

Abstract
Mitochondria sense, shape and integrate signals, and thus function as central players in cellular signal transduction. Ca2+ waves and redox reactions are two such intracellular signals modulated by mitochondria. Mitochondrial Ca2+ transport is of utmost physio-pathological relevance with a strong impact on metabolism and cell fate. Despite its importance, the molecular nature of the proteins involved in mitochondrial Ca2+ transport has been revealed only recently. Mitochondrial Ca2+ promotes energy metabolism through the activation of matrix dehydrogenases and down-stream stimulation of the respiratory chain. These changes also alter the mitochondrial NAD(P)H/NAD(P)+ ratio, but at the same time will increase reactive oxygen species (ROS) production. Reducing equivalents and ROS are having opposite effects on the mitochondrial redox state, which are hard to dissect. With the recent development of genetically encoded mitochondrial-targeted redox-sensitive sensors, real-time monitoring of matrix thiol redox dynamics has become possible. The discoveries of the molecular nature of mitochondrial transporters of Ca2+ combined with the utilization of the novel redox sensors is shedding light on the complex relation between mitochondrial Ca2+ and redox signals and their impact on cell function. In this review, we describe mitochondrial Ca2+ handling, focusing on a number of newly identified proteins involved in mitochondrial Ca2+ uptake and release. We further discuss our recent findings, revealing how mitochondrial Ca2+ influences the matrix redox state. As a result, mitochondrial Ca2+ is able to modulate the many mitochondrial redox-regulated processes linked to normal physiology and disease.
Keywords: Calcium transport, Signal transduction, Redox regulation, Mitochondria, Oxidation-reduction, Mitochondrial membrane transport proteins, Mitochondrial Ca2+ uniporter, Mitochondrial Na+/Ca2+ exchanger
Core tip: Deregulated redox signaling in mitochondria leads to mitochondrial dysfunction, associated with several disorders and disease states. Matrix Ca2+ rising can be linked through multiple pathways to the mitochondrial redox state. Here we describe recent progress in the field of mitochondrial Ca2+ handling. We further summarize how mitochondrial Ca2+ signals are influencing the mitochondrial redox state. This link between Ca2+ and redox signals is likely of central importance in the regulation of mitochondrial function in health and disease.
INTRODUCTION
Mitochondria are versatile multifunctional organelles best known for their contribution to cellular energy homeostasis. Mitochondria are also central regulators of cell fate. The involvement of mitochondria in a large number of biological processes is dependent on two unique characteristics. First, mitochondria are organelles able to sense and influence a number of intracellular signals (ions and small molecules, such as Ca2+, ATP, pH and the redox potential). Second, these organelles are very dynamic. Mitochondria undergo morphological changes (they fragment and fuse)[1,2], and they increase or decrease in the mass due to mitochondrial biogenesis[3] and mitophagy[4]. Finally, they can move within the cell[5]. Mitochondrial dynamics combined with their ability to control the fluxes of ions and small molecules makes this organelle a central player in signal transduction. Two of the signals strongly affected by mitochondria are Ca2+ and redox state.
Ca2+ is a key intracellular messenger that coordinates a vast repertoire of cellular functions, ranging from contraction, secretion and fertilization to the control of transcription, proliferation, several aspects of development as well as learning and memory[6-8]. Cells express a large number of proteins for the precise spatial and temporal control of Ca2+ rising[6]. Mitochondria efficiently contribute to the shaping of Ca2+ signals through Ca2+ uptake and release[9-12]. The associated matrix Ca2+ rises (transient or prolonged) act as a signal per se that can modulate energy metabolism and cell fate[12-16]. Although the basic properties of mitochondrial Ca2+ handling have been established several decades ago, the molecular identities of the mitochondrial Ca2+ transport systems have only been revealed over the last 6 years (see section on “Molecular identification of mitochondrial Ca2+ transporters”, below). The identification and study of these transporters has improved our understanding of the physio-pathological role of mitochondrial Ca2+ transport and provided researchers with new opportunities for molecular intervention.
Signals other than Ca2+ are generated/integrated in the mitochondrial matrix, notably redox reactions linked to the production of reactive oxygen species (ROS), and changes in the oxidation state of thiol groups in proteins (thiol switches). Redox reactions and associated changes can serve as cellular signals. Metabolites and proteins can activate specific cellular signaling pathways in a redox state-dependent manner[17,18]. Thiol switches in proteins are controlled by the balance of two opposite influences, oxidizing and reducing. ROS are able to shift the equilibrium to a more oxidized state. This is particularly relevant in mitochondria, which are a major source of ROS[19,20]. Such oxidation in mitochondria is counteracted by reducing systems that depend on the availability of NAD(P)H, which is generated by mitochondrial metabolism[18]. The oxidation/reduction of thiol groups in mitochondrial target proteins can modulate their activity, localization and stability. Such changes can regulate mitochondrial functions, including nutrient oxidation, oxidative phosphorylation, ROS production, mitochondrial permeability transition, cell death and mitochondrial morphology[21]. The study of redox switches in vivo has been a challenge[22]. However, the recent development of fluorescent protein redox sensors has revolutionized the study of redox processes in living cells. They allow real-time compartment-specific monitoring of thiol redox dynamics[22-25].
Excellent reviews have covered the mechanisms of ROS production[19], the importance of redox signals in the modulation of mitochondrial function, and how deregulation of these signals can lead to the development of various disease states[21,26,27]. The effect of Ca2+ on ROS generation and in the regulation of cellular energetics has also been reviewed recently[13,28]. Here we will review the transporters mediating and regulating the entry and extrusion of calcium across the inner membrane. We will further discuss the evidences linking mitochondrial Ca2+ rises to the modulation of the matrix redox signals.
MOLECULAR IDENTIFICATION OF MITOCHONDRIAL Ca2+ TRANSPORTERS
Mitochondria are equipped with sophisticated machinery mediating Ca2+ fluxes across the inner mitochondrial membrane (Figure 1). This system is composed of channels, exchangers, regulatory proteins and a poorly characterized matrix Ca2+ buffer system. In this section, we will focus on the recently identified Ca2+ channels and exchangers.
Figure 1.
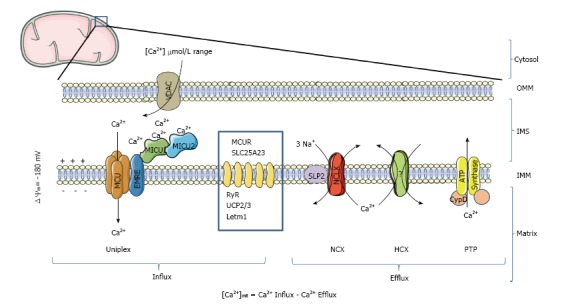
Ca2+ transport proteins of mitochondria. In mammalian mitochondria, the uptake of Ca2+ is mediated by the Ca2+-selective channel MCU, which is part of a high molecular weight protein complex called Uniplex. At least 4 additional proteins (MCUb, MICU1, MICU2 and EMRE) regulate MCU activity. Ca2+ is then extruded by a sodium/calcium exchange (NCX) or proton/calcium exchange (HCX). If the protein NCLX has been confirmed to be the mitochondrial NCX, which is down-regulated by the protein SLP-2, the molecular nature of the mitochondrial HCX is still debated. Dimers of mitochondrial ATP synthase have been proposed to form the PTP, a mitochondrial channel regulated by CypD, that facilitates PTP opening by desensitizing PTP to Ca2+. Besides being activated by Ca2+, PTP has also been proposed to act as a reversible fast Ca2+ release channel. Other non-MCU mitochondrial proteins with an indirect or debated effect on Ca2+ transport are represented in the blue square (MCUR; SLC25A23; ryanodine receptor, RyR; UCP2; UCP3; LETM1). OMM: Outer mitochondrial membrane; IMS: Inter-membrane space; IMM: Inner mitochondrial membrane; VDAC: Mitochondrial porin; PTP: Permeability transition pore; CypD: Cyclophilin D; MCU: Mitochondrial Ca2+ uniporter; MCUR1: Mitochondrial calcium uniporter regulator 1; MICU1: Mitochondrial Ca2+ uptake protein 1; MICU2: Mitochondrial Ca2+ uptake protein 2; EMRE: Essential MCU regulator.
