinetic studies. Intranuclear dynamics parameters (diffusion coefficient and the immobile fraction) were estimated using fluorescence recovery after photobleaching (FRAP) experiments and confocal microscopy. Molecular modeling experiments were performed to estimate the effects of mutations on NAD+ cofactor binding.
RESULTS: Using MALDI-TOF analysis, we identified novel phosphorylation sites within the NAD+ binding center of GAPDH at Y94, S98, and T99. Using polyclonal antibody specific to phospho-T99-containing peptide within GAPDH, we demonstrated accumulation of phospho-T99-GAPDH in the nuclear fractions of A549, HCT116, and SW48 cancer cells after cytotoxic stress. We performed site-mutagenesis, and estimated enzymatic properties, intranuclear distribution, and intranuclear mobility of GAPDH mutated variants. Site-mutagenesis at positions S98 and T99 in the NAD+ binding center reduced enzymatic activity of GAPDH due to decreased affinity to NAD+ (Km = 741 ± 257 μmol/L in T99I vs 57 ± 11.1 µmol/L in wild type GAPDH. Molecular modeling experiments revealed the effect of mutations on NAD+ binding with GAPDH. FRAP (fluorescence recovery after photo bleaching) analysis showed that mutations in NAD+ binding center of GAPDH abrogated its intranuclear interactions.
CONCLUSION: Our results suggest an important functional role of phosphorylated amino acids in the NAD+ binding center in GAPDH interactions with its intranuclear partners.
Keywords: NAD+, Binding site, Fluorescence recovery after photobleaching, Nuclear proteins, Mutation, Glyceraldehyde 3-phosphate dehydrogenase, Anticancer agents
Core tip: We detected the phosphorylated amino acid residues Y94, S98, T99 within the NAD+ binding center of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Substitution of these amino acids with non-phosphorylated alanine residues did not abrogate intranuclear localization of GAPDH. Instead, such mutations altered the molecular dynamics parameters of intranuclear GAPDH probably by hindering its interactions with yet to be identified nuclear biomolecules. Our molecular modeling experiments invoke an important structural feature -T99-E97 H-bond likely involved in stabilization of NAD+ binding center.
INTRODUCTION
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is an intriguing example of moonlighting proteins, which performs multiple functions in several unrelated cellular pathways[1]. Participation of GAPDH in diverse biochemical pathways inside distinct cellular compartments poses a problem from evolutionary point of view. Because energy metabolism of cancer cells relies mainly on glucose utilization via the glycolytic pathway, rather than on oxidative phosphorylation (the Warburg effect), GAPDH is important for glycolysis-dependent energy supply. Depletion of GAPDH arrests cell proliferation, and induces accelerated senescence in cancer cells[2-4].
In response to stress, GAPDH acts as a signaling molecule linking stress factors and cellular apoptotic machinery[5-7]. S-nitrosylation of Cys149 in the GAPDH catalytic center induces its intranuclear translocation and activation of apoptosis-related signaling cascade[5,8].
An additional level of complexity in regulating intracellular functions of GAPDH comes from the fact that multiple GAPDH isoforms are detected in cytosolic and nuclear fractions. Seo et al[9] identified multiple posttranslational modifications of GAPDH (19 types of modification on 42 sites), whose physiological role still remains to be elucidated.
Details of GAPDH-mediated intranuclear functions are incompletely known. Several lines of evidence suggest involvement of GAPDH in chemotherapy-induced DNA damage response. First, GAPDH demonstrates uracil-DNA-glycosylase activity contributing to DNA repair activity[10]. Viability of human leukemia cells treated with 6-mercaptopurine correlates with intranuclear level of GAPDH; the higher level of intranuclear GAPDH was detected in cells with lower viability[11]. Next, GAPDH was demonstrated to be a component of DNA-protein complexes formed on short DNA duplexes containing inserted modified nucleosides cytarabine, fluorouridine, and thioguanosine[12]. Finally, GAPDH binds DNA covalently linked to saframycin, a natural product with potent antiproliferative effect[13]. DNA alkylation with an antitumor drug S23906-1 results in chromatin binding with GAPDH and its protein partner HMGB1[14].
In the present study, we detected post-synthetic modifications in three amino acid residues encompassing the NAD+ binding center in GAPDH. In order to elucidate the functional significance of these modifications, we prepared the mutant forms of GAPDH, and assessed enzyme properties, intracellular localization, and intranuclear interactions of the mutated proteins. Our results indicate that NAD+ binding center in GAPDH is an important structural element which regulates GAPDH interactions with other nuclear components.
MATERIALS AND METHODS
Cell cultures, drug treatment and plasmids
Lung carcinoma cells A549 were obtained from the ATCC collection (ATCC, Manassas, VA). HCT116-1640 (p53+/+) and SW48-297 (p53+/+) cells were a generous gift from Dr. Vogelstein (John Hopkins University, Baltimore, MR). Cells were maintained in RPMI1640 medium (A549 cells; ATCC, Manassas, VA) or in McCoy’s A5 (HCT116 and SW48 cells; Lonza, Allendale, NJ) at 40%-80% confluence. Cells were treated with araC {cytarabine, cytosine arabinoside, 4-amino-1-[(2R,3S,4R,5R)-3,4-dihydroxy-5- (hydroxymethyl) oxolan-2-yl] pyrimidin-2-one} dissolved in water as 500-1000 × stock solutions; drug concentrations were determined spectrophotometrically (araC, Ε272 = 9259; The Merck Index, 2001). Human GAPDH cDNA was inserted in frame with enhanced green fluorescent protein (EGFP) into pcDNA3.1 as described earlier[3] and verified by sequencing. Site-mutagenesis of GAPDH ORF was performed using QuickChange Lightning Site-Directed Mutagenesis kit (Stratagene, CA), per manufacturer’s instructions. Oligonucleotides used for mutagenesis are listed in Table 1. Wild type and mutated GAPDH cDNA were inserted into pET28a plasmid, expressed in BL21 (DE3) Escherichia coli (E. coli) expression system (Novagen, WI) and purified using Ni-NTA chromatography.
Table 1.
Mutations introduced to glyceraldehyde 3-phosphate dehydrogenase cDNA ORF
| GAPDH variant | Mutagenic primers1 | Nucleotide change | Codon change |
| Y94A | CGATGCTGGCGCTGAGGCCGTCGTGGAGTCCACTGG | 280-281 | UAC- > GCC |
| CCAGTGGACTCCACGACGGCCTCAGCGCCAGCATCG | |||
| S98A | GAGTACGTCGTGGAGGCCACTGGCGTCTTCACC | 290 | UCC- > GCC |
| GGTGAAGACGCCAGTGGCCTCCACGACGTACTC | |||
| T99A | AGTACGTCGTGGAGTCCGCTGGCGTCTTCACCACC | 295 | ACU- > GCU |
| GGTGGTGAAGACGCCAGCGGACTCCACGACGTACT | |||
| T99I | GAGTACGTCGTGGAGTCCATTGGCGTCTTCA | 296 | ACU- > AUU |
| TGAAGACGCCAATGGACTCCACGACGTACTC |
Mutated nucleotides are indicated in bold font; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Two dimensional gel electrophoresis
Separation of cytosolic and nuclear fractions of cells was performed with NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific, Rockford, IL), per manufacturer’s instructions as described earlier[3]. The extracts (cytoplasmic fraction and nuclear extract) were snap-frozen on dry ice and stored at -80 °C.
