

Abstract
Global alterations in epigenetic landscape are now recognized as a hallmark of cancer. Epigenetic mechanisms such as DNA methylation, histone modifications, nucleosome positioning and non-coding RNAs are proven to have strong association with cancer. In particular, covalent post-translational modifications of histone proteins are known to play an important role in chromatin remodeling and thereby in regulation of gene expression. Further, histone modifications have also been associated with different aspects of carcinogenesis and have been studied for their role in the better management of cancer patients. In this review, we will explore and discuss how histone modifications are involved in cancer diagnosis, prognosis and treatment.
Keywords: Epigenetics, Cancer, Diagnosis, Prognosis, Histone post-translational modifications, Treatment
Core tip: The purpose of the review is to describe the potential of histone post-translational modifications in the field of cancer.
INTRODUCTION
Cancer is a manifestation of both genetic and epigenetic alterations leading to the genomic instability and thus affecting several classes of genes, such as oncogenes, tumor suppressor genes, apoptotic genes and DNA repair genes. The field of cancer genetics which include the study of point mutation, deletion, insertion, gene amplification, chromosomal deletion/inversion/translocation, and allelic loss/gain has got the attention of most cancer researchers in the last few decades. However, the appreciation of cancer epigenetics is more recent as several studies have now shown that in addition to numerous genetic alterations human cancers also harbor global epigenetic abnormalities[1,2].
Epigenetics, was initially defined by C. H. Waddington as “the causal interactions between genes and their products, which bring the phenotype into being”[3]. With time, the definition of epigenetics has evolved and is implicated in a wide variety of biological processes. The current definition is “the study of heritable changes in gene expression that occur independent of changes in the primary DNA sequence”. Epigenetic mechanisms include DNA methylation[4], noncoding RNA[5,6], histone variants[7] and histone post translational modifications (PTMs). These mechanisms together alter the local structural dynamics of chromatin to regulate the functioning of the genome, mostly by regulating its accessibility and compactness. All together, these mechanisms govern the chromatin architecture and gene function in various cell types, developmental and disease states[2,8-12]. Disruption in the proper maintenance of these heritable epigenetic mechanisms can result in activation or inhibition of various critical cell signaling pathways thus leading to disease states such as cancer[1,13]. Epigenetic mechanisms also cooperate with genetic alteration and work together at all stages of cancer development from initiation to progression[14]. Unlike genetic alterations, epigenetic changes are reversible in nature and can be potentially restored back to their original state by epigenetic therapy. These findings have inspired many studies aimed to understand the role of epigenetics in tumorigenesis and further explore its utility in cancer diagnosis, prognosis and therapy[15]. In recent years, research focus has been shifted to understand various post translational modifications for gaining deeper insights in to the functioning of histone/chromatin associated proteins. Information about the PTMs and the related modifying enzymes is available in the database HIstome: The Histone Infobase (http://www.actrec.gov.in/histome/)[16]. This review will discuss the role of histone post-translational modifications and its utility in cancer diagnosis, prognosis and treatment.
HISTONE PTMS: A DYNAMIC PROCESS
Histones are highly conserved and basic proteins with a globular C-terminal domain and an unstructured N-terminal tail[17]. They are also the most important proteins for converting a linear naked genome in to physiologically sensible architecture, chromatin. Nucleosomes are fundamental units of chromatin, consisting an octamer of H2A, H2B, H3 and H4 (two each) around which 146 base pairs of DNA is wrapped-. There are sequence variants of these histones which are expressed and incorporated into chromatin in a context dependent manner in normal and disease related processes. In cancer, histone H2A variants, H2A.1, H2A.Z and macroH2A have also been reported to express aberrantly[18-20]. Also, histones proteins can undergo a variety of PTMs some of which are methylation (me), acetylation (ac), ubiquitylation (ub), sumoylation (su) and phosphorylation (ph) on specific amino acid (Figure 1)[10]. Apart from these modifications, histones are also known to undergo homocysteinylation, crotonylation and glucosylation amongst others[21]. These histone modifications occur at several degrees, for example, methylation can be of monomethyl (me), dimethyl (me2) and trimethyl (me3).
Figure 1.
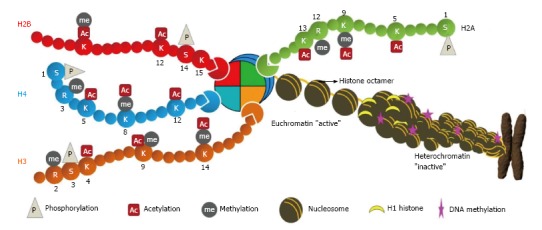
Chromatin architecture. The DNA is wrapped in two turns around histone octamers (nucleosomes) at intervals of about 200 bp along the DNA. Histones within the nucleosome (two each of H2A, H2B, H3 and H4) undergo numerous post-translational modifications at their N-terminal tail which protrudes from the nucleosome. Further folding of nucleosome with linker histone H1 creates a spiral structure, the heterochromatin leading to metaphase chromosome. These modifications directly regulate the chromatin structure and thus DNA-mediated cellular processes. The diagram indicates some modifications at specific residues: M: Methylation; A: Acetylation; P: Phosphorylation.
Histone PTMs are added and removed from histones by enzymes called “writers” and “erasers” respectively. Histone acetyltransferases (HATs), histone methyltransferases (HMTs) and histone kinases are the examples of “writers” which add acetyl, methyl and phosphoryl groups, whereas histone deacetylases (HDACs), histone demethylases (HDMs) and histone phosphatases are examples of “erasers” which remove acetyl, methyl and phosphoryl groups, respectively (Figure 2)[22-24]. Histone-modifying enzymes are also known to interact with each other as well as other chromatin related proteins thus influencing key cellular processes such as transcription, replication and repair[10].
Figure 2.
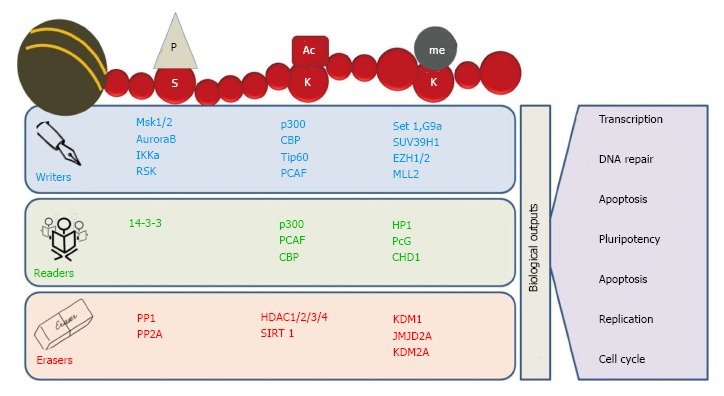
Readers, writers and erasers of chromatin marks. Histone modifications are highly dynamic in nature. The “writers” like histone acetyltransferases (HATs), histone methyltransferases (HMTs) and kinases add specific marks on specific amino acid residues on histone tails. These marks are identified by various proteins containing specific domains such as bromodomains, chromodomains and Tudor domain containing proteins called “readers”. The written marks are removed by “erasers” like histone deacetylases (HDACs), lysine demethylases (KDMs) and phosphatases. In addition, removal and identification of these post-translational modifications on histone tails regulate various biological processes, including transcription, DNA replication and DNA repair.