Mitochondrial Ca2+ uptake mechanisms
Mitochondrial Ca2+ uniporter: The mitochondrial Ca2+ uniporter (MCU) is the principal mediator of Ca2+ transport into the mitochondrial matrix. The MCU catalyzes the passive and unidirectional transport of Ca2+ across the inner mitochondrial membrane, a process that is driven by the electrochemical gradient (Δψmit) in energized mitochondria. The inside negative potential of -180 mV is generated as electrons transferred stepwise along the respiratory chain from electron donors inside the matrix to the end-acceptor molecular oxygen. Given the large electrical gradient across the inner mitochondrial membrane, the organelle has the potential to import large amounts of Ca2+. At a concentration of 100 nmol/L cytosolic Ca2+ in a resting cells and a mitochondrial potential of -180mV, the Nernst equation would predict that the equilibrium will only be reached at 100 mmol/L mitochondrial [Ca2+]. The result would be a gradient of six orders of magnitude between matrix and cytosolic calcium concentration. However, this matrix Ca2+ concentration is never reached under physiological conditions for three reasons: (1) the mitochondrial Ca2+ uptake mechanism displays low Ca2+ affinity, and the Kd has been estimated close to 10 μmol/L. Therefore efficient mitochondrial Ca2+ uptake only occurs in the μmol/L cytosolic [Ca2+] range, protecting the mitochondria from Ca2+ overload in resting cells; (2) mitochondria activate Ca2+ extrusion as soon as [Ca2+] rises in the mitochondrial matrix, following cell stimulation; and (3) the mitochondrial matrix contains a poorly defined high capacity Ca2+ buffer system, composed of PO4-, Ca2+ binding proteins and metabolites that significantly reduce free mitochondrial [Ca2+].
Electrophysiological recordings in mitoplasts have revealed the existence of an inward rectifying highly Ca2+-selective current across the inner mitochondrial membrane (IMCU)[29]. This current was shown to be reflecting mitochondrial Ca2+ uniport activity as it was blocked by two well-characterized pharmacological inhibitors of MCU, ruthenium red and Ru360[29]. This study was the first to define the electrophysiological properties of the MCU.
Following a long-lasting search for the proteins responsible for mitochondrial calcium uniport, two groups independently identified the essential component of the MCU in 2011[30,31]. The Mitocarta, the most complete compendium of mitochondrial proteins, was used as a starting point for the identification of MCU1 (mitochondrial Ca2+ uptake protein 1). Baughman et al[30] identified MCU1 on the basis of an integrative genomic approach combining whole-genome phylogenetic profiling, genome-wide co-expression analysis and organelle-wide protein co-expression analysis to predict proteins functionally related with MICU1. MICU1 had previously been identified as essential for mitochondrial Ca2+ uptake[32]. De Stefani et al[31] uncovered MCU1 by analyzing well-known and predicted characteristics of the mitochondrial uptake mechanism: the ubiquitous expression in mammalian cells, its absence in yeast (which lacks a ruthenium red-sensitive Ca2+ uptake mechanism), its presence in kinetoplastids (which express a ruthenium red-sensitive Ca2+ uptake mechanism), and the presence of two or more predicted transmembrane domains.
The MCU is part of a high molecular weight protein complex called Uniplex (Uniporter Complex, Figure 1). This complex is comprised of at least 5 different proteins: MCU1[30,31], MCUb[33], MICU1[32], MICU2[34,35] (mitochondrial Ca2+ uptake protein 2) and essential MCU regulator (EMRE)[36]. Less well established is the interaction with and functional relevance of 2 additional mitochondrial proteins: MCUR and SLC25A23. MCU1 constitutes the pore-forming subunit. This essential component of the uniporter is sufficient for uniporter activity[37]. MCU1 contains two α-helix trans-membrane domains connected by a loop in the inter-membrane space[30]. Biochemical evidence and computational modeling predict MCU1 forming a tetramer. The trans-membrane domains build the pore of the channel. The loops, facing the inter-membrane space, constitute the mouth of the channel, which also confers Ca2+ selectivity. Sequence analysis of MCU led to the identification of a new MCU1 paralogue, named MCUb. This protein can replace MCU1 subunits resulting in different MCU1/MCUb ratios as observed in different tissues. MCUb can be considered a dominant negative pore-forming version of MCU1. Its presence adds a regulatory mechanism that modulates the properties of the channel[33]. MICU1 and MICU2 are Ca2+-sensitive subunits of the complex[32,38]. Both carry EF-hand (helix-loop-helix) Ca2+ binding domains facing the inter-membrane space (Figure 1). Indirectly, these EF-hands sense cytosolic signals. MICU1 and MICU2 are likely to work as a gatekeeper defining the activation threshold of the channel, preventing the activity at resting Ca2+ levels (100 nmol/L) and triggering MCU activity when Ca2+ microdomains (several μmol/L) are generated close to the channel[39,40]. MICU1 has been shown to control cooperativity of Ca2+ uptake, a well-defined characteristic of the MCU. This regulatory mechanism favors the active state of the channel at high cytosolic [Ca2+][39]. The Ca2+ binding affinity of each active helix-loop-helix domain on MICU1 and MICU2 has been estimated to be in the range of 15-21 μmol/L. These values are consistent with the necessity to reach high Ca2+ microdomains as an essential requirement for full MCU activation[41]. The recently obtained crystal structure of MICU1 suggests that in the absence of Ca2+, the protein forms hexamers that inhibit MCU1. Conversely, in the presence of Ca2+, MICU1 undergoes a conformational change, forming multiple oligomers that activate MCU1[41]. Biochemical evidence suggests that MICU2 physically interacts with MICU1, which in turn interacts with MCU1. Functional interaction studies between MCU1, MICU1 and MICU2 suggest that both regulatory subunits contribute to MCU activation as a function of the amount of cytosolic [Ca2+]. At high cytosolic [Ca2+], the stimulatory effect of MICU1 drives the rapid response of mitochondria to cytosolic [Ca2+] rises. Conversely, at low cytosolic [Ca2+], MICU2 is required to prevent mitochondrial Ca2+ uptake[34,35]. EMRE[36] was discovered as part of the mitochondrial calcium uptake machinery by quantitative mass spectrometry, and was shown to mediate MCU1-MICU1 physical and functional interaction. When EMRE was missing, the interaction between MCU1 and MICU1/2 was disrupted, despite intact MCU1 oligomers and preserved MICU1-2 interactions[36]. EMRE knock-out and knock-down models display a strong decrease in the ability to take up Ca2+ in permeabilized mitochondria. Furthermore, IMCU Ca2+ current is virtually absent in mitoplasts from EMRE knocked-out cells. These results are in conflict with earlier results suggesting that MCU1 alone is sufficient for MCU activity[37].
A large variety of mitochondrial Ca2+ currents has been observed across different tissues and cell types[42]. Based on the current knowledge of the function of MCU components, it will be possible to study how their stoichiometry influences the variability of mitochondrial Ca2+ currents in different cell types.
