

Abstract
Cyclin A2 is an essential regulator of the cell division cycle through the activation of kinases that participate to the regulation of S phase as well as the mitotic entry. However, whereas its degradation by the proteasome in mid mitosis was thought to be essential for mitosis to proceed, recent observations show that a small fraction of cyclin A2 persists beyond metaphase and is degraded by autophagy. Its implication in the control of cytoskeletal dynamics and cell movement has unveiled its role in the modulation of RhoA activity. Since this GTPase is involved in both cell rounding early in mitosis and later, in the formation of the cleavage furrow, this suggests that cyclin A2 is a novel actor in cytokinesis. Taken together, these data point to this cyclin as a potential mediator of cell-niche interactions whose dysregulation could be taken as a hallmark of metastasis.
Keywords: Cyclin, Mitosis, Mesenchymal transition, Metastasis, Autophagy, RhoA, Phospholipase C
Core tip: Cyclin A2, as an essential regulator of the cell division cycle, is commonly associated to dividing cells and, like Ki67, is usually taken as a marker of cell proliferation. However, the level of this cyclin does not always correlate with the aggressiveness of the tumor, more particularly with respect to its invasiveness. Surprisingly, recent data suggest that it plays with RhoA also a role in the late phase of mitosis during which it is degraded by autophagy. Moreover, its dysregulation appears to be associated with the epithelial to mesenchymal transition.
INTRODUCTION
The cell division cycle is controlled by the coordinated expression of regulatory proteins whose degradation is orchestrated by specific and timely phosphorylation/dephosphorylation events. Cyclins, as binding and activating partners of cyclin-dependent kinases (CDK) constitute a key subset of the regulatory components of the cell cycle engine. In animal cells, cyclins A and B play a central role in the control of mitosis, with cyclin A being degraded before cyclin B by the proteasome, just after the nuclear envelope breakdown. In mammalian cells there are two A-type cyclins: Cyclin A1 that is specifically expressed in the testis, and cyclin A2, that is ubiquitously expressed. Accordingly, cyclin A2 is usually linked to cell proliferation and as such is often found expressed at a high level in human cancers[1]. However, the level of this cyclin does not always correlate with the aggressiveness of the tumor, more particularly with respect to its invasiveness[2].
A wealth of information has accumulated suggesting that some cell cycle regulators play a more general role in cellular transactions[3,4], and along these lines, cyclin A2 has been shown to participate in the control of cytoskeleton dynamics and cell motility[5].
CYCLIN A2: A LATE ACTOR OF CYTOKINESIS?
Using cyclin A2-EGFP to study its ubiquitylation by FRET, we recently demonstrated that it starts in foci in prometaphase and then spreads throughout the cell in metaphase[6]. Indeed, endogenous cyclin A2 colocalized within these structures with Cdc20, a key regulator of its ubiquitylation, as well as with active proteasome, as detected with DQ-ovalbumin[6]. This unprecedented observation revealed two unexpected aspects of cyclin A2 expression: (1) not all foci colocalized with Cdc20 or DQ-ovalbumin; and (2) cyclin A2 foci persisted until late mitosis, i.e., when its proteasomal degradation is thought to be complete. Since this cyclin is absent at the beginning of the G1 phase of the next round of cell division, this led us to see whether autophagy, another major intracellular proteolysis pathway, could also be involved in its degradation.
Autophagy is a degradation pathway that eliminates misfolded, damaged, or superfluous cell components, whether individual molecules or organelles, into recycled pools of biomolecules. The operating structures of autophagic degradation are the lysosomes that contain many hydrolases that break down proteins, lipids as well as nucleic acids[7]. Interestingly, some endogenous cyclin A2 foci colocalized with light chain 3-B protein (LC3-B), a marker of autophagosomes, in metaphase as well as with p62[6], a receptor for ubiquitylated proteins necessary for their degradation by selective autophagy[8]. Furthermore, our data suggest that, in prometaphase, cyclin A2 foci colocalized mainly with Cdc20 or activated proteasome, while in metaphase, they colocalized mainly with LC3-B, p62, or lysosomes[6].