The mechanism behind the regulation of key cellular processes by histone post-translational modifications is not fully understood; however, it can be generalized into two categories. First, the addition of any PTM on histone protein affects inter/intra-nucleosomal interactions and their binding to DNA by steric hindrance or charge interactions. Second, addition of these PTMs to histone proteins inhibits or facilitates the binding of various proteins to chromatin[10]. These mechanisms allow a vast range of flexibility in regulating chromatin dynamics and signaling transmission and thereby regulating the gene expression. As an example of first mechanism, histone acetylation is proposed to be associated with chromatin relaxation and transcription activation, H4K16ac inhibits the formation of compact 30 nm fibers and higher order chromatin structures[25,26]. As an example of second mechanism, evolutionarily conserved specialized proteins, termed “histone readers,” possess the ability to specifically bind certain histone modifications and affects a defined nuclear process such as transcription, DNA repair and replication, etc. (Figure 2). For example, through its evolutionary conserved chromodomain heterochromatin protein 1 recognize and gets recruited to H3K9me3 and leads to the formation of compact chromatin which in turn inhibits the access of the transcriptional machinery[27,28]. Moreover, the fact that there are different variants of each histone protein differing from few to many amino acids adds another level of complexity in functional aspects of histone PTMs. Such complicated and multilayered regulatory mechanisms of cellular processes through histone modifications have led to the hypothesis of “histone code” where a set of histone variants and modifications together perform a specific function[29]. However, due to its complexity histone code is still not fully understood[30]. Further, the status of one histone modification also regulates that of another by cross-talk and affects chromatin remodeling and gene expression. Cross-talk between H3S10ph and H3K14ac, H2Bub and H3K4me and H3K4ac and H3K4me3 and H3K14ac are few prominent examples regulating gene expression[31]. For example, acetylation of H3K18 and H3K23 by CBP (CREB binding protein) can promote the methylation of H3R17 by Coactivator-Associated Arginine Methyltransferase 1 (CARM1), resulting in activation of estrogene-responsive genes[32].
HISTONE PTMS IN CANCER
In cancer, several histone PTMs have been reported to be misregulated; however, their involvement in cancer pathophysiological characteristics like cellular transformation, angiogenesis and metastasis etc., is not well understood. Moreover, there are very few studies commenting on the cancer specific regulatory mechanism behind the alteration of histone PTMs. It has been a decade when global loss of H4K16ac and H4K20me3 was reported for their association with cancer and considered as a common hallmark of tumor cells[33]. However, still there are no reports of their direct involvement in cellular transformation or any other cancer characteristics. Despite of the awareness of hMOF (human Male absent Of First) and HDAC4 as writer and eraser of H4K16ac, it is a recent development that low expression of hMOF has been implicated for its loss in gastric cancer[34]. Moving on to histone methylation, Lin et al[35] showed histone lysine demethylase KDM1A mediated loss of H3K4me2 is associated with epithelial to mesenchymal transition (EMT) in human breast cancer cells. Loss of H3ac, H3K9me3 and H3S10ph is observed at the promoters of Sfrp2, Sfrp5 and Wnt5a during genistein induced development of colon cancer in the rat model system[36]. Alterations in methylation patterns of H3K9 and H3K27 are related to aberrant gene silencing in many cancers[37,38]. Tissue microarrays done to compare the levels of H2B ub1 levels in normal mammary epithelial tissue as well as benign, malignant, and metastatic breast cancer samples have clearly shown a sequential decrease in H2B monoubiquitination with breast cancer progression and metastasis in comparision with normal epithelia[39]. A very important discovery has been made in term of phosphorylation of H3S10 as the only histone marks directly associated with cellular transformation. The knockdown and mutant (S10A) of histone H3 suppressed LMP1-induced proliferation of nasopharyngeal carcinoma cell line CNE1[40]. H3S10P has been reported to increase and has been established as indispensable for cellular transformation[41,42]. Cellular transformation by v-src constitutively activated phosphorylation of histone H3 at Ser10 in a transformation-specific manner; while, non-transforming mutant of v-src did not activate H3 phosphorylation[43]. Further, Mitogen- and stress-activated kinase 1 (MSK1) has been shown to phosphorylate H3S10 in TPA and EGF mediated cellular transformation[44]. Unpublished data from our lab has also shown increase in H3S10ph in gastric cancer, which is regulated by p38-MAPK/MSK1 pathway.
It has now been clear that acetylation, methylation and phosphorylation of histones are the most studied histone marks. In cancer, most of the studies have been done for these modifications with respect to the identification of their enzymes, regulation, effect on cellular physiology and as well as molecular biological markers for the disease management. The National Institute of Health defines a biological marker (biomarker) as a biological molecule found in blood, other body fluids, or tissues that are an objective indicator of normal or abnormal process, or of a condition or disease[45]. From the next part of the review we will see how histone acetylation, methylation and phosphorylation can be exploited as biomarkers for cancer diagnosis, prognosis and treatment.
HISTONE PTMS IN CANCER DIAGNOSIS
Diagnosis of a disease majorly depends on the analysis of physical symptoms, body fluids and fecal samples. A sensitive and specific diagnostic marker is not only useful in early diagnosis, but also helps in assessing the risk of developing the disease. Advances in the technology have enabled investigators to isolate metabolites, proteins and DNA from body fluids and fecal material and correlate them with pathophysiological symptoms of diseases including cancer.
Decades of research have discovered a battery of markers for cancer diagnosis; however, only few could reach to clinics because of issues of sensitivity and specificity. Therefore, at one side there is a need to improve techniques and on the other hand discovery of new markers is of immense importance. The discovery of the presence of DNA in fecal and urine samples[46] and circulating nucleosomes in serum[47,48] has led to the foundation of identifying epigenetic markers such as DNA methylation and histone posttranslational modification for cancer diagnosis. Ahlquist et al[49] demonstrated the recovery of DNA from frozen fecal samples of colorectal cancer patients which was followed by other investigators showing matching DNA methylation patterns between DNA from tissue and fecal samples of gastric and colorectal cancer patients[50-52]. Methylation pattern of DNA isolated from urine samples was also used to diagnose bladder and prostate cancer[53-57]. All these methylation studies have successfully detected global hypomethylation and gene specific hypermethylation of DNA, as established from tissue based studies.
Presence of histone proteins is not known in fecal and urine samples; therefore, histone posttranslational modifications have been utilized as cancer diagnostic markers using circulating nucleosomes (cNUCs) in serum samples. Two histone methylation marks, H3K9me3 and H4K20me3, the hallmarks of pericentric heterochromatin[58], were investigated in circulating nucleosomes by subsequent studies. Gezer et al[59] investigated the correlation between the H3K9me3 and H4K20me3 of cNUCs in healthy subjects and patients with colorectal cancer (CRC) and multiple myeloma and found low levels of these PTMs in cancer. Sera of patients with malignant tumors including colorectal, lung, breast, ovarian, renal, prostate cancer, and lymphoma showed high level of nucleosome concentration compared with those of healthy persons and patients with benign diseases[60]. Further, the same group showed high level ALU115 DNA sequence associated H3K9Me in multiple myeloma patients compared to healthy individuals[61]. ChIP based analysis of circulating nucleosomes in serum samples by Gloria et al reported a low level of H3K9me3 and H4K20me3 in patients with colorectal, pancreatic, breast and lung cancer compared to healthy control[62,63]. Moreover, H3K9me3 and H4K20me3 have been found to be lower at the pericentromeric satellite II repeat in patients with CRC when compared with healthy controls or patients with multiple myeloma. In summary, identification of histone PTMs from serum isolated circulating nucleosomes have open the doors of immense possibility that blood samples collected by cancer patients can also be used for histone PTM based cancer diagnosis.
HISTONE PTMS IN CANCER PROGNOSIS
In cancer, to date, histones PTMs have been mostly studied for their potential as prognostic marker (Table 1). The first report in this area strongly suggested the utility of histone PTMs in cancer diagnosis and showed loss of H4K16ac and H4K20me3 in several cancers and establish these two marks as a hallmark of tumor and establishes the correlation of H4K16ac with tumor progression[33]. Further, loss of H4K20me3 is as also detected in various cancer animal models[64,65]. A study on prostate cancer showed a positive correlation of H3K18ac, H4K12ac and H4R3me2 with increasing tumor grade[66]. Another study on prostate cancer showed independently of other clinical and pathologic parameters, high rate of tumor recurrence in low-grade prostate carcinoma patients with low level of H3K4me2[66]. Loss of H3K4me2/me3 is reported in various neoplastic tissues such as non-small cell lung cancer, breast cancer, renal cell carcinoma and pancreatic adenocarcinoma serving as a predictor of clinical outcomes[67-72].