A number of additional proteins have been shown to be important for mitochondrial Ca2+ uptake[43-47]. These proteins are not likely part of the Uniplex, but rather may be involved in alternative mechanisms of mitochondrial Ca2+ uptake. Therefore, this group of proteins is discussed separately. A genome-wide siRNA screen, designed to detect new proteins involved in mitochondrial Ca2+ uptake[43,44], has identified leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) as a molecule able to mediate high affinity (200 nmol/L) mitochondrial Ca2+ uptake in exchange for H+. Several laboratories have been able to reproduce the mitochondrial Ca2+ uptake defect in LETM1 knock-down cells[48,49]. Furthermore, when reconstituted in liposomes, LETM1 mainly promotes electro-neutral Ca2+/2H+ exchange[50]. However, LETM1 was previously proposed to exchange K+ against H+[51,52]. Also, thermodynamic considerations would argue against electro-neutral Ca2+/2H+ exchange, as this should promote Ca2+ extrusion rather than uptake given the proton gradient (0.8 pH units more alkaline in the matrix) across the inner mitochondrial membrane[53]. Knockout of LETM1 causes mitochondrial dysfunction, swelling and depolarization, thus reducing the driving force for mitochondrial Ca2+ uptake. Depolarization of the inner mitochondrial membrane as a secondary consequence of LETM1 disruption may explain impaired MCU-mediated Ca2+ transport[51,52,54]. The role of LETM1 in mitochondrial ion homeostasis remains controversial. MCUR1 was originally reported to be a component of the Uniplex, contributing to ruthenium red-sensitive mitochondrial Ca2+ uptake[45]. However, MCUR1 was not identified in mass spectrometry experiments of the purified Uniplex, arguing against a direct role in mitochondrial Ca2+ uptake[36]. In the absence of MCUR1 expression, oxidative phosphorylation is impaired and cellular ATP levels lower, leading to activation of AMP kinase. Recent evidence suggests that MCUR1 may instead work as a cytochrome-c oxidase assembly factor[55]. The evidence suggests that loss of MCUR1 impairs respiratory function, leading to diminished Δψmit and thereby a reduction of mitochondrial Ca2+ uptake. Recently, the protein SLC25A23, a member of the mitochondrial Ca2+-dependent solute carrier family, previously considered to be an ATP-Mg/PO4- carrier, has been shown to physically interact and positively modulate MCU activity. Lack of SLC25A23, as well as overexpression of the SLC25A23 EF-hand mutants, was shown to reduce MCU activity[46]. SLC25A23 may therefore be a regulatory subunit of the Uniplex. UCP2 and UCP3 have also been proposed to be essential for mitochondrial Ca2+ uptake[47]. This is unlikely, since mitochondria isolated from tissues of the UCP2 and UCP3 knock-out mice displayed unaltered Ca2+ uptake[56]. Recently, impaired mitochondrial Ca2+ uptake has been confirmed in UCP3 knock-out cells[57]. Even though MCU activity did not rely on the UCP3 in intact cells, the lack of UCP3 decreased cytosolic ATP available for SERCA pumps. As a result, IP3-driven cytosolic and mitochondrial Ca2+ rises were reduced[57]. The contribution of UCP2 and UCP3 to mitochondrial calcium uptake is therefore via their impact on Ca2+ handling in the ER.
Studies in cardiac cells indicate that ryanodine receptors, one of the main endoplasmic reticulum Ca2+ release channels, contribute to an alternative mitochondrial Ca2+ uptake mechanism[58,59]. Consistent with this, localization of ryanodine receptors to mitochondria was demonstrated using electron microscopy and Western-blotting[58,59]. Pharmacological inhibition with ryanodine diminished inward Ca2+ current in mitoplasts insensitive to the MCU blocker Ru360[60]. Along the same line of evidence, single channel recordings in mitoplasts from HeLa cells after knock-down of MCU1 revealed a 2.5-fold increase in the occurrence of the extra-large conductance Ca2+ current[61]. These observations are consistent with an alternative and compensatory molecular mechanism for mitochondrial Ca2+ uptake.
Mitochondrial Ca2+ release mechanisms
Two main mechanisms have been proposed to account for mitochondrial Ca2+ release[62]: (1) Na+-dependent, mediated by a recently identified mitochondrial Na+/Ca2+ exchanger named NCLX; and (2) Na+-independent, probably mediated by a H+/Ca2+ exchanger. These two mechanisms operate to extrude Ca2+ during physiological mitochondrial [Ca2+] transients. A third mechanism, called permeability transition pore (PTP, see below) opening gets activated under specific physiopathological conditions when mitochondria experience Ca2+ overload for extended periods of time[62].
The mitochondrial Na+/Ca2+ exchange discovered by Carafoli et al[63] constitutes the main pathway for mitochondrial Ca2+ extrusion[64,65]. The stoichiometry of ion exchange has been estimated to be 3Na+/Ca2+. This electrogenic export mode favors Ca2+ extrusion in energized mitochondria[66]. Like MCU, NCLX is highly selective for Ca2+ when compared with other divalent ions, but less selective for Na+ that can be replaced by Li+[63,67]. Although NCLX has recently been thought to localize to the plasma membrane, electron microscopy and cell fractionation experiments clearly showed that NCLX is targeted to the mitochondrial inner membrane. Na+-dependent Ca2+ release was strongly reduced in NCLX knock-down cells, whereas NCLX overexpression enhanced it[68]. NCLX-driven Ca2+ extrusion is inhibited by 7-Chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP-37157), the most selective inhibitor of mitochondrial Na+/Ca2+ exchange. Taken together, the evidence led to the conclusion that NCLX encodes the so-called mitochondrial Na+/Ca2+ exchanger[68]. Several studies suggest the existence of regulatory mechanisms controlling mitochondrial Ca2+ export kinetics. The protein kinases PKC[69] and PINK1[70] were reported to modulate the activity of this ion exchanger. Stomatin-like protein 2 (SLP-2), which localizes to the inner mitochondrial membrane, was also shown to inhibit mitochondrial Na+/Ca2+ exchange[71]. CGP-37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, was used to demonstrate that this exchange mechanism contributes to shape cytosolic [Ca2+] transients[72], to mediate the Ca2+ transfer from the extracellular medium to the ER during IP3-driven Ca2+ signaling[73], and modulates the NAD(P)H redox state[74] and ATP production[72].
Mitochondria suspended in buffers devoid of Na+ retain their capacity to extrude Ca2+, pointing to a Na+-independent mechanism. This Ca2+ efflux pathway is catalyzed by a to date non-identified H+/Ca2+ exchanger (reviewed in Bernardi)[62]. LETM1 has been proposed recently to exchange Ca2+ against H+. LETM1 may therefore drive extrusion of Ca2+ from energized mitochondria[43,44]. In conflict with this interpretation, LETM1 expression in HeLa cells did not alter Ca2+ efflux rates, regardless of the amplitude of Ca2+ elevation reached during agonist stimulation[74]. These findings cast doubt on the Ca2+ exchanger function of LETM1.