Protein extracts were precipitated by acetone, and re-dissolved in 2D buffer containing 7 mol/L urea, 2 mol/L thiourea, 4% CHAPS, 40 mmol/L Tris, 60 mmol/L DTT, and 1 × protease inhibitor cocktail (Roche Applied Science). Further procedures for sample preparation, the first and the second dimension separations were performed according to the published protocol[15]. Briefly, samples were diluted to 120 μL with rehydration buffer, applied on Immobiline Drystrip (7 cm) with 3-11 or 6-11 pH range (Amersham Biosciences) overnight. Isoelectric focusing was performed by applying a voltage gradient as follows: 30 min to 250 V, 1 h to 1500 V, 1 h to 3000 V and then 3 h to 8000 V. After IEF, the strips were incubated for 30 min in equilibration buffer and treated with iodoacetamide. The second dimension (SDS-PAGE) was performed on 12% Mini-PROTEAN® TGX™ Precast Gels (456-1041, Biorad). Separated proteins were stained with SYPRO® Ruby protein gel stain (Life Technologies) or transferred onto the nitrocellulose membrane by electroblotting procedure[16]. Membranes were developed with mouse monoclonal anti-GAPDH antibody at 1:10000 dilution (Millipore, CA) or phospho-T99-GAPDH antibody (Rockland Immunochemicals, PA). Polyclonal rabbit anti-pT99 antibody was raised by immunization with KLH-conjugated phosphopeptide AEYVVES [pT] GVFT corresponding to the sequence 92-103 in human GAPDH. Antibody was further purified by affinity chromatography, and used at a 1:10000 dilution. Bands were visualized by treatment with secondary antibody - IRDye 800CW donkey anti-rabbit antibody or IRDye 680 goat anti-mouse antibody (Rockland, Gilbertsville, PA) at 1:10000 dilution, and quantified by Odyssey Infrared Imaging system (LI-COR Biosciences, Lincoln, NE) using two-color fluorescence detection at 700 and 800 nm.
Proteomics protocols
Preparation of protein samples and in-gel trypsin digestion: Protein spots were processed for gel electrophoresis-liquid chromatography-mass spectroscopy (GeLC-MS/MS) proteomics analysis. After visualization with SYPRO staining, gel images were acquired in an image reader FLA-5000 (FujiFILM) and analyzed densitometrically using the multi gauge v3.0 (FujiFILM).
The GAPDH spots were extracted and diced into - 1 mm × 1 mm. After destaining with 50% v/v Acetonitrile (ACN) in 25 mmol/L ammonium bicarbonate buffer (bicarbonate buffer), proteins within gel pieces were reduced with 10 mmol/L DTT in bicarbonate buffer, and alkylated by incubation with 50 mmol/L iodoacetamide. After gel dehydration with 100% ACN, the gel pieces were covered with approximately 40 μL of 12.5 μg/mL trypsin in bicarbonate buffer. In-gel digestion was performed at 37 °C for 12 h, trypsin was inactivated with formic acid at 2% final volume, and peptides were extracted and cleaned-up using C18 Tip column (ZipTips®) as previously described[17].
GeLC-MS/MS proteomics analysis: Peptides were dried in a vacuum centrifuge, then resuspended in 30 μL of 0.1% v/v TFA/H2O. Peptide samples were loaded onto 2 μg capacity peptide traps (CapTrap; Michrom Bio-resources) and separated using a C18 capillary column (15 cm 75 mm, Agilent) with an Agilent 1100 LC pump delivering mobile phase at 300 nL/min. Gradient elution using mobile phases A (1% ACN/0.1% formic acid, balance H2O) and B (80% ACN/0.1% formic acid, balance H2O) was as follows (percentages for B, balance A): Linear from 0 to 15% at 10 min, linear to 60% at 60 min, linear to 100% at 65 min. The nano ESI MS/MS (electrospray ionization mass spectrometry) was performed using a HCT Ultra ion trap mass spectrometer (Bruker). ESI was delivered using distal-coating spray Silica tip (id 20 μm, tip inner id 10 μm, New Objective, Ringoes, NJ). Mass spectra were acquired in positive ion mode, capillary voltage at -1200 V and active ion charge control trap scanning from 300 to 1500 m/z. Using an automatic switching between MS and MS/MS modes, MS/MS fragmentation was performed on the two most abundant ions on each spectrum using collision-induced dissociation with active exclusion (excluded after two spectra, and released after 2 min). The complete system was fully controlled by HyStar 3.2 software.
MS data analysis and post-translational modifications: Mass spectra processing was performed using BrukerDaltonics esquire 6.1 Data-Analysis (Version 3.4). The generated de-isotoped peak list was submitted to an in-house Mascot server 2.2.07 for searching against the Swiss-Prot database (Release 2011-06) (version 56.6, 536029 sequences). Mascot search parameters were set as follows: species, Homo sapiens (20413 sequences); enzyme, trypsin with maximum 2 missed cleavage sites. Post-translational modifications were analyzed specifically for GAPDH: Fixed modification, cysteine carboxymethylation; variable modifications: Methionine oxidation, phosphorylation of serine, threonine and tyrosine. All peptides matches were filtered using an ion score cutoff of 30.
Transfection with pEGFP-GAPDH and fluorescence recovery after photobleaching experiments
About 25000 cells/dish were seeded in 35 mm glass bottom Petri dishes (MatTek, MA) and transfected with pEGFP (Clontech, Palo Alto, CA) or pEGFP-GAPDH plasmid using FuGene6 transfection reagent (Roche, NJ). Alternatively, HCT116-4016 cells were transiently transfected by electroporation (Neon Transfection System, Invitrogen, CA). The next day, cells were treated with 1-10 μmol/L araC and incubated for 24 h.
After incubation, fluorescence recovery after photobleaching (FRAP) experiments were performed on a Leica TCS SP2 AOBS confocal microscope equipped with a 63 × 1.4 N.A. oil immersion objective at 37 °C, as described earlier[3]. Briefly, pre-bleaching plateau was defined by acquiring 20 single section images with 6 × zoom on an area 7 × 7 μm, with acquisition speed 287 msec/frame. Bleaching was performed with 3 pulses using the 458, 476, and 488 nm lines of Ar laser. Fluorescence recovery was monitored collecting 40 single-section images at 287 msec intervals with low laser intensity (5% of the bleach intensity with the single 488 laser line, detection 495-600 nm). Quantitative analysis was performed after background subtraction, correction for laser fluctuations and acquisition photobleaching, and normalization as described by Rabut and Ellenberg[18]. 5-10 cells were analyzed on each dish; all experiments were repeated 4-5 times. Diffusion coefficient D value was calculated using the equation D = 0.88*w2/(4t1/2) where w is a radius of bleached area, with the assumptions that the bleached area is a disc and that diffusion occurs only laterally[19,20]. The immobile fraction was calculated after correction for loss of signal due to photobleaching[18].