Table 1.
Global post-translational modifications of histones in cancer
| Histone PTM | Writer | Eraser | Function | Cancer Diagnosis/ Prognosis/ Treatment |
| H3K9ac | GCN-5 | SIRT-1; SIRT-6 | Transcription initiation | Diagnosis: ? |
| Prognosis: Lung, breast, ovarian | ||||
| Treatment: ? | ||||
| H3K18ac | CBP/p300 | ? | Transcription initiation and repression | Diagnosis: ? |
| Prognosis: Lung, prostate, breast, esophagus | ||||
| Treatment: ? | ||||
| H4K5ac | CBP/P300; HAT1; TIP60; HB01 | ? | Transcription activation | Diagnosis: ? |
| Prognosis: Lung | ||||
| Treatment: ? | ||||
| H4K8ac | TIP60; HB01 | ? | Transcription activation | Diagnosis: ? |
| Prognosis: Lung, | ||||
| Treatment: ? | ||||
| H4K16ac | TIP60; hMOF | SIRT-1; SIRT-2 | Transcription activation | Diagnosis: Colorectal |
| Prognosis: Lung, breast | ||||
| Treatment: ? | ||||
| H3K4me | SETD1A; SETD1B; ASH1L; MLL; MLL2; MLL3: MLL4; SETD7 | KDM1A; KDM1B; KDM5B; NO66 | Transcription activation | Diagnosis: ? |
| Prognosis: Prostate, kidney | ||||
| Treatment: ? | ||||
| H3K4me2 | SETD1A; SETD1B; MLL; MLL2; MLL3; MLL4; SMYD3 | KDM1A; KDM1B; KDM5A; KDM5B; KDM5C; KDM5D; NO66 | Transcription activation | Diagnosis: ? |
| Prognosis: Prostate, lung, kidney, breast, pancreatic, liver, | ||||
| Treatment: ? | ||||
| H3K4me3 | SETD1A; SETD1B; ASH1L; MLL; MLL2; MLL3; MLL4; SMYD3; PRMD9 | KDM2B; KDM5A; KDM5B; KDM5C; KDM5D; NO66 | Transcription elongation | Diagnosis: ? |
| Prognosis: Kidney, liver, prostate | ||||
| Treatment: ? | ||||
| H3K9me | SETDB1; G9a; EHMT1; PRDM2 | KDM3A; KDM3B§; PHF8; JHDM1D | Transcription initiation | Diagnosis: Myeloma |
| Prognosis: Kidney, pancreas, prostate | ||||
| Treatment: ? | ||||
| H3K9me2 | SUV39H1; SUV39H2; SETDB1; G9a; EHMT1; PRDM2 | KDM3A; KDM3B§; KDM4A; KDM4B; KDM4C; KDM4D; PHF8; KDM1A; JHDM1D | Transcription initiation and repression | Diagnosis: ? |
| Prognosis: Prostate, pancreas | ||||
| Treatment: ? | ||||
| H3K9me3 | SUV39H1; SUV39H2; SETDB1; PRDM2 | KDM3B§; KDM4A; KDM4B; KDM4C; KDM4D | Transcription initiation and repression | Diagnosis: Colorectal, myeloma, prostate, breast and lung |
| Prognosis: Lung, prostate, breast, leukemia, stomach | ||||
| Treatment: ? | ||||
| H3K27me | EZH2; EZH1 | JHDM1D | Transcription activation | Diagnosis: ? |
| Prognosis: Kidney | ||||
| Treatment: ? | ||||
| H3K27me3 | EZH2; EZH1 | KDM6A; KDM6B; | Transcription repression | Diagnosis: ? |
| Prognosis: Breast, pancreatic, ovarian, prostate, stomach, Esophagus, Liver | ||||
| Treatment: ? | ||||
| H4K20me3 | SUV420H1; SUV420H2 | ? | Transcription repression | Diagnosis: Colorectal, myeloma, prostate, breast and lung |
| Prognosis: Breast, lymphoma, colon, ovarian | ||||
| Treatment: ? |
PTM: Post translational modification.
Acetylation of histone H3K9 has shown ambiguous results with the increase in some and decrease in other cancers. Decrease of H3K9ac has been linked with tumor progression, histological grading and clinical stage in prostate and ovarian tumors, hence is coupled with a poor prognosis for these patients[66,73-75]. Patients with non-small cell lung adenocarcinoma exhibited better prognosis on the reduction of the H3K9ac expression level[68,76]. In contrast, increase in H3K9ac levels was reported in liver cancer[73]. Methylation of the same residue K9 of histone H3 requires loss of H3K9ac and is also linked to number of cancers. An association with the increase in methylation of H3K9 and aberrant gene silencing, has been found in many cancers[37,77] and its high level is associated with poor prognosis in gastric adenocarcinoma patients[77]. However, in patients with acute myeloid leukemia decrease in H3K9me3 has been found to be associated with better prognosis[78]. Decrease in H3K18ac is correlated with poor prognosis in prostate, pancreatic, lung, breast and kidney cancers[66,69,71]. It has also shown a strong correlation with tumor grade, signifying its importance in tumor progression[69]. In this regard, Kurdistani laboratory has confirmed that oncogenic transformation by the adenovirus protein E1a is associated with drastic changes in the global H3K18 acetylation pattern[79,80]. In addition, H3K18 hypoacetylation has been associated with an high risk of tumor recurrence in low-grade prostate cancer patients[66]. However, in contrast to this, low expression of H3K18ac has been correlated with a better prognosis for esophageal squamous cell carcinoma and glioblastoma patients[76,81]. This suggests that a single histone modification could predict differential prognosis in different cancers depending on it tissue specificity.
Another histone mark, H3K27me3 has been evaluated as a prognostic factor in patients with prostate, breast, ovarian, pancreatic and esophageal cancer[81-84], however, some of the results are perplexing and need further investigation. High level of H3K27me3 correlates with poor prognosis in esophageal cancers[81,84]. On the other hand H3K27me3 showed a negative correlation with overall survival time in breast, prostate, ovarian and pancreatic cancer patients[83]. Zhang et al[85] have identified many genes like oncogenes, tumor suppressor genes, cell cycle regulators, and genes involved in cell adhesion with significant differences in H3K27me3 pattern in gastric cancer samples in comparison to adjacent non-neoplastic gastric tissues. Further they were able to correlate changes in H3K27me3 to gene expression pattern of MMP15, UNC5B, and SHH. In non-small cell lung cancer enhanced H3K27me3 was correlated with longer overall survival (OS) and better prognosis. Moreover, both univariate and multivariate analyses indicated that H3K27me3 level was a significant and independent predictor of better survival[86]. Recently, a study showed K27M mutations of histone H3.3 variants in 31% pediatric glioblastoma tumors suggesting another level of complexity in alteration of histone PTMs in cancer which is independent of histone modifying enzymes[87]. Mass spectrometry based analysis showed high level of H3K27ac in colorectal cancer than the corresponding normal mucosa[88]. Immunohistochemical analysis on metachronous liver metastasis of colorectal carcinomas by Tamagawa et al[89] has correlated H3K4me2 and H3K9ac with the tumor histological type. In addition, lower levels of H3K4me2 correlated with a poor survival rate and also found to be an independent prognostic factor.
Recently DNA damage mark γH2AX also have shown its prognostic value. In triple negative breast tumors, high level of γH2AX was associated poor overall survival[90] and which was further found to be associated with shorter telomere length[91]. In colorectal cancer a high γH2AX expression in CRC tissues was associated with tumor stage and perineurial invasion. Furthermore, a high γH2AX expression was associated with poor distant metastasis-free survival (DMFS) and OS. Cox regression analysis also revealed that γH2AX was an independent predictor of DMFS and OS. A high γH2AX expression in CRC tissues is associated with a more malignant cancer behavior, as well as poor patient survival[92]. ELISA based analysis in glioblastoma multiformes tumors showed the high level of H3T6ph,H3S10p and H3Y41ph as signatures associated with a poor overall survival[93]. Increase in H3S10ph has been associated with poor prognosis in several cancers including glioblastoma multiformes[93], cutaneous nodular melanoma[94], cutaneous melanoma[95], breast cancer[96,97], esophageal squamous cell carcinoma[98], gastric cancer[99,100], melanoma[101] and nasopharyngeal carcinoma[40].