Permeability transition pore: The PTP is a Ca2+ and ROS-activated, voltage-dependent and cyclosporine A-sensitive channel located in the inner mitochondrial membrane. Opening of the permeability transition pore causes a sudden increase in the mitochondrial inner membrane permeability to solutes with molecular masses up to 1500 Dalton[75-77]. Opening of PTP leads to mitochondrial permeability transition, which plays an important role in intracellular death signaling and in events ranging from tissue damage upon infarction to muscle wasting in some forms of dystrophy[75]. Given that PTP activation occurs under several pathological conditions[14,75], the channel has been extensively characterized as a pharmacological target. The proteins forming the PTP channel have been recently rediscovered. The classical model envisioned a supramolecular complex spanning the double membrane system of mitochondria including the protein voltage-dependent anion channel (VDAC) in the outer membrane, the adenine nucleotide translocator in the inner membrane, cyclophilin D in the mitochondrial matrix, and also including additional proteins such as Bax. However, genetics studies have demonstrated that permeability transition and/or even single channel activity can be observed in mitochondria devoid of the proposed components of the PTP[78-83]. Recent evidence from the Bernardi laboratory has revolutionized our view of the PTP. They propose that the pore of the channel is formed by dimers of the ATP synthase[84] (Figure 1). To be activated by Ca2+, PTP has also been proposed to act as a reversible fast Ca2+ release channel[85]. Such transient opening of the PTP may be induced by physiological stimuli for fast mitochondrial Ca2+ release, preventing Ca2+ overload[86].
Reevaluation of the physiological role of mitochondrial Ca2+
Mitochondrial Ca2+ uptake has been related with a plethora of cell functions, including exocytosis, gene transcription, cell cycle regulation, respiration and cell death[12,14]. Much of the evidence linked to the function of mitochondrial Ca2+ was obtained over the last few decades using pharmacological tools. The identification of the molecular machinery governing mitochondrial Ca2+ fluxes has led to a large number of genetic studies that re-evaluate the role of mitochondrial Ca2+ in cell physiology/pathology. Furthermore, they have allowed for the first time their role to be studied in the context of the whole organism. Recently, an MCU1 knock-out mouse has been generated. Analysis of the mouse phenotype has led to new questions regarding the importance of mitochondrial Ca2+ uptake. Unexpectedly, the mouse is viable. It only displays limited impairment of muscle function, even during exercise. Contrary to expectations, MCU KO hearts were also not protected from the damage induced by ischemia/reperfusion[87,88]. It is worth mentioning that the mice analyzed were obtained from a mixed genetic population (CD1) background, because disruption of the MCU1 gene in pure C57/BL/6 inbred mice led to embryonic lethality[89]. Developmental defects were also observed in a Zebra fish knock-down model that showed defects in gastrula morphogenesis[90]. The results with MCU1 KO mice on a mixed genetic background suggest that in these animals, alternative mechanisms are able to compensate for the absence of MCU function. It has been postulated that alternative Ca2+ uptake mechanisms may be responsible for this compensation. This hypothesis is hard to reconcile with the absence of regulated Ca2+ uptake in mitochondria isolated from MCU1 KO mice[87]. Despite the negative results obtained in KO mice on a mixed genetic background, MCU activity seems to play an important role in muscle physiology. Manipulation of MCU1 expression in skeletal muscle cells in vivo revealed that MCU levels modulate muscle size. The phenomenon is linked to the PGC1α4 and on IGF1-AKT/PKB signaling pathways[91]. MCU-dominant negative transgenic mice showed similar heart rates compared to the wild type animals under resting conditions, but failed to increase the beating frequency upon physiological adrenergic stimulation[92]. Detailed assessment of hearts from mice lacking MCU revealed markedly impaired mitochondrial Ca2+ uptake. Surprisingly, the hearts of these animals appear to function relatively normally, even during stress[93]. MICU1 loss of function mutations in human fibroblasts led to a defect in mitochondrial Ca2+ homeostasis[94]. Patients carrying such mutations displayed neurological disorders and muscle disease.
MATRIX REDOX SIGNALING MODULATION BY MITOCHONDRIAL Ca2+ EXTRUSION
Energy metabolism and redox balance are regulated by mitochondrial Ca2+. There is a complex relationship between Ca2+ and redox signaling, as Ca2+ promotes both oxidizing and reducing biological processes. Matrix Ca2+ rises stimulate respiratory chain activity[95,96], which as a side-product also produces ROS[97]. ROS production occurs at complexes I, II and III of the respiratory chain and several flavoproteins in different cellular compartments[18]. In fact, it was shown that mitochondria are able to produce H2O2[98,99]. The peroxide is formed from dismutation of superoxide (O2•ˉ), which is generated within mitochondria[100,101]. By promoting ROS formation, matrix Ca2+ causes net oxidation of the mitochondrial redox state. On the other hand, several Ca2+-activated dehydrogenases of the mitochondrial matrix form reducing equivalents, therefore favoring reduction of mitochondrial redox couples. Notably, studies in Bristol in the 1960s and 1970s led to the recognition that mitochondrial Ca2+ promotes the supply of reducing equivalents in the form of NADH or FADH2[102-104]. Four Ca2+-activated mitochondrial dehydrogenases were described: FAD-glycerol phosphate dehydrogenase (located on the outer surface of the inner mitochondrial membrane; influenced by changes in cytoplasmic Ca2+ concentration), pyruvate dehydrogenase, NAD-isocitrate dehydrogenase and oxoglutarate dehydrogenase (the latter three located within mitochondria and regulated by changes in mitochondrial matrix Ca2+ concentration). Following early studies with isolated mitochondria, the results on Ca2+ regulation of mitochondrial metabolism were confirmed in situ[13]. Recent evidence on insulin-secreting INS-1E-cells demonstrates that the interplay between mitochondrial Ca2+ and matrix production of reducing equivalents may be even more complex[105]. Matrix calcium signals accelerate respiration and increase cytosolic ATP levels[16]. Under the same conditions, NAD(P)H levels increased rapidly to reach a new steady-state in both INS-1E cells and human pancreatic islets[105]. Surprisingly, this substrate-dependent increase of NAD(P)H was also observed when calcium signaling was prevented. The data is consistent with Ca2+-dependent control both at the level of dehydrogenases and the respiratory chain. The accelerated formation of reducing equivalents by dehydrogenases is balanced by enhanced oxidation of NADH and FADH2 by the respiratory chain. Such coordinated activation of oxidative metabolism and respiratory chain activity allows the respiratory rate to change several fold with only small or no alterations of the NAD(P)H/NAD(P)+ ratio[105]. These results underline the complex connection between matrix Ca2+ and the control of mitochondrial redox signaling.
The recently developed fluorescent green fluorescence protein (GFP)-based redox sensors can be used to further clarify the interplay between Ca2+ and redox regulations (see for a recent review describing in detail novel sensor variants and their utilization to understand redox biology in living cells)[22]. These roGFP sensors equilibrate predominantly with the glutathione redox couple (GSSG/2GSH)[22]. This redox balance depends on glutaredoxins, which catalyze thiol-disulfide exchange between the glutathione pool and the redox-sensitive protein[106]. By coupling human glutaredoxin to an roGFP, new redox sensors have been developed that can be used even in compartments lacking glutaredoxin activity[107]. Glutathione can be oxidized by superoxide radical, hydrogen peroxide and other oxidizing agents[108,109], therefore mitochondrial targeted roGFP1 was demonstrated to be in dynamic equilibrium with the mitochondrial redox status and to respond to membrane-permeant reductants and oxidants[24] (Figure 2).
Figure 2.
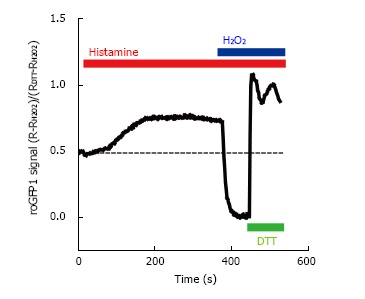
Ratiometric fluorescence intensity response of roGFP1 to histamine stimulation and exogenous H2O2 and DL-dithiothreitol in living HeLa cells. HeLa cells were transfected with the mitochondrial targeted redox sensor roGFP1. Cells were excited at 410 and 480 nm and emission was collected at 535 nm. The 410/480 fluorescence ratio (R) was normalized to the minimum of fluorescence (obtained after the addition of 1 mmol/L H2O2) and the maximum (after the addition of 10 mmol/L dithiothreitol, DL-dithiothreitol). The effect of intracellular Ca2+ was assessed by stimulating the cell with 100 μmol/L histamine. DTT: DL-Dithiothreitol.