Glycolytic assay
GAPDH glycolytic activity was measured by spectrophotometric assay at 340 nm as described earlier[21]. Briefly, the assay was carried out in 0.015 mol/L sodium pyrophosphate, 0.03 mol/L sodium arsenate (Sigma), pH 8.5, in the presence of 3.5 mmol/L DTT, 0.26 mmol/L NAD+ and 0-2 mmol/L glyceraldehyde-3-phosphate (Sigma), or 0.51 mmol/L glyceraldehyde-3-phosphate, and 0-2 mmol/L NAD+ catalyzed by wild type or mutated GAPDH expressed in E. coli. Human GAPDH (Sigma) was used as control.
Molecular modeling of the effects of mutation on NAD+ cofactor binding
Molecular modeling experiments were performed utilizing the Sybyl molecular modeling environment. The X-ray crystal structure of human GAPDH at 1.75 Angstrom resolution (1U8F.pdb) was obtained from the Protein Data Bank and preprocessed for virtual mutagenesis studies by initial extraction of the crystallographic water molecules from the tetramer, selection and extraction of the P chain monomer with its corresponding NAD+ cofactor, and final extraction of the cofactor as separate files. The resulting monomer was then subjected to a localized energy minimization (annealing) routine involving the region around the amino acid of interest (T99). The subset of residues within 8 Å of T99 were considered to be the “hot” region, while a radius of 14 Å comprised the “interesting” region for the annealing process, and the region was minimized to a gradient of 0.05 utilizing the AMBER7FF99 force field and charges. The resulting structure was saved, and T99 was mutated to Ala and to Ile, the resulting structures then undergoing the same local annealing process after substitution. The structures for the native T99, and the mutant forms T99A and T99I were prepared for submission to a canonical (NVT) molecular dynamics calculation utilizing the same force field and charges as above.
Statistical analysis
The statistical analyses were carried out using Student’s t test with Statistica software program (StatSoft, OK), and non-linear regression analysis with GraphPad Prizm 4.0 software (GraphPad software, CA). A P value < 0.05 was considered statistically significant. Data are presented as the mean ± SE.
RESULTS
Proteomics analysis of intranuclear GAPDH after genotoxic stress
Nuclear and the cytosolic fractions were extracted from either untreated A549 cells, or cells treated 1 μmol/L araC for 24 h. Nuclear and cytosolic GAPDH isoforms were detected by one- or two-dimensional Western blot with anti-GAPDH antibodies. Because intranuclear GAPDH in untreated cells was below detection level[3], we analyzed the nuclear fraction of A549 cells after treatment with araC. Figure 1A demonstrates 2D gel separation (pH range 3-11) of the nuclear (upper part) and the cytosolic (bottom part) fractions of A549 cells treated with araC. Western blot analysis of cytosol revealed the presence of multiple GAPDH isoforms in cytosolic fraction. Only basic isoform accumulated in the nuclear fraction of treated A549 cells (Figure 1A). 2D separation (pH range 6-11) of the cytosolic fraction from untreated A549 cells is shown on (Figure 1B). The left panel shows the gel stained with SYPRO® Ruby, and the right image depicts proteins detected with anti-GAPDH antibody. Proteomics analysis of spot 1 and spot 2 on Figure 1B identified a peptide WGDAGAEYVVESTGVFTTMEK corresponding to the amino acid sequence 87-107 of human GAPDH. Further analysis of this peptide resulted in identification of phosphorylated amino acids at position 94, 98 and 99 as revealed by MS detection of the precursor ion 850.51 m/z (MS1) (Figure 2A), and the subsequent fragmentation ions (MS2) (Figure 2B).
Figure 1.
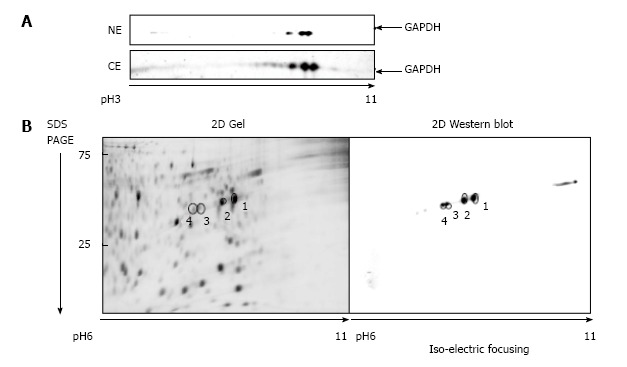
Glyceraldehyde 3-phosphate dehydrogenase isoforms in A549 cells. A: 2D gel separation (pH range 3-11) of proteins from the nuclear (NE) and the cytosolic (CE) fraction of A549 cells after 1 μmol/L araC treatment for 24 h. The membrane was developed with anti-GAPDH antibody as described in “Materials and Methods”; B: Proteins from the cytosolic fraction were separated by 2D gel (pH range 6-11). Separated proteins were stained with SYPRO® Ruby (left panel), or developed with anti-GAPDH antibody (right panel). GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; 2D: Two dimensional.
Figure 2.
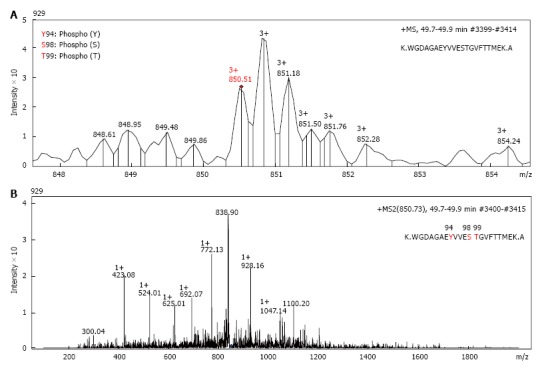
The tandem mass spectrometry (MS/MS) profile of glyceraldehyde 3-phosphate dehydrogenase peptides. Proteomics characterization of a basic GAPDH isoform (spot 1 from Figure 1B) from the cytosolic fraction of A549 cells by mass spectroscopy. A: Spectrum profile of the precursor ion 850.51 m/z (MS 1) selected for further fragmentation; B: Spectrum profile of the fragmentation ions (MS 2) of the peak 850.51 m/z from panel A. Retention time and number of scans where the peptide was detected are shown in the right top part of each panel. GAPDH: Glyceraldehyde 3-phosphate. dehydrogenase.
The presence of phosphorylated T99-GAPDH in the cytosolic and nuclear fractions of treated A549 cells was confirmed with anti-phospho-T99-GAPDH antibody (Figure 3). In cytosol of araC- treated A549 cells, we detected the basic forms of GAPDH with phosphorylated T99 (Figure 3A). We also observed accumulation of basic isoforms in the nuclear fraction of treated A549 cells (Figure 3B upper image). Consecutive immuno staining of 2D gel with anti-GAPDH antibody and anti-phospho-T99-GAPDH antibody revealed phosphorylated T99 in the spots co-localized with the basic GAPDH isoforms in the nuclear fraction (Figure 3B merged image).
Figure 3.