HISTONE PTM’S IN CANCER TREATMENT
Reversible nature of epigenetic changes or mechanisms has drawn major attention of scientific community to study the molecular mechanism regulating the alteration in epigenetic marks, specifically the histone post-translational modifications. Such efforts have led to the discovery of several histone modifying enzymes[102] and their chemical inhibitors[103] which has emerged as an attractive strategy in cancer treatment. Targeting these enzymes can reactivate epigenetically silenced tumor-suppressor genes by modulating the levels of histone posttranslational modifications[104]. Further, these drugs have also given additional advantage in the area of combinatorial chemotherapy[105,106].
Histone acetyl-transferases and histone deacetylases as the targets
Loss of histone acetylation has a strong correlation with aberrant gene silencing in cancer. Treatment with HDAC inhibitors reactivate silenced tumor suppressor genes by increasing histone acetylation levels and act as anti-tumorigenic agent by promoting growth arrest, apoptosis and cell differentiation[107]. Additionally, HDACi have shown their potential in reversing chemoresistance and induce antiproliferative effects on a number of cancer cell lines[108-113]. However, the question still remains whether the promise shown in the above studies by HDAC inhibitors are mainly due to their potency to alter epigenetic mechanisms or mere its effect on key cellular growth regulatory pathways.
Initial results upon treatment with HDACi like valproic acid and phenylbutyrate, as a single agent against hematologic malignancies were not encouraging[81]. However, the field showed much promise with the development of more potent HDACi such as the class-specific inhibitors (entinostat and romidepsin) and the pan HDAC inhibitors (vorinostat, belinostat and panobinostat). The field however gained boost when in a landmark Phase IIb multicenter trial, Yu et al[82] have shown vorinostat as effective treatment modality for refractory cutaneous T-cell lymphoma. Further, in Phase II multi-institutional trial, romidepsin has also been shown to have significant and durable efficacy against cutaneous T-cell lymphoma[83]. Due to their great successes in many studies, HDACi romidepsin and vorinostat have been approved by FDA as the treatment regime of cutaneous T-cell lymphoma, and romidepsin also for the treatment of relapsed peripheral T-cell lymphoma[84]. Since then many other HDACi have been under study of phase I and/or II trials as monotherapy, including belinostat, panobinostat, entinostat, chidamide, SB939 and LAQ824 in various cancers like ovarian, lung, soft tissue carcinoma, non-small-cell lung and breast[114-121]. However, unlike that of earlier success in treatment of lymphomas the majority of the results among solid tumor patients have been disappointing. In spite of achieving only intermittent anecdotal clinical responses, HDACi been related with severe toxicities.
Interactions between different epigenetic mechanisms have led to the foundation of research on combinatorial approach of cancer treatment using epigenetic drugs. Indeed, combinations of DNA methyltransferase and histone deacetylase inhibitors appear to synergize effectively in the reactivation of epigenetically silenced genes[107,122-124]. Such combinatorial approaches of cancer treatment have been found to be more effective than treatment with a single therapeutic agent. For example, treatment with 5-Aza-CdR and trichostatin-A in combination led to the derepression of certain putative tumor suppressor genes unlike individual treatments[107]. Pre-treatment of HDAC inhibitor SAHA relaxes the chromatin sensitizes cells to DNA damage induced by Topoisomerase II inhibitor[125]. Similarly pretreatment of valproic acid act in synergy with epirubicine and reduces the tumor volume in breast cancer mouse model[126].
Furthermore, synergistic activity of decitabine and HDACi sodium phenylbutyrate was shown to decrease the lung cancer formation by more than 50% in comparison with decitabine alone in a murine model based study by Belinsky et al[124]. The same group also reported that the combination of HDACi entinostat with the DNMTi azacitidine was able to decrease tumor size and reduce the growth of K-ras/p53 mutant lung adenocarcinomas orthotopic engrafted in immunocompromised nude rats[127]. In another case HDACi sodium butyrate reduces the cell proliferation of MCF-7 cell when combine with vitamin-A[128].
Histone methyl-transferases and histone demethylases as the targets
Studies on histone methylation and their modifiers have been slow. Only few histone methylases (HMT) and demethylases (HDM) and their inhibitors have been discovered. However, studies on histone methylation could be more fruitful for their therapeutic potential because the less redundancy in HMTs and HDM compared to HATs and HDACs in targeting specific amino acid residue of histone[129]. This property of HMTs and HDMs provides exciting opportunities with more tailored treatment, while potentially minimizing side effects.
LSD1/KDM1 was among the first identified histone demethylases selectively targeting H3K4me1 and H3K4me2[130] and mediate gene repression. LSD1 has been reported to be overexpressed in many cancers like brain, breast, and prostate, thus thought to be a promising target for drug therapy[130-132]. Small molecules such as SL11144 and tranylcypromine have been developed to inhibit LSD1[133,134], Since then have shown to restore expression many silenced tumor suppressors like secreted frizzled-related protein and GATA transcription factors in many cancer cell lines. They have also been shown to possess antitumor activity in a study involving neuroblastoma xenografts model[132]. However, similar to HDACi, HDM and HMT inhibitors also have off-target effects on H3K9me2 and DNMT1 thus limiting their use[135] and further in-depth studies are required. EZH2 is another methyltransferase responsible for H3K27me3 leads to gene silencing by promoting DNA methylation[136]. EZH2 is overexpressed in head and neck, breast, and prostate cancers[137] and can be targeted by a hydrolase inhibitor called 3-deazaneplanocin A (DZNep). It induces differentiation as well as apoptosis in cancer cell lines and xenografts by countering EZH2 and inhibiting H3K27 trimethylation[138,139], while sparing normal cells.
Histone kinases and phosphatases as the targets
Compared to histone acetylation and methylation, the effort of regulating histone phosphorylation by targeting kinases and phosphatases for therapeutic uses is new. High level of several histone H3 phosphorylations such as H3S10ph, H3T6ph has been reported in a number of cancers. Unpublished data from our lab shows increase of H3S10ph in cisplatin resistance gastric cancer cell lines AGS and KATOIII. Our observation further supported the finding that p38 MAPK pathway mediated increase in H3S10ph in response to cisplatin treatment[140] in HeLa and MCF7 cells. Pacaud et al[93] recently reported that the kinase inhibitors like Enzastaurin (PKC-beta inhibitor), AZD1152 (Aurora-B inhibitor) and AZD1480 (Jak2 inhibitor) increases the cell death of TMZ-Irrad resistant GBM and decreases H3T3ph, H3S10ph and H3Y41ph respectively. Further, H89 (MSK1 inhibitor) treatment reduces the TPA and EGF mediated cellular transformation and by decreasing H3S10ph[44]. All these studies represent the potential of regulating histone phosphorylation for therapeutic use in cancer; however, these observations need to be further explored.