We have recently used the mitochondrially-targeted roGFP probe to study the link between mitochondrial Ca2+ signals and matrix redox state. The kinetics of mitochondrial Ca2+ transients were analyzed by focusing on the rate of mitochondrial Ca2+ extrusion[74]. Following agonist-induced Ca2+ mobilization, maximal mitochondrial Ca2+ efflux rates were calculated as a function of the signal amplitude. A large heterogeneity of matrix Ca2+ extrusion rates was observed. Thus, only single-cell analysis is able to capture the complexity of this biological process. Manipulation of the mitochondrial Na+/Ca2+ exchanger (NCLX)[68] expression has a strong impact on agonist-induced matrix Ca2+ transients. These experimental conditions were also used to assess the effect of mitochondrial Ca2+ signals on the matrix redox state (Figure 2). During HeLa cell stimulation with the agonist histamine, calcium is mobilized from the ER, leading to a mitochondrial calcium rise. Concomitant with the Ca2+ rise, the mitochondrial redox state was increasingly reduced as measured with a mitochondrially-targeted roGFP1. These changes likely reflect a shift of the mitochondrial glutathione pool towards the reduced form. By promoting mitochondrial Ca2+ extrusion in cells overexpressing NCLX, histamine-induced redox changes were completely prevented. This effect was reverted by blocking NCLX activity using CGP-37157[68,74]. Consistent with these results, NCLX expression was able to limit histamine-induced mitochondrial NAD(P)H production in HeLa cells, and this response was fully restored by CGP-37157. We conclude that the calcium rises induced by histamine stimulate matrix dehydrogenases, as reflected by the increased NAD(P)H/NAD(P)+ ratio. These changes favor the formation of reduced glutathione measured by an increase of the roGFP1 signal. Mitochondrial Ca2+ is also a powerful activator of respiratory chain complexes. The associated acceleration of ROS production should have a net oxidizing effect on the mitochondria. Our findings demonstrate that the reducing effects are dominant during physiological calcium mobilization. Furthermore, our data establish a causal relationship between NCLX activity and matrix redox state (Figure 3).
Figure 3.
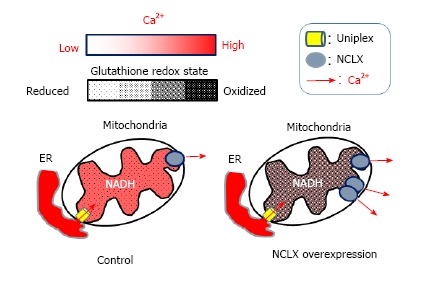
Proposed model for the modulation of the matrix redox signaling by mitochondrial Ca2+. After cell stimulation, Ca2+ released from the endoplasmic reticulum (ER) enters in mitochondria by the mitochondrial uniplex (left), stimulating matrix Ca2+-dependent dehydrogenases, which increase NADH levels and promoting a reduction of the mitochondrial glutathione pool. In cells over-expressing the mitochondrial Na+/Ca2+ exchanger (NCLX, right) Ca2+ extrusion is more efficient and therefore Ca2+-dependent dehydrogenases less activated. Such lowering the mitochondrial Ca2+ response blunts NADH formation and prevents matrix redox changes of the mitochondrial glutathione pool. NADH: Reduced form of nicotinamide-adenine dinucleotide; NCLX: Mitochondrial Na+/Ca2+ exchanger.
REDOX REGULATION OF MITOCHONDRIAL FUNCTION
Redox control of mitochondrial proteins is an important topic in cell physiology and pathology because many mitochondrial functions are linked to redox reactions[21]. An increasing number of publications demonstrate a role for redox signals in the control of mitochondrial functions, including nutrient oxidation, oxidative phosphorylation, ROS production, mitochondrial permeability transition, mitochondrial morphology and cell death (reviewed in[21]). A number of reviews have covered the role of redox switches in the control of specific mitochondrial functions[21,110-113]. They highlight the fact that mitochondria harbor a unique environment that promotes thiol modifications and redox signaling. The mitochondrial proteome is very rich in protein thiols. The total concentration of such thiol groups was estimated to be in the range of 60-90 mmol/L[114]. In addition, as previously mentioned, mitochondria are a very important source for ROS (notably superoxide anion radical and hydrogen peroxide), reduced glutathione and NAD(P)H, which are required for oxidation/reduction reactions. Importantly, redox potentials are strongly influenced by pH, and mitochondria are able to dynamically regulate mitochondrial proton gradient during cytosolic and mitochondrial Ca2+ elevations[115]. Changes in mitochondrial pH seem to play an important role in physiological and pathological situations such as apoptosis, neurotransmission, pancreatic beta cells activation and insulin secretion[116,117]. Interestingly, spontaneous flashes of alkalinization have been reported in the mitochondrial matrix of living cells[118]; they can even spread between contiguous mitochondria, but their potential impact on redox potential has not yet been studied. Since the redox potential of any redox reaction involving H+ is pH-dependent, it is likely that matrix pH influences redox reactions. By extension, pH flashes could promote localized matrix redox reactions. Given the described properties, mitochondria represent a perfect microenvironment to promote redox signaling via cysteine oxidation reactions.
Several types of redox modifications have been observed in mitochondria. These modifications include S-oxidation (sulfenylation and sulfinylation), S-glutathionylation and S-nitrosylation[21]. A proteomic method has been recently developed to profile quantitatively free cysteine thiol groups based on their intrinsic reactivity in situ[119]. It is noteworthy that among the 50 most reactive cellular cysteine residues listed, 19 were found in mitochondrial proteins. The most reactive mitochondrial cysteines were found in aldehyde dehydrogenases. Other examples of enzymes that undergo physiologically-relevant thiol switches have been reviewed elsewhere[18]. They include mitochondrial thiolases, creatine kinase, aconitase, homoaconitase and branched chain aminotransferase. The importance of mitochondrial thiol switches and the role of “physiological” ROS production to trigger those switches has also been highlighted by Riemer et al[18]. For instance, the phenotypes of knock-out mice for several mitochondrial redox-regulating enzymes (superoxide dismutases 1 and 2; glutaredoxins 1 and 2; glutathione peroxidases 1 and 4; thioredoxin 2; thioredoxin reductases 1 and 2; peroxiredoxin 3) were reviewed. The observed phenotypes in these animals range from embryonic lethality, developmental aberrations and neurodegeneration to impaired signal transduction.
Mitochondrial proteins involved in oxidative metabolism and energy production are primary targets regulated by reactive cysteines. A subunit of pyruvate dehydrogenase, which links glycolysis to the citric acid cycle, is reversibly inactivated by hydrogen peroxide[120]. The activity of several tricarboxylic acid cycle enzymes (aconitase, isocitrate dehydrogenase, ketoglutarate dehydrogenase and succinyl-CoA synthetase) is modulated by redox reactions as well[121-124]. In addition, mitochondrial respiratory chain complexes are a target of thiol-base redox regulation[125].
Glutathionylation of uncoupling protein 2 (UCP2) and UCP3, two mitochondrial protein paralogues of UCP1, has been proposed to modulate proton leak[126-128]. Interestingly, UCP2 and UCP3 modulate the activity of sarco/endoplasmic reticulum Ca2+ ATPases by decreasing mitochondrial ATP production[57], revealing an additional link between Ca2+ and redox signals.