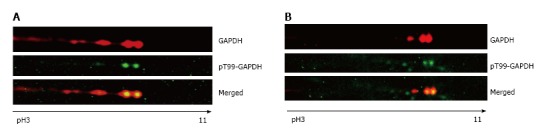
Phospho-T99-glyceraldehyde 3-phosphate dehydrogenase was detected in nuclei of A549 cells after genotoxic stress. A: 2D gel analysis of the cytosolic fraction extracted from A549 cells. Only a section of the membrane containing GAPDH isoforms is shown. Membranes were developed with anti-GAPDH antibody or anti-phospho-T99-GAPDH antibody in untreated and araC-treated cells. pH gradient is indicated under the bottom image; B: The nuclear fraction from A549 cells after treatment with araC. Only a section of the membrane containing GAPDH isoforms is shown. Membranes were developed with anti-GAPDH antibody (top image) or anti-phosphoT99-GAPDH antibody (bottom image). pH gradient is indicated under the bottom image. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; 2D: Two dimensional.
Site-specific mutagenesis of NAD+ binding domain in GAPDH polypeptide
Phosphorylated amino acid residues identified in our proteomics experiments are localized within the NAD+ binding center of GAPDH, as demonstrated by high-resolution structural analysis of human GAPDH[22]. To elucidate the functional role of newly found phosphorylated amino acids in NAD+ binding domain of GAPDH in chemotherapy-induced stress response, we prepared plasmids coding for wild type- and mutated forms of GAPDH polypeptide with mutations that prevented phosphorylation at position 94, 98, and 99 of GAPDH.
Plasmids coding for (His)-tagged GAPDH variants Y94A, S98A, T99A, and T99I were used for heterologous expression of mutated GAPDH polypeptides in a bacterial expression system. Enzymatic activity of purified GAPDH proteins was assayed in vitro. Constructs coding for GAPDH variants Y94A, S98A, T99A, and T99I in-frame with EGFP cDNA under CMV promoter were generated from pEGFP-GAPDH as described earlier[3], and these constructs were used in transfection experiments with human lung carcinoma cells A549, human colorectal carcinoma cells HCT 116-4016, and SW48-297.
Mutations in NAD+ binding domain of GAPDH inhibit glycolytic activity
Enzymatic properties of wild type and mutated forms of GAPDH were assessed spectrophotometrically using the varying concentrations of glyceraldehyde-3-phosphate or NAD+[21]. Results of these experiments are shown on Figure 4, and summarized in Table 2. All GAPDH mutant proteins had affinity for glyceraldehyde-3-phosphate close to that of wild type enzyme. In contrast, mutated polypeptides manifested decreased affinity to NAD+ compared with wild type protein (Table 2). T99I variant had the lowest affinity to NAD+, its Km (NAD+) was increased by more than an order of magnitude compared with wild type GAPDH (741 ± 257 vs 57 ± 11.1 μmol/L). This low binding of NAD+ explains why T99I variant has low saturation under conditions of experiment (i.e., at NAD+ concentration 260 μmol/L), as depicted on Figure 4A. Extremely low binding of T99I-GAPDH to NAD+ coenzyme accounts for decreased velocity of glycolytic reaction catalyzed by this mutated enzyme (Figure 4).
Figure 4.
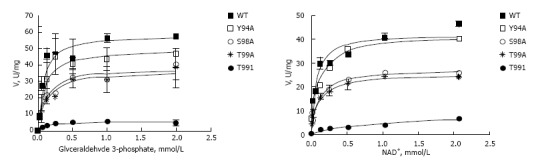
Kinetic analysis of wild type and mutated forms of Hisx6-tagged glyceraldehyde 3-phosphate dehydrogenase prepared in BL21 (DE3) Escherichia coli expression system. Conditions for the glycolytic assay are indicated in “Materials and Methods”. A: Reaction was performed with 0.26 mmol/L NAD+ and varying concentrations of D-glyceraldehyde-3-phosphate (0-2 mmol/L); B: Reaction was performed with 0.51 mmol/L D-glyceraldehyde-3-phosphate and varying concentrations of NAD+ (0-2 mmol/L). Vmax and Km were calculated by non-linear regression analysis with GraphPad Prizm 4.0 as described in Materials and Methods. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Table 2.
Kinetic parameters of wild type and mutated glyceraldehyde 3-phosphate dehydrogenase variants
| GAPDH variant | Km (G3P) (μmol/L) | Km (NAD+) (μmol/L) |
| Wild type | 77 ± 25.9 | 57 ± 11.1 |
| Y94A | 87 ± 33.1 | 97.4 ± 11 |
| S98A | 100 ± 36.7 | 101 ± 7.2 |
| T99A | 112 ± 44.7 | 105 ± 17.7 |
| T99I | 106 ± 38.3 | 741 ± 257 |
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Mutations in NAD+ binding domain do not affect nuclear accumulation of EGFP-GAPDH in response to genotoxic stress
As we demonstrated earlier, endogenous GAPDH or EGFP-GAPDH fusion polypeptides localized predominantly in cytoplasm of untreated A549, SW620, and DLD1 cultured cells[3,21]. To assess the effect of mutation in NAD+ binding center on intracellular localization of GAPDH, we used confocal microscopy and image analysis to compare GAPDH distribution between cytoplasmic and nuclear compartments in HCT116-4016 and SW48-297 cells expressing EGFP-GAPDH (wild type), or mutated fusion proteins. After genotoxic stress caused by araC treatment, wild type EGFP-GAPDH as well as all mutated fusion proteins accumulated in the nuclei. Figure 5A shows the distribution of EGFP-GAPDH wild type, T99A and T99I mutated forms in HCT116-4016 cells before and after treatment. The basal (i.e., before the genotoxic stress) intranuclear level of mutant forms was higher compared to wild type form (up to 15% vs 2.3%). Neither mutation at positions 94, 98, or 99 prevented GAPDH nuclear accumulation after genotoxic stress. In HCT116-4016 cells, T99I demonstrated the most prominent nuclear accumulation (45% of total EGFP-GAPDH, Figure 5B). Similar results were received in p53-proficient SW48-297 cells (not shown).
Figure 5.
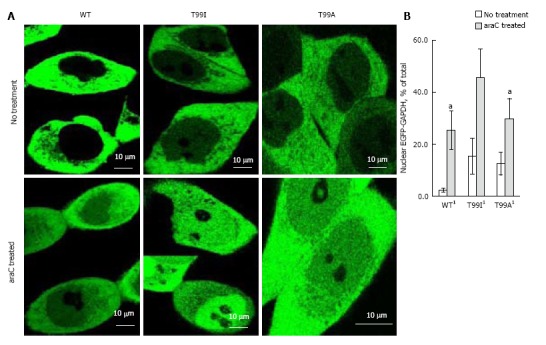
Nuclear accumulation of wild type and mutated enhanced green fluorescent protein-glyceraldehyde 3-phosphate dehydrogenase in live HCT116-4016 cells after transient transfections with plasmids coding for enhanced green fluorescent protein fusions with WT-glyceraldehyde 3-phosphate dehydrogenase, T99I-glyceraldehyde 3-phosphate dehydrogenase, and T99A-glyceraldehyde 3-phosphate dehydrogenase polypeptides. A: Distribution of EGFP-GAPDH variants in HCT116-4016 cells before (upper images) and after (lower images) treatment with 1 µmol/L araC for 24 h; B: Quantitative analysis of images shown in A was performed using ImageJ 1.48 v software (NIH, United States). For statistical evaluation, pixel analysis of 20-25 cells was performed for each experiment (mean ± SE). 1Significant (P < 0.03) alteration in nuclear GFP-GAPDH between cells treated with araC and untreated cells. EGFP: Enhanced green fluorescent protein; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; GFP: Green fluorescent protein.