Despite of all this progress in the utilization of histone PTMs in chemotherapeutic interventions, a very little is known about their utility in monitoring the response to chemotherapy. For this purpose, levels of cNUCs and their modifications can be utilized. Because, circulating nucleosomes in serum are a result of apoptosis of actively dividing cells; therefore, after chemotherapy/radiotherapy increase in the circulating nucleosomes correlates with progressive disease and decrease was associated with disease regression. Increase in the concentration of serum nucleosomes has been shown at 24-72 h after the first application of chemotherapy and 6-24 h after the start of radiotherapy[60]. Thus, the concentration of nucleosomes in serum might be a useful tool for monitoring the biochemical responses during antitumor therapy, particularly for the early estimation of therapeutic efficacy. Histone modifications such as H4K16ac for example, can be utilized in this regard as its loss has been reported in several cancers and also chemosensitize cancer cells[33,69,141]. Histone modifications like H3K27me3 have indeed showed perplexing results when analyzed with respect to various cancers. This can be attributed to tissue type, and indeed histone PTMs are known to be showing their abundance in a tissue specific manner[142]. This might be as because many writers and erasers utilize co-factors or substrates like acetyl CoA, SAM, NAD+, FAD+ or ATP which are crucial metabolites in core pathways of intermediary metabolism[143]. The cellular concentrations of these metabolites fluctuate with the metabolic status of the cells and thus, the activity of these enzymes gets affected thus the histone PTMs.
CONCLUSION AND FUTURE DIRECTIONS
The role of histone modifications in governing cellular functions has been not yet fully understood. However, with increased research over the past decade, all the organisms studied so far (from yeast to man) have bought to light the importance of chromatin environment especially histone PTMs in development and disease. These observations have revolutionized the field of epigenetics and have challenged the old hypothesis of the genetic code being the sole determinant of the pathophysiology of any disease. In cancer, especially this is further established with the discovery of small molecule inhibitors targeting histone modifying enzymes, which can restore the expression of various genes to normal and can induce apoptosis of transformed cells. The best studied examples of these drugs are HDACi, which have proven to be highly effective anticancer drugs, thus are in clinics. Although the exact nature of the mechanism by which these drugs act is not understood yet, still these drugs are faring better against cancer. Future studies need to be directed more towards understanding these mechanisms and increasing the potency of these drugs. Though many histone PTMs are known to change during cancer, less is understood regarding the significance and mechanistic details of the change observed. Much of the work done in this direction has been hindered due to technical limitations. However with the advent of new technologies, and also decrease in the cost of high throughput technologies like ChIP-seq and TMA amongst other global approaches, it is a matter of time we have more knowledge of these mechanisms. Also, new targets for development of more potent drugs need to be explored by careful understanding of an already existing chromatin atlas of various cancer cell lines and tissues. Further work in the next decade may gain deeper understanding of the global patterns of histone posttranslational modifications and their corresponding changes which will hopefully reveal many molecular targets that can be employed as new weapons in long fought battle against cancer.
ACKNOWLEDGMENTS
The authors would like to thank Asmita Sharda and Prathamesh Amnekar, members of “Epigenetics and Chromatin Biology Group” for their contribution in editing and figures of the manuscript. SAK was supported by DBT, India and DR is supported by CSIR, India for their doctoral fellowships.
Footnotes
P- Reviewer: Cui YP, Freire-De-Lima CG, Hong YR, Pajares MA S- Editor: Song XX L- Editor: A E- Editor: Lu YJ
Conflict-of-interest statement: No potential conflicts of interest relevant to this article were reported.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 2, 2015
First decision: June 18, 2015
Article in press: October 8, 2015
References
- 1.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- 4.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 5.Castelo-Branco G, Bannister AJ. The epigenetics of cancer: from non-coding RNAs to chromatin and beyond. Brief Funct Genomics. 2013;12:161–163. doi: 10.1093/bfgp/elt020. [DOI] [PubMed] [Google Scholar]
- 6.Beckedorff FC, Amaral MS, Deocesano-Pereira C, Verjovski-Almeida S. Long non-coding RNAs and their implications in cancer epigenetics. Biosci Rep. 2013;33:e00061. doi: 10.1042/BSR20130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E. Histone variants: emerging players in cancer biology. Cell Mol Life Sci. 2014;71:379–404. doi: 10.1007/s00018-013-1343-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 14.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 15.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 16.Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. HIstome-a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic acids research. 2011;40:D337–D342. doi: 10.1093/nar/gkr1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 18.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khare SP, Sharma A, Deodhar KK, Gupta S. Overexpression of histone variant H2A.1 and cellular transformation are related in N-nitrosodiethylamine-induced sequential hepatocarcinogenesis. Exp Biol Med (Maywood) 2011;236:30–35. doi: 10.1258/ebm.2010.010140. [DOI] [PubMed] [Google Scholar]
- 20.Rangasamy D. Histone variant H2A.Z can serve as a new target for breast cancer therapy. Curr Med Chem. 2010;17:3155–3161. doi: 10.2174/092986710792231941. [DOI] [PubMed] [Google Scholar]
- 21.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 24.Baek SH. When signaling kinases meet histones and histone modifiers in the nucleus. Mol Cell. 2011;42:274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 25.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 26.Shogren-Knaak M, Peterson CL. Switching on chromatin: mechanistic role of histone H4-K16 acetylation. Cell Cycle. 2006;5:1361–1365. doi: 10.4161/cc.5.13.2891. [DOI] [PubMed] [Google Scholar]
- 27.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 28.Daujat S, Zeissler U, Waldmann T, Happel N, Schneider R. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J Biol Chem. 2005;280:38090–38095. doi: 10.1074/jbc.C500229200. [DOI] [PubMed] [Google Scholar]
- 29.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 30.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 33.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 34.Zhu L, Yang J, Zhao L, Yu X, Wang L, Wang F, Cai Y, Jin J. Expression of hMOF, but not HDAC4, is responsible for the global histone H4K16 acetylation in gastric carcinoma. Int J Oncol. 2015;46:2535–2545. doi: 10.3892/ijo.2015.2956. [DOI] [PubMed] [Google Scholar]
- 35.Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. 2010;29:4896–4904. doi: 10.1038/onc.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Li Q, Chen H. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis. 2013;34:1756–1763. doi: 10.1093/carcin/bgt129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen CT, Weisenberger DJ, Velicescu M, Gonzales FA, Lin JC, Liang G, Jones PA. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2’-deoxycytidine. Cancer Res. 2002;62:6456–6461. [PubMed] [Google Scholar]