Characterized first as a general recycling process for defective structures, autophagy is now proposed to participate in regular cellular regulatory pathways. Accordingly, our observation showing that it plays a complementary role in cyclin A2 degradation to prevent its accumulation at the end of mitosis, points to a potential novel and unexpected function for this cyclin late in cytokinesis. Indeed, formation of the mitotic cleavage furrow depends upon the activity of RhoA via its exchange factors such as Ect2[9] and GEF-H1 that localizes to the mitotic apparatus[10,11]. Our previous observation unveiled the potentiating effect of cyclin A2 on RhoA GTP loading by its exchange factors[5]. Thus, formation of the contractile ring, which is dependent upon local activation of the GTPase, would appear to result from its concomitant interaction with Ect2 and/or microtubule-activated GEF-H1 and cortical cyclin A2.
Finally, autophagy has already been shown to participate in abscission of the cleavage furrow after telophase and to promote the degradation of active RhoA[12]. This suggests that cyclin A2 may be degraded by autophagy with the GTPase (in the same complex?) when localized to cortical membranes, whereas degradation of its soluble fraction would occur mainly through proteasomal activity (Figure 1).
Figure 1.
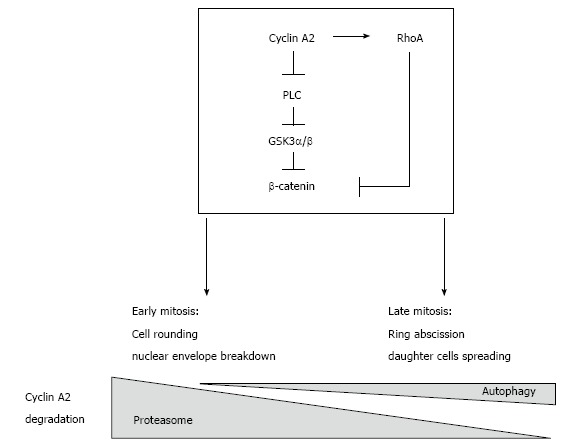
Presumptive regulatory network involving cyclin A2 and RhoA in the control of mitosis and epithelial to mesenchymal transition. Cyclin A2 degradation by the proteasome in mid mitosis is essential for mitosis to proceed. However, recent observations show that a small fraction of cyclin A2 persists beyond metaphase and is degraded by autophagy. Cyclin A2 participates in the modulation of RhoA activity; since this GTPase is involved in both cell rounding early in mitosis and later, in the formation of the cleavage furrow, this suggests that cyclin A2 plays also a role in cytokinesis. Taken together, these data point to this cyclin as a potential mediator of cell-niche interactions, its down regulation leading to EMT via PLC and RhoA converging pathways. EMT: Epithelial to mesenchymal transition; PLC: Phospholipase C.
EPITHELIAL TO MESENCHYMAL TRANSITION AND CYCLIN A2 DYSREGULATION
The epithelial to mesenchymal transition (EMT) is a normal biological process that is important in organogenesis during early development and in wound healing. It has recently become a leading concept to explain metastasis[13]. This process entails the trans differentiation of epithelial cells to mesenchymal cells, with an associated increase in invasive properties. Cyclin A2 depletion in fibroblasts leads to an increase in cell motility and cooperates with oncogenic transformation to increase invasiveness in collagen matrices[5,14], a phenomenon that is associated with its cytoplasmic localization and independent of its association with the CDKs. When performed in epithelial cells, cyclin A2 knockdown induces an EMT in RasV12-transformed mammary epithelial cells, and increases their invasiveness in vitro as well as in chicken embryos[14] (discussed in 15). Interestingly, these cells form tumorospheres and are more resistant to anoikis under non-adherent conditions. Whereas the Rho GTPases, RhoA and RhoC, were shown to be responsible for the invasion phenotype, with RhoA involved in the maintenance of cell-cell junctions, they do not fully account for the observed EMT phenotype[15].
Oncogenic EMT, which is thought to arise during tumorigenesis, is generally characterized by a decrease and/or a delocalization of epithelial markers (E-cadherin, occludin), upregulation of mesenchymal markers (N-cadherin, vimentin) and transcriptional factors (Zeb1/2, Slug, Snail, Twist1/2, etc.), as well as functional attributes such as increase in migration, invasion, cell scattering and resistance to anoikis[16].
Multiple signalling pathways are known to be dysregulated during tumorigenesis and EMT. Among them, the WNT pathway[17] has been shown to play a major role in metastasis and promotion of EMT in breast cancer, in which expression of WNT ligands or suppression of their inhibitors induces EMT and metastasis[18]. The canonical WNT signalling pathway is initiated by binding of a WNT ligand to a frizzled (FZD) receptor and its associated co-receptors, low-density lipoprotein receptor–related proteins 5/6. This inactivates glycogen synthase kinase-3β (GSK-3β) via phosphorylation, followed by the release, dephosphorylation, and nuclear translocation of β-catenin, where it binds to T-cell factor/lymphocyte enhancer factor components and activate transcription of target genes, such as the mesenchymal marker, fibronectin[19].