Mutations within the NAD+ binding domain alter intranuclear mobility of GAPDH
FRAP technique provides important information about dynamic properties of fluorescent proteins in the cytoplasmic and nuclear compartments of the live cells[3,23]. After short-term photo bleaching of the preselected spots inside the cytoplasmic or nuclear compartments of the cells, recovery of fluorescence intensity in the cytosolic and nuclear compartments was measured as described in “Materials and Methods”. Recovery of fluorescence in the nuclear and cytosolic fractions of EGFP-GAPDH in HCT116-4016, and SW48-297 occurred differently depending on the nature of mutation in the NAD+ binding center. As shown on Figure 6, recovery of the fluorescent signal in cytosol for all EGFP-GAPDH fusion proteins was fast and reached plateau after approximately 7.5 msec (Figure 6B, open symbols). The rate of fluorescence recovery in the nuclei of cells expressing mutant EGFP-GAPDH fusion proteins was similar to fluorescence recovery in cytosol (Figure 6B, closed symbols). In contrast, recovery of wild type EGFP-GAPDH was slow, and didn’t reach plateau even after 12.5 msec (Figure 6B). Importantly, only wild type nuclear EGFP-GAPDH demonstrated about 10 times lower D value and about 3 time higher immobile fraction (1-Mf) compared with cytosolic EGFP-GAPDH (Table 3).
Figure 6.
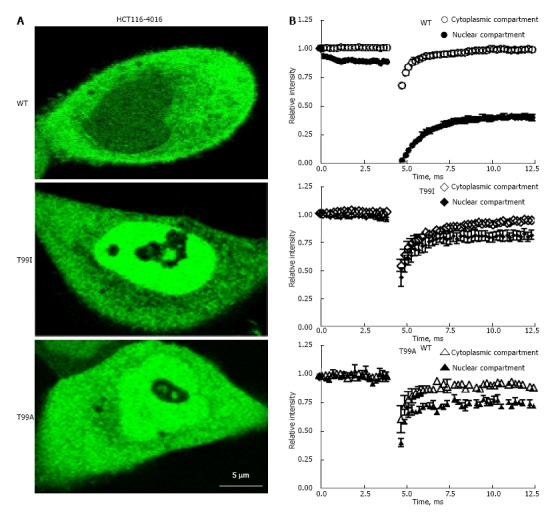
Intranuclear mobility of wild type and mutated variants of enhanced green fluorescent protein-glyceraldehyde 3-phosphate dehydrogenase in HCT116-4016 cells after genotoxic stress. A: FRAP analysis of fluorescent fusion protein EGFP-GAPDH in subcellular compartments of HCT116-4016 cells. Confocal images were collected from transfected cells expressing WT-EGFP-GAPDH (WT), EGFP-GAPDH-T99I (T99I), and EGFP-GAPDH-T99A (T99A) after treatment with 1 μmol/L araC for 24 h; B: Recovery rate of wild type and mutant EGFP-GAPDH fusion proteins after photo bleaching. Open symbols, cytoplasmic compartment, and closed symbols, nuclear compartment. The dynamic parameters mobile fraction (1-Mf), half-time of equilibration t(1/2) (s), and diffusion coefficients D were calculated from FRAP experiments, as described in “Materials and Methods” and summarized in Table 3. Where not seen, the error bars are smaller than the data point symbols. EGFP: Enhanced green fluorescent protein; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; GFP: Green fluorescent protein; FRAP: Fluorescence recovery after photobleaching.
Table 3.
Fluorescence recovery after photobleaching analysis of dynamic properties of wild type and mutated enhanced green fluorescent protein-glyceraldehyde 3-phosphate dehydrogenase
| Protein | Compartment | (1-Mf) | t1/2, s | D, μm2/s |
| EGFP-GAPDH-WT | cyto | 0.18 ± 0.038 | 0.19 ± 0.055 | 27.17 ± 1.632 |
| EGFP-GAPDH-Y94A | cyto | 0.17 ± 0.129 | 0.09 ± 0.014 | 43.76 ± 7.158 |
| EGFP-GAPDH-S98A | cyto | 0.20 ± 0.009 | 0.12 ± 0.016 | 29.15 ± 3.885 |
| EGFP-GAPDH-T99A | cyto | 0.11 ± 0.062 | 0.11 ± 0.023 | 35.52 ± 8.133 |
| EGFP-GAPDH-T99I | cyto | 0.17 ± 0.06 | 0.22 ± 0.041 | 17.46 ± 1.733 |
| EGFP-GAPDH-WT | nuclei | 0.62 ± 0.048 | 1.41 ± 0.033 | 2.91 ± 0.331 |
| EGFP-GAPDH-Y94A | nuclei | 0.23 ± 0.127 | 0.09 ± 0.034 | 39.05 ± 6.291 |
| EGFP-GAPDH-S98A | nuclei | 0.35 ± 0.136 | 0.13 ± 0.018 | 26.57 ± 3.747 |
| EGFP-GAPDH-T99A | nuclei | 0.38 ± 0.120 | 0.11 ± 0.027 | 33.01 ± 9.209 |
| EGFP-GAPDH-T99I | nuclei | 0.29 ± 0.028 | 0.21 ± 0.059 | 14.97 ± 1.855 |
Cyto: Cytoplasmic compartment; nuclei: Nuclear compartment; EGFP: Enhanced green fluorescent protein; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; FRAP: Fluorescence recovery after photobleaching.
Molecular modeling of the effects of mutation on NAD+ cofactor binding
To probe the impact of site-directed mutagenesis on the affinity of GAPDH for NAD+, we performed molecular modeling experiments. The X-ray crystal structure of human GAPDH at 1.75 Angstrom resolution (1U8F.pdb) was obtained from the Protein Data Bank (Figure 7A)[22]. Because of the high degree of conserved secondary structure near the region of interest, only the loop containing amino acid at position 99 was subjected to the dynamics calculation, the rest of the protein being held fixed as an aggregate. We determined the loop region to extend from E97 (end of the beta sheet) through T104 (first residue in the attached alpha helix). The in vacuo dynamics calculation was then submitted starting with a random initial velocity for a total duration of 100 ps at 300 K for the native and each mutant construct.
Figure 7.