- 38.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 39.Prenzel T, Begus-Nahrmann Y, Kramer F, Hennion M, Hsu C, Gorsler T, Hintermair C, Eick D, Kremmer E, Simons M, et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011;71:5739–5753. doi: 10.1158/0008-5472.CAN-11-1896. [DOI] [PubMed] [Google Scholar]
- 40.Li B, Huang G, Zhang X, Li R, Wang J, Dong Z, He Z. Increased phosphorylation of histone H3 at serine 10 is involved in Epstein-Barr virus latent membrane protein-1-induced carcinogenesis of nasopharyngeal carcinoma. BMC Cancer. 2013;13:124. doi: 10.1186/1471-2407-13-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, Davie JR. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J Biol Chem. 1999;274:24914–24920. doi: 10.1074/jbc.274.35.24914. [DOI] [PubMed] [Google Scholar]
- 42.Choi HS, Choi BY, Cho YY, Mizuno H, Kang BS, Bode AM, Dong Z. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer Res. 2005;65:5818–5827. doi: 10.1158/0008-5472.CAN-05-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tange S, Ito S, Senga T, Hamaguchi M. Phosphorylation of histone H3 at Ser10: its role in cell transformation by v-Src. Biochem Biophys Res Commun. 2009;386:588–592. doi: 10.1016/j.bbrc.2009.06.082. [DOI] [PubMed] [Google Scholar]
- 44.Kim HG, Lee KW, Cho YY, Kang NJ, Oh SM, Bode AM, Dong Z. Mitogen- and stress-activated kinase 1-mediated histone H3 phosphorylation is crucial for cell transformation. Cancer Res. 2008;68:2538–2547. doi: 10.1158/0008-5472.CAN-07-6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Gruttola VG, Clax P, DeMets DL, Downing GJ, Ellenberg SS, Friedman L, Gail MH, Prentice R, Wittes J, Zeger SL. Considerations in the evaluation of surrogate endpoints in clinical trials. summary of a National Institutes of Health workshop. Control Clin Trials. 2001;22:485–502. doi: 10.1016/s0197-2456(01)00153-2. [DOI] [PubMed] [Google Scholar]
- 46.Machiels BM, Ruers T, Lindhout M, Hardy K, Hlavaty T, Bang DD, Somers VA, Baeten C, von Meyenfeldt M, Thunnissen FB. New protocol for DNA extraction of stool. Biotechniques. 2000;28:286–290. doi: 10.2144/00282st05. [DOI] [PubMed] [Google Scholar]
- 47.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646–650. [PubMed] [Google Scholar]
- 48.Matei DE, Nephew KP. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol Oncol. 2010;116:195–201. doi: 10.1016/j.ygyno.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahlquist DA, Skoletsky JE, Boynton KA, Harrington JJ, Mahoney DW, Pierceall WE, Thibodeau SN, Shuber AP. Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Gastroenterology. 2000;119:1219–1227. doi: 10.1053/gast.2000.19580. [DOI] [PubMed] [Google Scholar]
- 50.Leung WK, To KF, Man EP, Chan MW, Bai AH, Hui AJ, Chan FK, Lee JF, Sung JJ. Detection of epigenetic changes in fecal DNA as a molecular screening test for colorectal cancer: a feasibility study. Clin Chem. 2004;50:2179–2182. doi: 10.1373/clinchem.2004.039305. [DOI] [PubMed] [Google Scholar]
- 51.Leung WK, To KF, Man EP, Chan MW, Hui AJ, Ng SS, Lau JY, Sung JJ. Detection of hypermethylated DNA or cyclooxygenase-2 messenger RNA in fecal samples of patients with colorectal cancer or polyps. Am J Gastroenterol. 2007;102:1070–1076. doi: 10.1111/j.1572-0241.2007.01108.x. [DOI] [PubMed] [Google Scholar]
- 52.Li WH, Zhang H, Guo Q, Wu XD, Xu ZS, Dang CX, Xia P, Song YC. Detection of SNCA and FBN1 methylation in the stool as a biomarker for colorectal cancer. Dis Markers. 2015;2015:657570. doi: 10.1155/2015/657570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scher MB, Elbaum MB, Mogilevkin Y, Hilbert DW, Mydlo JH, Sidi AA, Adelson ME, Mordechai E, Trama JP. Detecting DNA methylation of the BCL2, CDKN2A and NID2 genes in urine using a nested methylation specific polymerase chain reaction assay to predict bladder cancer. J Urol. 2012;188:2101–2107. doi: 10.1016/j.juro.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 54.Chung W, Bondaruk J, Jelinek J, Lotan Y, Liang S, Czerniak B, Issa JP. Detection of bladder cancer using novel DNA methylation biomarkers in urine sediments. Cancer Epidemiol Biomarkers Prev. 2011;20:1483–1491. doi: 10.1158/1055-9965.EPI-11-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoque MO, Begum S, Topaloglu O, Chatterjee A, Rosenbaum E, Van Criekinge W, Westra WH, Schoenberg M, Zahurak M, Goodman SN, et al. Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J Natl Cancer Inst. 2006;98:996–1004. doi: 10.1093/jnci/djj265. [DOI] [PubMed] [Google Scholar]
- 56.Reinert T. Methylation markers for urine-based detection of bladder cancer: the next generation of urinary markers for diagnosis and surveillance of bladder cancer. Adv Urol. 2012;2012:503271. doi: 10.1155/2012/503271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olkhov-Mitsel E, Zdravic D, Kron K, van der Kwast T, Fleshner N, Bapat B. Novel multiplex MethyLight protocol for detection of DNA methylation in patient tissues and bodily fluids. Sci Rep. 2014;4:4432. doi: 10.1038/srep04432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 59.Gezer U, Mert U, Ozgur E, Yoruker EE, Holdenrieder S, Dalay N. Correlation of histone methyl marks with circulating nucleosomes in blood plasma of cancer patients. Oncol Lett. 2012;3:1095–1098. doi: 10.3892/ol.2012.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holdenrieder S, Stieber P, Bodenmüller H, Busch M, Fertig G, Fürst H, Schalhorn A, Schmeller N, Untch M, Seidel D. Nucleosomes in serum of patients with benign and malignant diseases. Int J Cancer. 2001;95:114–120. doi: 10.1002/1097-0215(20010320)95:2<114::aid-ijc1020>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 61.Deligezer U, Akisik EE, Erten N, Dalay N. Sequence-specific histone methylation is detectable on circulating nucleosomes in plasma. Clin Chem. 2008;54:1125–1131. doi: 10.1373/clinchem.2007.101766. [DOI] [PubMed] [Google Scholar]
- 62.Leszinski G, Gezer U, Siegele B, Stoetzer O, Holdenrieder S. Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Res. 2012;32:2199–2205. [PubMed] [Google Scholar]
- 63.Gezer U, Ustek D, Yörüker EE, Cakiris A, Abaci N, Leszinski G, Dalay N, Holdenrieder S. Characterization of H3K9me3- and H4K20me3-associated circulating nucleosomal DNA by high-throughput sequencing in colorectal cancer. Tumour Biol. 2013;34:329–336. doi: 10.1007/s13277-012-0554-5. [DOI] [PubMed] [Google Scholar]
- 64.Tryndyak VP, Kovalchuk O, Pogribny IP. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins. Cancer Biol Ther. 2006;5:65–70. doi: 10.4161/cbt.5.1.2288. [DOI] [PubMed] [Google Scholar]
- 65.Bagnyukova TV, Tryndyak VP, Montgomery B, Churchwell MI, Karpf AR, James SR, Muskhelishvili L, Beland FA, Pogribny IP. Genetic and epigenetic changes in rat preneoplastic liver tissue induced by 2-acetylaminofluorene. Carcinogenesis. 2008;29:638–646. doi: 10.1093/carcin/bgm303. [DOI] [PubMed] [Google Scholar]
- 66.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 67.Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol. 2009;174:1619–1628. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barlési F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol. 2007;25:4358–4364. doi: 10.1200/JCO.2007.11.2599. [DOI] [PubMed] [Google Scholar]
- 69.Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009;69:3802–3809. doi: 10.1158/0008-5472.CAN-08-3907. [DOI] [PubMed] [Google Scholar]
- 70.Ellinger J, Kahl P, Mertens C, Rogenhofer S, Hauser S, Hartmann W, Bastian PJ, Büttner R, Müller SC, von Ruecker A. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int J Cancer. 2010;127:2360–2366. doi: 10.1002/ijc.25250. [DOI] [PubMed] [Google Scholar]
- 71.Manuyakorn A, Paulus R, Farrell J, Dawson NA, Tze S, Cheung-Lau G, Hines OJ, Reber H, Seligson DB, Horvath S, et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J Clin Oncol. 2010;28:1358–1365. doi: 10.1200/JCO.2009.24.5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rajendran G, Shanmuganandam K, Bendre A, Muzumdar D, Goel A, Shiras A. Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J Neurooncol. 2011;104:483–494. doi: 10.1007/s11060-010-0520-2. [DOI] [PubMed] [Google Scholar]