The matrix redox state also influences mitochondrial function through sirtuins, a class of NAD+-dependent deacetylases having beneficial health effects[129]. Among the seven members of this family, SIRT3, SIRT4 and SIRT5 are found in the mitochondria, where they govern mitochondrial processes[130]. Also, some mitochondrial transport systems have been reported to be regulated by the redox potential. For example, the carnitine/acylcarnitine carrier, required for the transport of fatty acid into mitochondria, is regulated by glutathionylation[131].
Redox regulation of mitochondrial proteins plays a crucial role also in pathology, as ROS promote the opening of the mitochondrial permeability transition pore[14,75,132]. The role of thiol oxidation during mitochondrial permeability transition has been carefully characterized[133,134]. For example, redox-active compounds belonging to the polyphenol family are able to modulate both PTP single-channel activity and PTP-dependent colloidosmotic swelling of isolated mitochondria[135]. Mitochondrial morphology has a strong impact on metabolism and cell death decisions, and this process is also modulated by redox reactions. Chronic ROS exposure promotes mitochondrial fragmentation[136]. Treatment with sublethal amounts of H2O2 or other acute stresses induces hyperfusion and can be prevented using antioxidants[137]. A recent genome-wide screen using RNA interference has identified ROMO1 as an essential redox-regulator, which is required for mitochondrial fusion and normal cristae morphology[138].
Deregulated mitochondrial redox signaling is associated with several diseases and condition[21]. The involvement of mitochondrial thiol oxidation has been reported in cardiovascular diseases[139]. Redox proteomics of hearts subjected to ischemia/reperfusion indicates major changes in the redox state of thiol groups in mitochondrial proteins, including components of electron transport complexes and enzymes involved in lipid metabolism[139]. Moreover, mitochondrial PTP opening has been demonstrated to be a causative event in reperfusion damage of the heart[140]. Mitochondrial redox signals have been implicated also in neurodegenerative disorders, and deregulation of glutaredoxin-1 and thioredoxin-1 have been proposed to be important events in Alzheimer’s disease pathogenesis[141]. A well-known feature of Parkinson’s diseases is an imbalanced redox state[142]. Acting on mitochondrial redox signals has been suggest as an approach to attenuate oxidative stress in dopaminergic neurons of the substantia nigra in individuals with Parkinson’s disease. ROS production and associated mitochondrial dysfunction may also play an important role during progression of type 2 diabetes[143]. The relevance of redox signaling in the development of type 2 diabetes has been highlighted recently[144]. In pancreatic beta cells, mitochondria are particularly important as they link nutrient metabolism to down-stream signals essential for insulin secretion. In this cell type the identification of mitochondrial proteins controlled by redox state may lead to the identification of novel signaling pathways modulating insulin secretion. Finally aging and age-related diseases in general are influenced by intracellular free radicals[145]. Disruption of mitochondrial redox signals seems to contribute to ageing[146,147] and the redox state of protein thiol group has been proposed to play key role in this process[148].
CONCLUSION
Several disorders and disease states are associated with deregulated redox signaling, including cardiovascular and neurodegenerative diseases, insulin resistance, obesity, diabetes and aging (discussed in[21]). Novel approaches are needed to rescue cellular function due to deregulated redox signaling. Polyphenols[149] are a good example as they have anti-oxidant properties and should prevent free radical damage, and thereby potentially normalize redox signaling. However, as discussed by Visioli et al[149], “basic and clinical science is showing that the reality is much more complex than this and that several issues, notably content in foodstuff, bioavailability, or in vivo antioxidant activity are yet to be resolved”. Mitochondria constitute an optimal target to face those issues because they drive/modulate their functions by redox reactions. The ability of mitochondrial Ca2+ to modulate matrix redox state offers potential novel strategies for the manipulation of the mitochondrial redox state. Several natural compounds are known to modulate mitochondrial Ca2+ transport[150]. Such compounds affecting mitochondrial Ca2+ handling may have beneficial health effects by rescuing mitochondrial redox-related functions.
Footnotes
P- Reviewer: Andrea R, Lawen A S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
Conflict-of-interest statement: The authors are employees of NIHS which is part of Nestle Group.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 10, 2015
First decision: July 31, 2015
Article in press: October 13, 2015
References
- 1.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 2.Cereghetti GM, Scorrano L. The many shapes of mitochondrial death. Oncogene. 2006;25:4717–4724. doi: 10.1038/sj.onc.1209605. [DOI] [PubMed] [Google Scholar]
- 3.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813:1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 8.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 9.Pizzo P, Drago I, Filadi R, Pozzan T. Mitochondrial Ca² homeostasis: mechanism, role, and tissue specificities. Pflugers Arch. 2012;464:3–17. doi: 10.1007/s00424-012-1122-y. [DOI] [PubMed] [Google Scholar]
- 10.Santo-Domingo J, Demaurex N. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta. 2010;1797:907–912. doi: 10.1016/j.bbabio.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- 12.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 13.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gouriou Y, Demaurex N, Bijlenga P, De Marchi U. Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie. 2011;93:2060–2067. doi: 10.1016/j.biochi.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50:222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Wiederkehr A, Szanda G, Akhmedov D, Mataki C, Heizmann CW, Schoonjans K, Pozzan T, Spät A, Wollheim CB. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 2011;13:601–611. doi: 10.1016/j.cmet.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Deponte M, Lillig CH. Enzymatic control of cysteinyl thiol switches in proteins. Biol Chem. 2015;396:401–413. doi: 10.1515/hsz-2014-0280. [DOI] [PubMed] [Google Scholar]
- 18.Riemer J, Schwarzländer M, Conrad M, Herrmann JM. Thiol switches in mitochondria: operation and physiological relevance. Biol Chem. 2015;396:465–482. doi: 10.1515/hsz-2014-0293. [DOI] [PubMed] [Google Scholar]
- 19.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins Y, Chouchani ET, James AM, Menger KE, Cochemé HM, Murphy MP. Mitochondrial redox signalling at a glance. J Cell Sci. 2012;125:801–806. doi: 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- 21.Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014;2:123–139. doi: 10.1016/j.redox.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarzlander M, Dick TP, Meye AJ, Morgan B. Dissecting Redox Biology using Fluorescent Protein Sensors. Antioxid Redox Signal. 2015;Apr 13:Epub ahead of print. doi: 10.1089/ars.2015.6266. [DOI] [PubMed] [Google Scholar]
- 23.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 24.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 25.Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J. 2001;20:5853–5862. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 27.Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 29.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 30.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014;15:299–307. doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. 2014;53:726–737. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovács-Bogdán E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD, Mootha VK. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci USA. 2014;111:8985–8990. doi: 10.1073/pnas.1400514111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca²+ uniporter. Cell Metab. 2013;17:976–987. doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Yang X, Li S, Wang Z, Liu Y, Feng J, Zhu Y, Shen Y. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. EMBO J. 2014;33:594–604. doi: 10.1002/embj.201386523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3:1317. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci USA. 2013;110:E2249–E2254. doi: 10.1073/pnas.1308558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, et al. SLC25A23 augments mitochondrial Ca²+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell. 2014;25:936–947. doi: 10.1091/mbc.E13-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol. 2007;9:445–452. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waldeck-Weiermair M, Jean-Quartier C, Rost R, Khan MJ, Vishnu N, Bondarenko AI, Imamura H, Malli R, Graier WF. Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J Biol Chem. 2011;286:28444–28455. doi: 10.1074/jbc.M111.244517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cárdenas C, Shanmughapriya S, Rajan S, Vallem S, Chen X, Foskett JK, et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. 2014;28:4936–4949. doi: 10.1096/fj.14-256453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai MF, Jiang D, Zhao L, Clapham D, Miller C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol. 2014;143:67–73. doi: 10.1085/jgp.201311096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimmer KS, Navoni F, Casarin A, Trevisson E, Endele S, Winterpacht A, Salviati L, Scorrano L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum Mol Genet. 2008;17:201–214. doi: 10.1093/hmg/ddm297. [DOI] [PubMed] [Google Scholar]