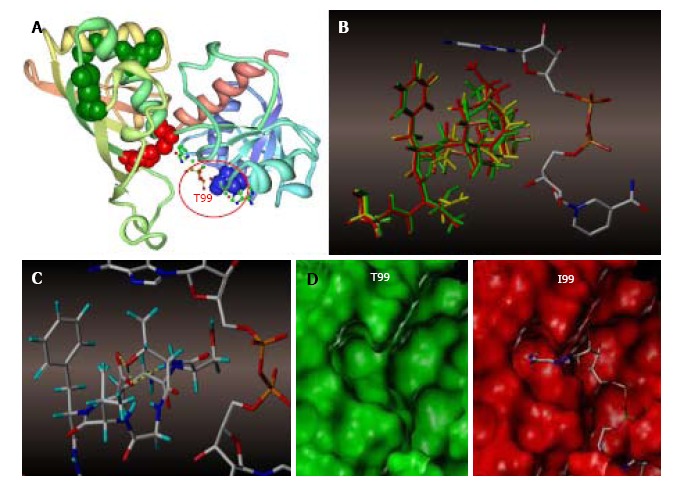
Molecular modeling of NAD+ binding site in human glyceraldehyde 3-phosphate dehydrogenase. A: NAD+ binding site within a GAPDH polypeptide (circled). Only one of four GAPDH subunits is shown. T99 is shown as a space-filled amino acid; B: The results of the trajectory calculations, with the average structure for each simulation shown overlaid for T99 (green), A99 (yellow), and I99 (red), along with the NAD+ molecule bound in the active cleft. Isoleucine side chain (red) of the T99I polypeptide acquires the closest position relative to the purine ring; C: In the native T99, an intramolecular H-bond between T99 and E97 (highlighted yellow) is likely to exist which holds the loop in a restricted conformation. This hydrogen bond could be a stabilizing factor permitting NAD+ to fully occupy the upper region of the cleft; D: Side-by-side comparison of T99 (green) and I99 (red) NAD+ binding site surfaces. In I99, the average structure places the terminus of the side chain well within the Van der Waals radii of the adenine ring of NAD+, thereby displacing or blocking the cofactor from its initial association with GAPDH. GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
The results of the trajectory calculations are shown in Figure 7B, with the average structure for each simulation being shown overlaid in green (T99), yellow (T99A), and red (T99I), along with the cofactor as initially bound in the active cleft. In the native T99, an intramolecular H-bond between T99 and E97 is likely to exist, and this H-bond was maintained throughout the simulation. This H bond has a stabilizing effect holding the loop in a restricted conformation, thereby permitting the cofactor to fully occupy the upper region of the cleft as illustrated by Figure 7C. Mutation of T99 to A99 removes the stabilizing H-bond, and the resultant loop is able to expand somewhat into the cleft and potentially affect the binding affinity of the cofactor to GAPDH. This hypothesis is most clearly demonstrated in the mutation to I99, where the average structure places the terminus of the sidechain well within the Van der Waals radii of the adenine ring of NAD+, thereby displacing or blocking the cofactor from its initial association with GAPDH, as illustrated by the surfaces shown in Figure 7D.
DISCUSSION
Multiple GAPDH isoforms characterized by different isoelectric points have long been known to exist in human and non-human tissues, as summarized in[24]. Because no kinetic differences were detected among the GAPDH isozymes, their role remains poorly understood. Nuclear isoforms of GAPDH are differentially regulated during apoptosis indicating important, yet to be characterized intranuclear functions[25]. In line with these data, our observations (Figure 1) also indicated the presence of post-translational modifications in GAPDH polypeptide[9]. Proteolytic digestion and subsequent MS analysis of the basic GAPDH forms with pI 8.3 revealed phospho amino acids within peptide 87-107, which were further identified as Y94, S98, and T99[26]. While multiple phosphorylation sites in GAPDH have been predicted or experimentally demonstrated, their functional role still remains obscure.
Western blot analysis with anti-GAPDH or anti-phosphoT99 antibody revealed two basic phosphorylated GAPDH isoforms in cytosol of the cells (Figure 3A). Consistently, GAPDH was excluded from the nuclei of untreated cells, but after treatment with araC, we found GAPDH phosphorylated at position T99 in the nuclei (Figure 3B).
Peptide 87-107 is a part of NAD+ binding center, as demonstrated by X-ray analysis[22]. Specifically, T99 is involved in non-polar interactions with the adenine residue of NAD+, while S98 forms electrostatic interactions with 5’-0-phosphate adjacent to nicotinamide nucleoside. The same NAD+ binding center is also involved in binding of GAPDH with RNA[27].
In order to characterize the role of phosphorylated amino acids in NAD+ binding center, we performed alanine scanning by consecutive substituting Y94, S98, and T99 with Ala. Additionally, we prepared T99I mutated GAPDH. T99I mutation results from a single nucleotide variation rs11549329 found in NCBI dbSNP database, which is likely a rare mutation.
Kinetic parameters of wild type and four mutated GAPDH variants were quite different depending on the nature of mutation. The most dramatic changes were detected in T99I variant where Km for NAD+ was increased by more than an order of magnitude compared with wild type GAPDH. In line with these findings, the rate of the reaction catalyzed by T99I was very low (Figure 4). Mutation T99A had less effect on GAPDH parameters, with Km (NAD+) about 2 times higher, and Km (G3P) about 1.5 times higher compared with wild type GAPDH. Mutation S98A at position adjacent to T99 showed an effect similar to that of T99A. Interestingly, Y94A mutation only slightly affected kinetics of the enzyme, while its effect on molecular dynamics parameters of nuclear GAPDH was quite prominent (Tables 2 and 3). The results of the molecular modeling experiments were in good agreement with the observed changes in enzyme kinetics of the mutated polypeptides (Figure 7).
Intracellular distribution of wild type and mutant GAPDH fused to EGFP was assessed in p53-proficient HCT116-4016 and SW48-297 cells using confocal microscopy and image analysis. Unexpectedly, mutations in the NAD+ binding center changed distribution of fused EGFP-GAPDH proteins between cytoplasmic and nuclear compartments. Consistent with our previous results, wild type EGFP-GAPDH was excluded from nuclei of untreated cells, and showed intranuclear accumulation following genotoxic stress. In contrast, T99A and T99I mutant proteins were already notable in the nuclei of untreated cells. Upon incubation with araC, significantly higher levels of intranuclear T99A, and T99I GAPDH were observed far exceeding the level of wild type intranuclear GAPDH under stress conditions (Figure 5B). Both mutated proteins contained non-phosphorylated amino acids in the NAD+ binding center, and manifested decreased affinity to NAD+. It remains to be elucidated what molecular events alter the intracellular distribution of the fusion proteins. Earlier, we demonstrated that GAPDH export from the nucleus was facilitated by CRM1[21]. We hypothesize that the redistribution of mutated GAPDH between the cytoplasmic and nuclear compartments could result from less effective interaction of mutated GAPDH polypeptides with the CRM1-mediated nuclear export system.