- 73.Bai X, Wu L, Liang T, Liu Z, Li J, Li D, Xie H, Yin S, Yu J, Lin Q, et al. Overexpression of myocyte enhancer factor 2 and histone hyperacetylation in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:83–91. doi: 10.1007/s00432-007-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mohamed MA, Greif PA, Diamond J, Sharaf O, Maxwell P, Montironi R, Young RA, Hamilton PW. Epigenetic events, remodelling enzymes and their relationship to chromatin organization in prostatic intraepithelial neoplasia and prostatic adenocarcinoma. BJU Int. 2007;99:908–915. doi: 10.1111/j.1464-410X.2006.06704.x. [DOI] [PubMed] [Google Scholar]
- 75.Zhen L, Gui-lan L, Ping Y, Jin H, Ya-li W. The expression of H3K9Ac, H3K14Ac, and H4K20TriMe in epithelial ovarian tumors and the clinical significance. Int J Gynecol Cancer. 2010;20:82–86. doi: 10.1111/IGC.0b013e3181ae3efa. [DOI] [PubMed] [Google Scholar]
- 76.Liu BL, Cheng JX, Zhang X, Wang R, Zhang W, Lin H, Xiao X, Cai S, Chen XY, Cheng H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol Biomarkers Prev. 2010;19:2888–2896. doi: 10.1158/1055-9965.EPI-10-0454. [DOI] [PubMed] [Google Scholar]
- 77.Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol. 2008;15:1968–1976. doi: 10.1245/s10434-008-9927-9. [DOI] [PubMed] [Google Scholar]
- 78.Müller-Tidow C, Klein HU, Hascher A, Isken F, Tickenbrock L, Thoennissen N, Agrawal-Singh S, Tschanter P, Disselhoff C, Wang Y, et al. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood. 2010;116:3564–3571. doi: 10.1182/blood-2009-09-240978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK. Epigenetic reprogramming by adenovirus e1a. Science. 2008;321:1086–1088. doi: 10.1126/science.1155546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. Adenovirus small e1a alters global patterns of histone modification. Science. 2008;321:1084–1085. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tzao C, Tung HJ, Jin JS, Sun GH, Hsu HS, Chen BH, Yu CP, Lee SC. Prognostic significance of global histone modifications in resected squamous cell carcinoma of the esophagus. Mod Pathol. 2009;22:252–260. doi: 10.1038/modpathol.2008.172. [DOI] [PubMed] [Google Scholar]
- 82.Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R, Wang X, Ghosh D, Shah RB, et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007;67:10657–10663. doi: 10.1158/0008-5472.CAN-07-2498. [DOI] [PubMed] [Google Scholar]
- 83.Wei Y, Xia W, Zhang Z, Liu J, Wang H, Adsay NV, Albarracin C, Yu D, Abbruzzese JL, Mills GB, et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog. 2008;47:701–706. doi: 10.1002/mc.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He LR, Liu MZ, Li BK, Rao HL, Liao YJ, Guan XY, Zeng YX, Xie D. Prognostic impact of H3K27me3 expression on locoregional progression after chemoradiotherapy in esophageal squamous cell carcinoma. BMC Cancer. 2009;9:461. doi: 10.1186/1471-2407-9-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang L, Zhong K, Dai Y, Zhou H. Genome-wide analysis of histone H3 lysine 27 trimethylation by ChIP-chip in gastric cancer patients. J Gastroenterol. 2009;44:305–312. doi: 10.1007/s00535-009-0027-9. [DOI] [PubMed] [Google Scholar]
- 86.Chen X, Song N, Matsumoto K, Nanashima A, Nagayasu T, Hayashi T, Ying M, Endo D, Wu Z, Koji T. High expression of trimethylated histone H3 at lysine 27 predicts better prognosis in non-small cell lung cancer. Int J Oncol. 2013;43:1467–1480. doi: 10.3892/ijo.2013.2062. [DOI] [PubMed] [Google Scholar]
- 87.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 88.Karczmarski J, Rubel T, Paziewska A, Mikula M, Bujko M, Kober P, Dadlez M, Ostrowski J. Histone H3 lysine 27 acetylation is altered in colon cancer. Clin Proteomics. 2014;11:24. doi: 10.1186/1559-0275-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tamagawa H, Oshima T, Shiozawa M, Morinaga S, Nakamura Y, Yoshihara M, Sakuma Y, Kameda Y, Akaike M, Masuda M, et al. The global histone modification pattern correlates with overall survival in metachronous liver metastasis of colorectal cancer. Oncol Rep. 2012;27:637–642. doi: 10.3892/or.2011.1547. [DOI] [PubMed] [Google Scholar]
- 90.Nagelkerke A, van Kuijk SJ, Sweep FC, Nagtegaal ID, Hoogerbrugge N, Martens JW, Timmermans MA, van Laarhoven HW, Bussink J, Span PN. Constitutive expression of γ-H2AX has prognostic relevance in triple negative breast cancer. Radiother Oncol. 2011;101:39–45. doi: 10.1016/j.radonc.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 91.Nagelkerke A, van Kuijk SJ, Martens JW, Sweep FC, Hoogerbrugge N, Bussink J, Span PN. Poor prognosis of constitutive γ-H2AX expressing triple-negative breast cancers is associated with telomere length. Biomark Med. 2015;9:383–390. doi: 10.2217/bmm.15.2. [DOI] [PubMed] [Google Scholar]
- 92.Lee YC, Yin TC, Chen YT, Chai CY, Wang JY, Liu MC, Lin YC, Kan JY. High expression of phospho-H2AX predicts a poor prognosis in colorectal cancer. Anticancer Res. 2015;35:2447–2453. [PubMed] [Google Scholar]
- 93.Pacaud R, Cheray M, Nadaradjane A, Vallette FM, Cartron PF. Histone H3 phosphorylation in GBM: a new rational to guide the use of kinase inhibitors in anti-GBM therapy. Theranostics. 2015;5:12–22. doi: 10.7150/thno.8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ladstein RG, Bachmann IM, Straume O, Akslen LA. Prognostic importance of the mitotic marker phosphohistone H3 in cutaneous nodular melanoma. J Invest Dermatol. 2012;132:1247–1252. doi: 10.1038/jid.2011.464. [DOI] [PubMed] [Google Scholar]
- 95.Tetzlaff MT, Curry JL, Ivan D, Wang WL, Torres-Cabala CA, Bassett RL, Valencia KM, McLemore MS, Ross MI, Prieto VG. Immunodetection of phosphohistone H3 as a surrogate of mitotic figure count and clinical outcome in cutaneous melanoma. Mod Pathol. 2013;26:1153–1160. doi: 10.1038/modpathol.2013.59. [DOI] [PubMed] [Google Scholar]
- 96.Skaland I, Janssen EA, Gudlaugsson E, Klos J, Kjellevold KH, Søiland H, Baak JP. Validating the prognostic value of proliferation measured by Phosphohistone H3 (PPH3) in invasive lymph node-negative breast cancer patients less than 71 years of age. Breast Cancer Res Treat. 2009;114:39–45. doi: 10.1007/s10549-008-9980-x. [DOI] [PubMed] [Google Scholar]
- 97.Skaland I, Janssen EA, Gudlaugsson E, Klos J, Kjellevold KH, Søiland H, Baak JP. Phosphohistone H3 expression has much stronger prognostic value than classical prognosticators in invasive lymph node-negative breast cancer patients less than 55 years of age. Mod Pathol. 2007;20:1307–1315. doi: 10.1038/modpathol.3800972. [DOI] [PubMed] [Google Scholar]
- 98.Nakashima S, Shiozaki A, Ichikawa D, Komatsu S, Konishi H, Iitaka D, Kubota T, Fujiwara H, Okamoto K, Kishimoto M, et al. Anti-phosphohistone H3 as an independent prognostic factor in human esophageal squamous cell carcinoma. Anticancer Res. 2013;33:461–467. [PubMed] [Google Scholar]
- 99.Uguen A, Conq G, Doucet L, Talagas M, Costa S, De Braekeleer M, Marcorelles P. Immunostaining of phospho-histone H3 and Ki-67 improves reproducibility of recurrence risk assessment of gastrointestinal stromal tumors. Virchows Arch. 2015;467:47–54. doi: 10.1007/s00428-015-1763-2. [DOI] [PubMed] [Google Scholar]
- 100.Takahashi H, Murai Y, Tsuneyama K, Nomoto K, Okada E, Fujita H, Takano Y. Overexpression of phosphorylated histone H3 is an indicator of poor prognosis in gastric adenocarcinoma patients. Appl Immunohistochem Mol Morphol. 2006;14:296–302. doi: 10.1097/00129039-200609000-00007. [DOI] [PubMed] [Google Scholar]
- 101.Nielsen PS, Riber-Hansen R, Jensen TO, Schmidt H, Steiniche T. Proliferation indices of phosphohistone H3 and Ki67: strong prognostic markers in a consecutive cohort with stage I/II melanoma. Mod Pathol. 2013;26:404–413. doi: 10.1038/modpathol.2012.188. [DOI] [PubMed] [Google Scholar]