- 52.Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, Wiesenberger G, Schweyen RJ. The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf-Hirschhorn syndrome. J Biol Chem. 2004;279:30307–30315. doi: 10.1074/jbc.M403607200. [DOI] [PubMed] [Google Scholar]
- 53.Nowikovsky K, Pozzan T, Rizzuto R, Scorrano L, Bernardi P. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the pathophysiology of LETM1. J Gen Physiol. 2012;139:445–454. doi: 10.1085/jgp.201110757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hasegawa A, van der Bliek AM. Inverse correlation between expression of the Wolfs Hirschhorn candidate gene Letm1 and mitochondrial volume in C. elegans and in mammalian cells. Hum Mol Genet. 2007;16:2061–2071. doi: 10.1093/hmg/ddm154. [DOI] [PubMed] [Google Scholar]
- 55.Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015;21:109–116. doi: 10.1016/j.cmet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP, Gunter TE, Nicholls DG, Bernardi P, Lemasters JJ, Brand MD. UCPs--unlikely calcium porters. Nat Cell Biol. 2008;10:1235–1237; author reply 1237-1240. doi: 10.1038/ncb1108-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Marchi U, Castelbou C, Demaurex N. Uncoupling protein 3 (UCP3) modulates the activity of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J Biol Chem. 2011;286:32533–32541. doi: 10.1074/jbc.M110.216044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276:21482–21488. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- 59.Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta. 2005;1717:1–10. doi: 10.1016/j.bbamem.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 60.Ryu SY, Beutner G, Kinnally KW, Dirksen RT, Sheu SS. Single channel characterization of the mitochondrial ryanodine receptor in heart mitoplasts. J Biol Chem. 2011;286:21324–21329. doi: 10.1074/jbc.C111.245597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bondarenko AI, Jean-Quartier C, Parichatikanond W, Alam MR, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca(2+) uniporter (MCU)-dependent and MCU-independent Ca(2+) channels coexist in the inner mitochondrial membrane. Pflugers Arch. 2014;466:1411–1420. doi: 10.1007/s00424-013-1383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 63.Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C. The release of calcium from heart mitochondria by sodium. J Mol Cell Cardiol. 1974;6:361–371. doi: 10.1016/0022-2828(74)90077-7. [DOI] [PubMed] [Google Scholar]
- 64.Puskin JS, Gunter TE, Gunter KK, Russell PR. Evidence for more than one Ca2+ transport mechanism in mitochondria. Biochemistry. 1976;15:3834–3842. doi: 10.1021/bi00662a029. [DOI] [PubMed] [Google Scholar]
- 65.Brierley GP, Baysal K, Jung DW. Cation transport systems in mitochondria: Na+ and K+ uniports and exchangers. J Bioenerg Biomembr. 1994;26:519–526. doi: 10.1007/BF00762736. [DOI] [PubMed] [Google Scholar]
- 66.Baysal K, Jung DW, Gunter KK, Gunter TE, Brierley GP. Na(+)-dependent Ca2+ efflux mechanism of heart mitochondria is not a passive Ca2+/2Na+ exchanger. Am J Physiol. 1994;266:C800–C808. doi: 10.1152/ajpcell.1994.266.3.C800. [DOI] [PubMed] [Google Scholar]
- 67.Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004;279:25234–25240. doi: 10.1074/jbc.M401229200. [DOI] [PubMed] [Google Scholar]
- 68.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang F, He XP, Russell J, Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Da Cruz S, De Marchi U, Frieden M, Parone PA, Martinou JC, Demaurex N. SLP-2 negatively modulates mitochondrial sodium-calcium exchange. Cell Calcium. 2010;47:11–18. doi: 10.1016/j.ceca.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 72.Hernández-SanMiguel E, Vay L, Santo-Domingo J, Lobatón CD, Moreno A, Montero M, Alvarez J. The mitochondrial Na+/Ca2+ exchanger plays a key role in the control of cytosolic Ca2+ oscillations. Cell Calcium. 2006;40:53–61. doi: 10.1016/j.ceca.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 73.Malli R, Frieden M, Trenker M, Graier WF. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J Biol Chem. 2005;280:12114–12122. doi: 10.1074/jbc.M409353200. [DOI] [PubMed] [Google Scholar]
- 74.De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem. 2014;289:20377–20385. doi: 10.1074/jbc.M113.540898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 76.Zoratti M, De Marchi U, Biasutto L, Szabò I. Electrophysiology clarifies the megariddles of the mitochondrial permeability transition pore. FEBS Lett. 2010;584:1997–2004. doi: 10.1016/j.febslet.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 77.Zoratti M, Szabò I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim Biophys Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 78.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 79.De Marchi U, Basso E, Szabò I, Zoratti M. Electrophysiological characterization of the Cyclophilin D-deleted mitochondrial permeability transition pore. Mol Membr Biol. 2006;23:521–530. doi: 10.1080/09687860600907644. [DOI] [PubMed] [Google Scholar]
- 80.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim Biophys Acta. 2006;1757:590–595. doi: 10.1016/j.bbabio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 82.De Marchi U, Campello S, Szabò I, Tombola F, Martinou JC, Zoratti M. Bax does not directly participate in the Ca(2+)-induced permeability transition of isolated mitochondria. J Biol Chem. 2004;279:37415–37422. doi: 10.1074/jbc.M314093200. [DOI] [PubMed] [Google Scholar]
- 83.Campello S, De Marchi U, Szabò I, Tombola F, Martinou JC, Zoratti M. The properties of the mitochondrial megachannel in mitoplasts from human colon carcinoma cells are not influenced by Bax. FEBS Lett. 2005;579:3695–3700. doi: 10.1016/j.febslet.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 84.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 86.Bernardi P, von Stockum S. The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell Calcium. 2012;52:22–27. doi: 10.1016/j.ceca.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herzig S, Maundrell K, Martinou JC. Life without the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1398–1400. doi: 10.1038/ncb2891. [DOI] [PubMed] [Google Scholar]
- 89.Murphy E, Pan X, Nguyen T, Liu J, Holmström KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun. 2014;449:384–385. doi: 10.1016/j.bbrc.2014.04.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prudent J, Popgeorgiev N, Bonneau B, Thibaut J, Gadet R, Lopez J, Gonzalo P, Rimokh R, Manon S, Houart C, et al. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat Commun. 2013;4:2330. doi: 10.1038/ncomms3330. [DOI] [PubMed] [Google Scholar]
- 91.Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015;10:1269–1279. doi: 10.1016/j.celrep.2015.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. doi: 10.1038/ncomms7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178–182. doi: 10.1016/j.yjmcc.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014;46:188–193. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]
- 95.Potter VR, Siekevitz P, Simonson HC. Latent adenosinetriphosphatase activity in resting rat liver mitochondria. J Biol Chem. 1953;205:893–908. [PubMed] [Google Scholar]
- 96.Lindberg O, Ernster L. Manganese, a co-factor of oxidative phosphorylation. Nature. 1954;173:1038–1039. doi: 10.1038/1731038a0. [DOI] [PubMed] [Google Scholar]
- 97.Jensen PK. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochim Biophys Acta. 1966;122:157–166. doi: 10.1016/0926-6593(66)90057-9. [DOI] [PubMed] [Google Scholar]