β-catenin is also known to be regulated independently of the WNT pathway, through inactivation of GSK-3β and/or delocalization of E-cadherin, which ultimately liberates β-catenin to undergo dephosphorylation and nuclear translocation. Some reports have shown that WNT-independent β-catenin regulation can occur through transduction components such as extracellular signal-regulated kinases 1 and 2, protein kinase A, protein kinase B (AKT1), as well as phospholipases A and C isoforms. Recently, phospholipase C (PLC) isoforms have been shown to be major players in EMT, mainly in metastatic breast cancer cells, due to their roles in regulating cytoskeletal organization, differentiation and other signal transduction pathways[20]. Interestingly, isoforms of PLC are known to be activated by Rho GTPases[21,22], and PLC-γ is also regulated by the E-cadherin/β-catenin complex at the plasma membrane[23].
Our recent observations suggest that inhibition of the β-catenin pathway and/or PLC might lead to a reversion of EMT induced by cyclin A2 depletion in Ras-transformed cells, with a concomitant increase in E-cadherin expression and localization to cell membrane, as well as decrease in mesenchymal traits in the same cellular context[24]. Thus, EMT induced by cyclin A2 depletion could well be dependent on PLC and β-catenin activation.
Interestingly, a previous report showed that the integrity of adherens junctions results from the balance of the antagonistic activities of RhoA and RhoC[25], a low level of cyclin A2 displacing the equilibrium toward the escape of β-catenin from the junction. That the two processes are linked or belong to synergistic pathways remains to be established. In any case, a down regulation of cyclin A2 expression appears to promote EMT and invasive properties. Accordingly, cyclin A2 protein levels have been shown to be much lower in human samples obtained from metastatic sites in comparison to matched primary colon adenocarcinomas, which suggest that a decreased expression level of cyclin A2 could be linked to cancer metastasis[25].
CONCLUSION
Metastasis relies on the acquisition by the candidate cell of invasive properties based on morphological changes that allow it to modify its interactions with its neighbours and, more generally, to reinterpret cues emanating from its surrounding niche. Mitosis offers such an opportunity to a cell embedded within an epithelial structure to change its fate. Indeed, while it is associated to a drastic change in cell shape, it also entails dramatic alterations of intracellular structures such as chromosome condensation and nuclear envelope breakdown. It is thus not surprising to find that dysregulation of cyclin A2 and RhoA, two major actors of its control, involved in both its early step, cell rounding, and then later, the spreading of the two daughter cells, be instrumental in giving rise to cells that escape niche controls. Interestingly, recent studies on circulating tumor cells have revealed that a large number of them harboured a phenotype that supports the idea that they were generated through an EMT-like mechanism[26]. Thus, promoting a mechanism such as EMT is nothing else but another example of how a cancer cell co-opts fundamental cellular mechanisms that are important in the early development or in wound healing.
ACKNOWLEDGMENTS
We thank Robert Hipskind and Daniel Fisher for fruitful discussions.
Footnotes
P- Reviewer: Carter WG, Wang YQ, Zhang WZ, Zhou SH S- Editor: Tian YL L- Editor: A E- Editor: Lu YJ
Supported by Agence Nationale de la Recherche (08-BLAN-0037-02), Fondation ARC pour la Recherche sur le Cancer, and Gefluc; CNRS/Région Languedoc-Roussillon and Fondation pour la Recherche Médicale (Loukil A); the French Ministry of Education and Research and Fondation pour la Recherche Médicale (Bendris N); and the Canadian Institutes of Health Research, La Ligue Contre le Cancer and the Fondation de France (Cheung CT).