- 102.Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Espino PS, Drobic B, Dunn KL, Davie JR. Histone modifications as a platform for cancer therapy. J Cell Biochem. 2005;94:1088–1102. doi: 10.1002/jcb.20387. [DOI] [PubMed] [Google Scholar]
- 105.Balch C, Nephew KP. Epigenetic targeting therapies to overcome chemotherapy resistance. Adv Exp Med Biol. 2013;754:285–311. doi: 10.1007/978-1-4419-9967-2_14. [DOI] [PubMed] [Google Scholar]
- 106.Li SY, Sun R, Wang HX, Shen S, Liu Y, Du XJ, Zhu YH, Jun W. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J Control Release. 2015;205:7–14. doi: 10.1016/j.jconrel.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 107.Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 108.Cacan E, Ali MW, Boyd NH, Hooks SB, Greer SF. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS One. 2014;9:e87455. doi: 10.1371/journal.pone.0087455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hehlgans S, Storch K, Lange I, Cordes N. The novel HDAC inhibitor NDACI054 sensitizes human cancer cells to radiotherapy. Radiother Oncol. 2013;109:126–132. doi: 10.1016/j.radonc.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 110.Hubaux R, Vandermeers F, Crisanti MC, Kapoor V, Burny A, Mascaux C, Albelda SM, Willems L. Preclinical evidence for a beneficial impact of valproate on the response of small cell lung cancer to first-line chemotherapy. Eur J Cancer. 2010;46:1724–1734. doi: 10.1016/j.ejca.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 111.Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001;61:8492–8497. [PubMed] [Google Scholar]
- 112.Louis M, Rosato RR, Brault L, Osbild S, Battaglia E, Yang XH, Grant S, Bagrel D. The histone deacetylase inhibitor sodium butyrate induces breast cancer cell apoptosis through diverse cytotoxic actions including glutathione depletion and oxidative stress. Int J Oncol. 2004;25:1701–1711. [PubMed] [Google Scholar]
- 113.Said TK, Moraes RC, Sinha R, Medina D. Mechanisms of suberoylanilide hydroxamic acid inhibition of mammary cell growth. Breast Cancer Res. 2001;3:122–133. doi: 10.1186/bcr284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dizon DS, Blessing JA, Penson RT, Drake RD, Walker JL, Johnston CM, Disilvestro PA, Fader AN. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;125:367–371. doi: 10.1016/j.ygyno.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tarhini AA, Zahoor H, McLaughlin B, Gooding WE, Schmitz JC, Siegfried JM, Socinski MA, Argiris A. Phase I trial of carboplatin and etoposide in combination with panobinostat in patients with lung cancer. Anticancer Res. 2013;33:4475–4481. [PMC free article] [PubMed] [Google Scholar]
- 116.Cassier PA, Lefranc A, Amela EY, Chevreau C, Bui BN, Lecesne A, Ray-Coquard I, Chabaud S, Penel N, Berge Y, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer. 2013;109:909–914. doi: 10.1038/bjc.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, Varella-Garcia M, Bunn PA, Hirsch FR. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. 2012;30:2248–2255. doi: 10.1200/JCO.2011.38.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ, Trepel JB. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–2135. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dong M, Ning ZQ, Xing PY, Xu JL, Cao HX, Dou GF, Meng ZY, Shi YK, Lu XP, Feng FY. Phase I study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother Pharmacol. 2012;69:1413–1422. doi: 10.1007/s00280-012-1847-5. [DOI] [PubMed] [Google Scholar]
- 120.Zorzi AP, Bernstein M, Samson Y, Wall DA, Desai S, Nicksy D, Wainman N, Eisenhauer E, Baruchel S. A phase I study of histone deacetylase inhibitor, pracinostat (SB939), in pediatric patients with refractory solid tumors: IND203 a trial of the NCIC IND program/C17 pediatric phase I consortium. Pediatr Blood Cancer. 2013;60:1868–1874. doi: 10.1002/pbc.24694. [DOI] [PubMed] [Google Scholar]
- 121.de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, Karavasilis V, Mita M, Shaw H, Workman P, Kaye S, et al. Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008;14:6663–6673. doi: 10.1158/1078-0432.CCR-08-0376. [DOI] [PubMed] [Google Scholar]
- 122.Shi H, Wei SH, Leu YW, Rahmatpanah F, Liu JC, Yan PS, Nephew KP, Huang TH. Triple analysis of the cancer epigenome: an integrated microarray system for assessing gene expression, DNA methylation, and histone acetylation. Cancer Res. 2003;63:2164–2171. [PubMed] [Google Scholar]
- 123.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61:7025–7029. [PubMed] [Google Scholar]
- 124.Belinsky SA, Klinge DM, Stidley CA, Issa JP, Herman JG, March TH, Baylin SB. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003;63:7089–7093. [PubMed] [Google Scholar]
- 125.Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem. 2004;92:223–237. doi: 10.1002/jcb.20045. [DOI] [PubMed] [Google Scholar]
- 126.Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. In vivo synergy between topoisomerase II and histone deacetylase inhibitors: predictive correlates. Mol Cancer Ther. 2005;4:1993–2000. doi: 10.1158/1535-7163.MCT-05-0194. [DOI] [PubMed] [Google Scholar]
- 127.Belinsky SA, Grimes MJ, Picchi MA, Mitchell HD, Stidley CA, Tesfaigzi Y, Channell MM, Liu Y, Casero RA, Baylin SB, et al. Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res. 2011;71:454–462. doi: 10.1158/0008-5472.CAN-10-3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andrade FO, Nagamine MK, Conti AD, Chaible LM, Fontelles CC, Jordão Junior AA, Vannucchi H, Dagli ML, Bassoli BK, Moreno FS, et al. Efficacy of the dietary histone deacetylase inhibitor butyrate alone or in combination with vitamin A against proliferation of MCF-7 human breast cancer cells. Braz J Med Biol Res. 2012;45:841–850. doi: 10.1590/S0100-879X2012007500103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mack GS. To selectivity and beyond. Nat Biotechnol. 2010;28:1259–1266. doi: 10.1038/nbt.1724. [DOI] [PubMed] [Google Scholar]
- 130.Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 131.Lim S, Janzer A, Becker A, Zimmer A, Schüle R, Buettner R, Kirfel J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512–520. doi: 10.1093/carcin/bgp324. [DOI] [PubMed] [Google Scholar]
- 132.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 133.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wu Y, Steinbergs N, Murray-Stewart T, Marton LJ, Casero RA. Oligoamine analogues in combination with 2-difluoromethylornithine synergistically induce re-expression of aberrantly silenced tumour-suppressor genes. Biochem J. 2012;442:693–701. doi: 10.1042/BJ20111271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 136.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2010;21:209–220. doi: 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 138.Gannon OM, Merida de Long L, Endo-Munoz L, Hazar-Rethinam M, Saunders NA. Dysregulation of the repressive H3K27 trimethylation mark in head and neck squamous cell carcinoma contributes to dysregulated squamous differentiation. Clin Cancer Res. 2013;19:428–441. doi: 10.1158/1078-0432.CCR-12-2505. [DOI] [PubMed] [Google Scholar]
- 139.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang D, Lippard SJ. Cisplatin-induced post-translational modification of histones H3 and H4. J Biol Chem. 2004;279:20622–20625. doi: 10.1074/jbc.M402547200. [DOI] [PubMed] [Google Scholar]
- 141.Füllgrabe J, Hajji N, Joseph B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 2010;17:1238–1243. doi: 10.1038/cdd.2010.58. [DOI] [PubMed] [Google Scholar]
- 142.Garcia BA, Thomas CE, Kelleher NL, Mizzen CA. Tissue-specific expression and post-translational modification of histone H3 variants. J Proteome Res. 2008;7:4225–4236. doi: 10.1021/pr800044q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]