In our previous studies, we estimated the diffusion coefficient and the immobile fraction of GAPDH inside the living A549 cells[3]. In the present study, we performed FRAP analysis of live HCT116-4016 and SW48-297 cells treated with araC. The results of these experiments in HCT116-4016 are shown on Figure 6, and summarized in Table 3. Similarly to our experiments on intracellular distribution of wild type and mutated polypeptides, we detected profound difference in molecular dynamics characteristics of GAPDH variants. After photo bleaching, the wild type GAPDH fusion protein demonstrated ten-fold lower mobility in the nuclear compartment of HCT116-4016 cells under genotoxic stress, compared to cytoplasm. The fraction of immobile wild type EGFP-GAPDH was 0.62 ± 0.048 in the nuclei of araC-treated cells vs 0.18 ± 0.055 in cytoplasmic compartment. This observation parallels our findings for wild type EGFP-GAPDH in the A549 cell line[3]. Our results indicate that, inside the nucleus of the stressed cells, GAPDH loses it mobility, and significant fraction of polypeptides remains immobile presumably due to strong binding to as yet unidentified macromolecule or macrostructure. In parallel to our findings, intranuclear GAPDH in dexamethasone-treated S49 cells was found resistant to extraction with high salt or DNase treatment, consistent with our FRAP experiments[28]. David-Cordonnier’s group reported binding of GAPDH with chromatin following DNA alkylation with antitumor drug S23906-1[14].
The immobile fraction was notably lower in Y94A, S98A, T99A, and T99I (Table 3). All four mutated variants had similar diffusion coefficients in cytoplasmic and nuclear compartments, in contrast to wild type nuclear GAPDH which had lower D and higher (1-Mf) values. These results indicate that mutated polypeptides are involved in less tight intranuclear interactions than their wild type counterpart, with as yet unknown nuclear components (Table 3). We conclude that the phosphorylated amino acids or functionality of NAD+ binding center is important for GAPDH intranuclear immobilization.
Tight interaction of intranuclear GAPDH with nuclear component(s) is an intriguing observation, considering the fact that multiple stress factors promote intranuclear accumulation of this protein. At least two groups of intranuclear proteins, sirtuins and ADP-ribosyl transferases, which control stress response and DNA repair, are NAD+-dependent. Both groups are present in the nucleus, and require NAD+ for their activity. Importantly, GAPDH was demonstrated to directly interact with NAD+-dependent Sir2 histone deacetylase, and poly (ADP-ribose) polymerase-1[29,30]. Therefore, we speculate that GAPDH could provide a nuclear NAD+ reservoir, and deliver it to these proteins following the stress stimulus.
T99I variant is especially interesting as it is not an entirely artificial construct. The cDNA clone containing missense rs11549329 leading to T99I mutation was prepared from melanotic melanoma cells (GenBank: BM904848.1). This mutated variant manifests very low affinity to NAD+ (Figure 4 and Table 2), high level of intranuclear accumulation (Figure 5), and a modest immobile fraction after genotoxic stress (Figure 6). Treatment with genotoxic drug does not alter mobility (diffusion coefficient) of this mutated form inside the nucleus or cytosol (Table 3).
Molecular modeling of NAD+ binding site in human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) showed differential effects of amino acid changes at position 99 (Figure 7A). Interestingly, our molecular modeling experiment suggested the existence of a hydrogen bond between T99 and E97. Such intramolecular H-bond is likely to stabilize the loop extending from E97 (end of the beta sheet) through T104 (first residue in the attached alpha helix). This H-bond suffices to hold the loop in a restricted conformation, thereby permitting the cofactor to fully occupy the upper region of the cleft (Figure 7C).
Though T99A mutation does not prevent NAD+ binding, decreased T99A GAPDH catalytic activity is best explained by the disruption of such T99-E97 intramolecular H-bond. Mutation of T99A removes the stabilizing H-bond, and the resultant loop is able to expand somewhat into the cleft and potentially affect the binding affinity of the cofactor to GAPDH. This hypothesis has been supported by our kinetic experiments with mutated GAPDH proteins in vitro, as Km of the mutant forms did decrease by 2-10 fold (Table 2). Further confirmation for T99-E97 interaction requires additional testing, for example by mutational scanning at E97. Finally, analysis of the average potential energies of the dynamics trajectories also indicates that the system exhibits decreased stabilization (higher average potential energy and total energy) upon mutation and concomitant expansion of the loop into the cleft (Table 4).
Table 4.
Average potential energy, kinetic energy, total energy and temperature calculated from dynamic simulations
| Polypeptide variant | Average potential (kcal) | Average kinetic energy (kcal) | Average total energy (kcal) | Average temperature (°C) |
| T99 | -5021.8 | 97.1 | -4924.7 | 293.4 |
| A99 | -5007.3 | 93.7 | -4913.6 | 293.7 |
| I99 | -4997.8 | 101.4 | -4896.4 | 293.3 |
Our model predicts T99I to have the strongest effect on NAD+ binding and catalytic activity of GAPDH. Indeed, insertion of Ile at position 99 brings its side chain in close proximity with adenine of the cofactor (Figure 7). Modeling of the van der Waals surfaces inside the NAD+ binding center shows sterically unacceptable interaction so that such interaction either displaces, or blocks cofactor binding by T99I GAPDH.
In conclusion, we detected the phosphorylated amino acid residues Y94, S98, T99 within the NAD+ binding center of GAPDH. Substitution of these amino acids with non-phosphorylated alanine residues did not abrogate intranuclear localization of GAPDH. Instead, such mutations altered the molecular dynamics parameters of intranuclear GAPDH probably by hindering its interactions with yet to be identified nuclear biomolecules. Our molecular modeling experiments invoke an important structural feature -T99-E97 H-bond likely involved in stabilization of NAD+ binding center.
ACKNOWLEDGMENTS
We thank Dr. Magid Abou-Gharbia (The Moulder Center for Drug Discovery Research, Temple University) for support in preparation of anti-phospho GAPDH antibody; Dr. Patrick J. Piggot and Dr. Bettina A Buttaro (Temple University School of Medicine) for their help in performing confocal microscopy and FRAP experiments; Nilay Shah for his help in preparation of cells producing mutated GAPDH polypeptides.
COMMENTS
Background
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is an important enzyme of the glycolytic pathway, and an intriguing moonlighting protein involved in multiple, seemingly unrelated biochemical processes. It is known to exist in multiple isoforms, biological role and physiological significance of which remain enigmatic. Distribution of GAPDH between cellular compartments reveals complex patterns, and is regulated by partially characterized mechanisms.
Research frontiers
Among multiple functions ascribed to GAPDH, its contribution to the nuclear processes remains obscure. The role of intranuclear GAPDH isoforms in maintaining the integrity of genome is a new direction of study.
Innovations and breakthrough
This is the first study in which the role of phosphorylation sites in a NAD+ binding center of GAPDH has been explored by site-mutagenesis, and the dynamics parameters of nuclear GAPDH have been directly measured in fluorescence recovery after photobleaching experiments.
Applications
These results indicate an unexpected, important role phosphorylated amino acids within the NAD+ binding center play in intranuclear functions of GAPDH, and suggest a single mechanism by which GAPDH contributes to formation and functioning of its multiple binding partners.
Terminology
Fluorescence recovery after photobleaching is an optical technique for quantifying the diffusion of the fluorescently labeled biological molecules inside the living cells.
Peer-review
In this study, Phadke et al investigate the phosphorylated amino acid residues Y94, S98, T99 within the NAD+ binding center of glyceraldehyde 3-phosphate dehydrogenase. Substitution of these amino acids with non-phosphorylated alanine residues did not abrogate intranuclear localization of GAPDH. Instead, such mutations altered the molecular dynamics parameters of intranuclear GAPDH probably by hindering its interactions with yet to be identified nuclear biomolecules. Molecular modeling experiments suggest an important structural feature -T99-E97 H-bond likely involved in stabilization of NAD+ binding center.