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 14, 2015
First decision: May 13, 2015
Article in press: October 13, 2015
References
- 1.Bendris N, Loukil A, Cheung C, Arsic N, Rebouissou C, Hipskind R, Peter M, Lemmers B, Blanchard JM. Cyclin A2: a genuine cell cycle regulator? Biomol Concepts. 2012;3:535–543. doi: 10.1515/bmc-2012-0027. [DOI] [PubMed] [Google Scholar]
- 2.Blanchard JM. To be or not to be a proliferation marker? Oncogene. 2014;33:954–955. doi: 10.1038/onc.2013.19. [DOI] [PubMed] [Google Scholar]
- 3.Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: an oncogenic function for CDK inhibitors? Nat Rev Cancer. 2004;4:948–955. doi: 10.1038/nrc1501. [DOI] [PubMed] [Google Scholar]
- 4.Bendris N, Lemmers B, Blanchard JM. Cell cycle, cytoskeleton dynamics and beyond: the many functions of cyclins and CDK inhibitors. Cell Cycle. 2015;14:1786–1798. doi: 10.1080/15384101.2014.998085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadéa G, Bouquier N, Bibeau F, Lemmers B, Blanchard JM. A novel function for Cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol. 2012;196:147–162. doi: 10.1083/jcb.201102085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loukil A, Zonca M, Rebouissou C, Baldin V, Coux O, Biard-Piechaczyk M, Blanchard JM, Peter M. High-resolution live-cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J Cell Sci. 2014;127:2145–2150. doi: 10.1242/jcs.139188. [DOI] [PubMed] [Google Scholar]
- 7.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 9.Kamijo K, Ohara N, Abe M, Uchimura T, Hosoya H, Lee JS, Miki T. Dissecting the role of Rho-mediated signaling in contractile ring formation. Mol Biol Cell. 2006;17:43–55. doi: 10.1091/mbc.E05-06-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jordan SN, Canman JC. Rho GTPases in animal cell cytokinesis: an occupation by the one percent. Cytoskeleton (Hoboken) 2012;69:919–930. doi: 10.1002/cm.21071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, Ilie M, Rubera I, Tauc M, Barale S, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013;73:4311–4322. doi: 10.1158/0008-5472.CAN-12-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ombrato L, Malanchi I. The EMT universe: space between cancer cell dissemination and metastasis initiation. Crit Rev Oncog. 2014;19:349–361. doi: 10.1615/critrevoncog.2014011802. [DOI] [PubMed] [Google Scholar]
- 14.Bendris N, Cheung CT, Leong HS, Lewis JD, Chambers AF, Blanchard JM, Lemmers B. Cyclin A2, a novel regulator of EMT. Cell Mol Life Sci. 2014;71:4881–4894. doi: 10.1007/s00018-014-1654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bendris N, Arsic N, Lemmers B, Blanchard JM. Cyclin A2, Rho GTPases and EMT. Small GTPases. 2012;3:225–228. doi: 10.4161/sgtp.20791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiery JP, Lim CT. Tumor dissemination: an EMT affair. Cancer Cell. 2013;23:272–273. doi: 10.1016/j.ccr.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 18.DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69:5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gradl D, Kühl M, Wedlich D. The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol Cell Biol. 1999;19:5576–5587. doi: 10.1128/mcb.19.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abalsamo L, Spadaro F, Bozzuto G, Paris L, Cecchetti S, Lugini L, Iorio E, Molinari A, Ramoni C, Podo F. Inhibition of phosphatidylcholine-specific phospholipase C results in loss of mesenchymal traits in metastatic breast cancer cells. Breast Cancer Res. 2012;14:R50. doi: 10.1186/bcr3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bourdon DM, Wing MR, Edwards EB, Sondek J, Harden TK. Quantification of isozyme-specific activation of phospholipase C-beta2 by Rac GTPases and phospholipase C-epsilon by Rho GTPases in an intact cell assay system. Methods Enzymol. 2006;406:489–499. doi: 10.1016/S0076-6879(06)06037-X. [DOI] [PubMed] [Google Scholar]
- 22.Seifert JP, Zhou Y, Hicks SN, Sondek J, Harden TK. Dual activation of phospholipase C-epsilon by Rho and Ras GTPases. J Biol Chem. 2008;283:29690–29698. doi: 10.1074/jbc.M805038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Z, Bikle DD. The recruitment of phosphatidylinositol 3-kinase to the E-cadherin-catenin complex at the plasma membrane is required for calcium-induced phospholipase C-gamma1 activation and human keratinocyte differentiation. J Biol Chem. 2007;282:8695–8703. doi: 10.1074/jbc.M609135200. [DOI] [PubMed] [Google Scholar]
- 24.Cheung CT, Bendris N, Paul C, Hamieh A, Anouar Y, Hahne M, Blanchard JM, Lemmers B. Cyclin A2 modulates EMT via β-catenin and phospholipase C pathways. Carcinogenesis. 2015;36:914–924. doi: 10.1093/carcin/bgv069. [DOI] [PubMed] [Google Scholar]
- 25.Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408–415. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- 26.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]