- 98.Loschen G, Flohé L, Chance B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett. 1971;18:261–264. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 99.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Loschen G, Azzi A, Richter C, Flohé L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 101.Forman HJ, Kennedy JA. Role of superoxide radical in mitochondrial dehydrogenase reactions. Biochem Biophys Res Commun. 1974;60:1044–1050. doi: 10.1016/0006-291x(74)90418-5. [DOI] [PubMed] [Google Scholar]
- 102.Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128:161–163. doi: 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176:899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Marchi U, Thevenet J, Hermant A, Dioum E, Wiederkehr A. Calcium co-regulates oxidative metabolism and ATP synthase-dependent respiration in pancreatic beta cells. J Biol Chem. 2014;289:9182–9194. doi: 10.1074/jbc.M113.513184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ostergaard H, Tachibana C, Winther JR. Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol. 2004;166:337–345. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 108.Wefers H, Sies H. Oxidation of glutathione by the superoxide radical to the disulfide and the sulfonate yielding singlet oxygen. Eur J Biochem. 1983;137:29–36. doi: 10.1111/j.1432-1033.1983.tb07791.x. [DOI] [PubMed] [Google Scholar]
- 109.Finley JW, Wheeler EL, Witt SC. Oxidation of glutathione by hydrogen peroxide and other oxidizing agents. J Agric Food Chem. 1981;29:404–407. doi: 10.1021/jf00104a045. [DOI] [PubMed] [Google Scholar]
- 110.Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal. 2012;16:476–495. doi: 10.1089/ars.2011.4289. [DOI] [PubMed] [Google Scholar]
- 111.Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 112.Piantadosi CA. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim Biophys Acta. 2012;1820:712–721. doi: 10.1016/j.bbagen.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yin F, Sancheti H, Cadenas E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid Redox Signal. 2012;17:1714–1727. doi: 10.1089/ars.2012.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Requejo R, Hurd TR, Costa NJ, Murphy MP. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010;277:1465–1480. doi: 10.1111/j.1742-4658.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Poburko D, Santo-Domingo J, Demaurex N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J Biol Chem. 2011;286:11672–11684. doi: 10.1074/jbc.M110.159962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santo-Domingo J, Demaurex N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J Gen Physiol. 2012;139:415–423. doi: 10.1085/jgp.201110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wiederkehr A, Park KS, Dupont O, Demaurex N, Pozzan T, Cline GW, Wollheim CB. Matrix alkalinization: a novel mitochondrial signal for sustained pancreatic beta-cell activation. EMBO J. 2009;28:417–428. doi: 10.1038/emboj.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Santo-Domingo J, Giacomello M, Poburko D, Scorrano L, Demaurex N. OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J. 2013;32:1927–1940. doi: 10.1038/emboj.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yan LJ, Sumien N, Thangthaeng N, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic Res. 2013;47:123–133. doi: 10.3109/10715762.2012.752078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bulteau AL, Lundberg KC, Ikeda-Saito M, Isaya G, Szweda LI. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc Natl Acad Sci USA. 2005;102:5987–5991. doi: 10.1073/pnas.0501519102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kil IS, Park JW. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J Biol Chem. 2005;280:10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 123.McLain AL, Cormier PJ, Kinter M, Szweda LI. Glutathionylation of α-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic Biol Med. 2013;61:161–169. doi: 10.1016/j.freeradbiomed.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 125.Dröse S, Brandt U, Wittig I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim Biophys Acta. 2014;1844:1344–1354. doi: 10.1016/j.bbapap.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 126.Mailloux RJ, Seifert EL, Bouillaud F, Aguer C, Collins S, Harper ME. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J Biol Chem. 2011;286:21865–21875. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mailloux RJ, Fu A, Robson-Doucette C, Allister EM, Wheeler MB, Screaton R, Harper ME. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. J Biol Chem. 2012;287:39673–39685. doi: 10.1074/jbc.M112.393538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mailloux RJ, Xuan JY, Beauchamp B, Jui L, Lou M, Harper ME. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J Biol Chem. 2013;288:8365–8379. doi: 10.1074/jbc.M112.442905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133–171. doi: 10.1016/j.freeradbiomed.2012.10.525. [DOI] [PubMed] [Google Scholar]
- 130.Pirinen E, Lo Sasso G, Auwerx J. Mitochondrial sirtuins and metabolic homeostasis. Best Pract Res Clin Endocrinol Metab. 2012;26:759–770. doi: 10.1016/j.beem.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Giangregorio N, Palmieri F, Indiveri C. Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim Biophys Acta. 2013;1830:5299–5304. doi: 10.1016/j.bbagen.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 132.Vercesi AE, Kowaltowski AJ, Grijalba MT, Meinicke AR, Castilho RF. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. 1997;17:43–52. doi: 10.1023/a:1027335217774. [DOI] [PubMed] [Google Scholar]
- 133.Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem. 1996;271:6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 134.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 135.De Marchi U, Biasutto L, Garbisa S, Toninello A, Zoratti M. Quercetin can act either as an inhibitor or an inducer of the mitochondrial permeability transition pore: A demonstration of the ambivalent redox character of polyphenols. Biochim Biophys Acta. 2009;1787:1425–1432. doi: 10.1016/j.bbabio.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 136.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–915. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Norton M, Ng AC, Baird S, Dumoulin A, Shutt T, Mah N, Andrade-Navarro MA, McBride HM, Screaton RA. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Sci Signal. 2014;7:ra10. doi: 10.1126/scisignal.2004374. [DOI] [PubMed] [Google Scholar]
- 139.Kumar V, Kleffmann T, Hampton MB, Cannell MB, Winterbourn CC. Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radic Biol Med. 2013;58:109–117. doi: 10.1016/j.freeradbiomed.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 140.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 141.Akterin S, Cowburn RF, Miranda-Vizuete A, Jiménez A, Bogdanovic N, Winblad B, Cedazo-Minguez A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006;13:1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- 142.Chinta SJ, Andersen JK. Redox imbalance in Parkinson’s disease. Biochim Biophys Acta. 2008;1780:1362–1367. doi: 10.1016/j.bbagen.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12:537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sánchez-Gómez FJ, Espinosa-Díez C, Dubey M, Dikshit M, Lamas S. S-glutathionylation: relevance in diabetes and potential role as a biomarker. Biol Chem. 2013;394:1263–1280. doi: 10.1515/hsz-2013-0150. [DOI] [PubMed] [Google Scholar]
- 145.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 146.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 147.Van Remmen H, Jones DP. Current thoughts on the role of mitochondria and free radicals in the biology of aging. J Gerontol A Biol Sci Med Sci. 2009;64:171–174. doi: 10.1093/gerona/gln058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Moosmann B. Respiratory chain cysteine and methionine usage indicate a causal role for thiyl radicals in aging. Exp Gerontol. 2011;46:164–169. doi: 10.1016/j.exger.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 149.Visioli F, De La Lastra CA, Andres-Lacueva C, Aviram M, Calhau C, Cassano A, D’Archivio M, Faria A, Favé G, Fogliano V, et al. Polyphenols and human health: a prospectus. Crit Rev Food Sci Nutr. 2011;51:524–546. doi: 10.1080/10408391003698677. [DOI] [PubMed] [Google Scholar]
- 150.Montero M, Lobatón CD, Hernández-Sanmiguel E, Santodomingo J, Vay L, Moreno A, Alvarez J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem J. 2004;384:19–24. doi: 10.1042/BJ20040990. [DOI] [PMC free article] [PubMed] [Google Scholar]