Footnotes
P- Reviewer: Hong YR, Sesti F, Shihori T S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
Supported by The National Cancer Institute, No. R01-CA104729; Jayne Haines Center for Pharmacogenomics and Drug Safety of Temple University School of Pharmacy and Temple University Summer Research Award (to Evgeny Krynetskiy).
Conflict-of-interest statement: To the best of our knowledge, no conflict of interest exists.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 30, 2015
First decision: August 20, 2015
Article in press: September 30, 2015
References
- 1.Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J Cell Biochem. 2005;95:45–52. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- 2.Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S, Ramotar D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Biol Chem. 2008;283:30632–30641. doi: 10.1074/jbc.M801401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phadke MS, Krynetskaia NF, Mishra AK, Krynetskiy E. Glyceraldehyde 3-phosphate dehydrogenase depletion induces cell cycle arrest and resistance to antimetabolites in human carcinoma cell lines. J Pharmacol Exp Ther. 2009;331:77–86. doi: 10.1124/jpet.109.155671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phadke M, Krynetskaia N, Mishra A, Krynetskiy E. Accelerated cellular senescence phenotype of GAPDH-depleted human lung carcinoma cells. Biochem Biophys Res Commun. 2011;411:409–415. doi: 10.1016/j.bbrc.2011.06.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakajima H, Amano W, Fujita A, Fukuhara A, Azuma YT, Hata F, Inui T, Takeuchi T. The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J Biol Chem. 2007;282:26562–26574. doi: 10.1074/jbc.M704199200. [DOI] [PubMed] [Google Scholar]
- 7.Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 8.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 9.Seo J, Jeong J, Kim YM, Hwang N, Paek E, Lee KJ. Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J Proteome Res. 2008;7:587–602. doi: 10.1021/pr700657y. [DOI] [PubMed] [Google Scholar]
- 10.Meyer-Siegler K, Mauro DJ, Seal G, Wurzer J, deRiel JK, Sirover MA. A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc Natl Acad Sci USA. 1991;88:8460–8464. doi: 10.1073/pnas.88.19.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krynetski EY, Krynetskaia NF, Gallo AE, Murti KG, Evans WE. A novel protein complex distinct from mismatch repair binds thioguanylated DNA. Mol Pharmacol. 2001;59:367–374. doi: 10.1124/mol.59.2.367. [DOI] [PubMed] [Google Scholar]
- 12.Krynetski EY, Krynetskaia NF, Bianchi ME, Evans WE. A nuclear protein complex containing high mobility group proteins B1 and B2, heat shock cognate protein 70, ERp60, and glyceraldehyde-3-phosphate dehydrogenase is involved in the cytotoxic response to DNA modified by incorporation of anticancer nucleoside analogues. Cancer Res. 2003;63:100–106. [PubMed] [Google Scholar]
- 13.Xing C, LaPorte JR, Barbay JK, Myers AG. Identification of GAPDH as a protein target of the saframycin antiproliferative agents. Proc Natl Acad Sci USA. 2004;101:5862–5866. doi: 10.1073/pnas.0307476101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lenglet G, Depauw S, Mendy D, David-Cordonnier MH. Protein recognition of the S23906-1-DNA adduct by nuclear proteins: direct involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Biochem J. 2013;452:147–159. doi: 10.1042/BJ20120860. [DOI] [PubMed] [Google Scholar]
- 15.Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S. Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol. 2008;38:541–550. doi: 10.1165/rcmb.2007-0221OC. [DOI] [PubMed] [Google Scholar]
- 16.Krynetskaia NF, Phadke MS, Jadhav SH, Krynetskiy EY. Chromatin-associated proteins HMGB1/2 and PDIA3 trigger cellular response to chemotherapy-induced DNA damage. Mol Cancer Ther. 2009;8:864–872. doi: 10.1158/1535-7163.MCT-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan X, Kelsen SG, Clarkson AB, Ji R, Merali S. SILAC analysis of oxidative stress-mediated proteins in human pneumocytes: new role for treacle. Proteomics. 2010;10:2165–2174. doi: 10.1002/pmic.201000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabut G, Ellenberg J. Photobleaching techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, iFRAP, and FLIP. In: Goldman RD and Spector DL., editor. Cold Spring Harbor. New York: Cold Spring Harbor Laboratory Press; 2005. pp. 101–126. [DOI] [PubMed] [Google Scholar]
- 19.Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kappel C, Eils R. Fluorescence recovery after photobleaching with Leica TCS SP2. USA: Confocal Application Letter; 2004. pp. 2–12. [Google Scholar]
- 21.Brown VM, Krynetski EY, Krynetskaia NF, Grieger D, Mukatira ST, Murti KG, Slaughter CA, Park HW, Evans WE. A novel CRM1-mediated nuclear export signal governs nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase following genotoxic stress. J Biol Chem. 2004;279:5984–5992. doi: 10.1074/jbc.M307071200. [DOI] [PubMed] [Google Scholar]
- 22.Jenkins JL, Tanner JJ. High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr. 2006;62:290–301. doi: 10.1107/S0907444905042289. [DOI] [PubMed] [Google Scholar]
- 23.Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 24.Seidler NW. Target for diverse chemical modifications. Adv Exp Med Biol. 2013;985:179–206. doi: 10.1007/978-94-007-4716-6_6. [DOI] [PubMed] [Google Scholar]
- 25.Saunders PA, Chen RW, Chuang DM. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J Neurochem. 1999;72:925–932. doi: 10.1046/j.1471-4159.1999.0720925.x. [DOI] [PubMed] [Google Scholar]
- 26.Krynetskiy E, Krynetskaia N, Phadke M, Mishra A, Gothe S, Barrero C, Merali S. Targeting the intranuclear functions of GAPDH: phosphoThr99-GAPDH is translocated into nucleus of A549 cells after genotoxic stress. The FASEB J. 2014;28:Suppl 739.6. [Google Scholar]
- 27.Nagy E, Rigby WF. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold) J Biol Chem. 1995;270:2755–2763. doi: 10.1074/jbc.270.6.2755. [DOI] [PubMed] [Google Scholar]
- 28.Sawa A, Khan AA, Hester LD, Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci USA. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakajima H, Kubo T, Ihara H, Hikida T, Danjo T, Nakatsuji M, Shahani N, Itakura M, Ono Y, Azuma YT, et al. Nuclear-translocated Glyceraldehyde-3-phosphate Dehydrogenase Promotes Poly(ADP-ribose) Polymerase-1 Activation during Oxidative/Nitrosative Stress in Stroke. J Biol Chem. 2015;290:14493–14503. doi: 10.1074/jbc.M114.635607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ringel AE, Ryznar R, Picariello H, Huang KL, Lazarus AG, Holmes SG. Yeast Tdh3 (glyceraldehyde 3-phosphate dehydrogenase) is a Sir2-interacting factor that regulates transcriptional silencing and rDNA recombination. PLoS Genet. 2013;9:e1003871. doi: 10.1371/journal.pgen.1003871. [DOI] [PMC free article] [PubMed] [Google Scholar]