

Abstract
CaMKII is a remarkably complex protein kinase, known to have a fundamental role in synaptic plasticity and memory formation. Further, CaMKII has also been suggested to be a tau kinase. CaMKII dysregulation may therefore be a modulator of toxicity in Alzheimer’s disease, a dementia characterised by aberrant calcium signalling, synapse and neuronal loss, and impaired memory. Here, we first examine the evidence for CaMKII dysregulation in Alzheimer’s patients and draw parallels to findings in disease models which recapitulate key aspects of the disease. We then put forward the hypothesis that these changes critically contribute to neurodegeneration and memory impairment in Alzheimer’s disease.
Keywords: CaMKII, Alzheimer’s disease, Post-mortem brain, Autophosphorylation, Memory, Tau
Background
The most common form of adult dementia, Alzheimer’s disease (AD) is characterised by progressive loss of selective cognitive functions, particularly those related to memory. It was in the early 20th century that Alois Alzheimer first described the presence of ‘positive’ lesions such as senile plaques (SPs) and neurofibrillary tangles (NFTs) in the brain of a patient suffering from dementia (for a translation see [1]). However, it was not until the mid- to late 1980s that these were found to comprise aggregated amyloid-β (Aβ) peptides [2–4] and hyperphosphorylated tau protein [5–8], respectively. Although AD pathogenesis is yet to be fully elucidated, it has been posited that the Aβ peptide is central to disease onset. The “amyloid cascade” hypothesis suggests that Aβ, resulting from aberrant cleavage of the amyloid precursor protein (APP) by β- and γ-secretases, can aggregate into a toxic species, leading to a series of events that culminate in AD pathology [9, 10]. New evidence suggests alternative proteolytic pathways of APP by η-secretases can lead to production of a toxic amyloid-η (Aη) species that can also contribute to AD pathology [11].
While SPs and NFTs are useful diagnostic markers during post-mortem examination, it is actually the occurrence of ‘negative’ lesions such as synaptic loss, which precedes neuronal loss, that best correlates with the advancement of cognitive decline. Several reports have noted the progressive loss of synaptic boutons and other synaptic elements in brains of patients with symptoms ranging from mild cognitive impairment (MCI) to early-mild AD [12–17]. Hippocampal and cortical regions show the most marked loss of these features, reflecting their importance in processes of memory formation and storage. The precise mechanism by which synaptic dysfunction occurs in the AD brain is unknown; in vitro studies have shown that Aβ oligomers can directly bind to synaptic sites [18] and reduce long-term potentiation (LTP) [19–21], while facilitating long-term depression (LTD) [22]. Aβ oligomers can compromise synaptic function at both pre- and post-synaptic sites, but their early targets may in fact be excitatory post-synapses [23], where they can alter several signalling pathways.
There is significant evidence that intracellular calcium (Ca2+) homeostasis is disrupted in both sporadic and familial forms of AD, and can exacerbate Aβ formation and promote tau hyperphosphorylation (for reviews see [24, 25]). Additionally Aβ can influence cellular pathways involved in Ca2+ buffering, compromising the ability of neurons to respond to excitotoxic challenge [26], suggestive of a pathogenic feed-forward cycle where Aβ and Ca2+ can concomitantly impair synaptic morphology, trigger neuronal apoptosis, and eventually lead to deterioration of cognition [27]. The key players in such a pathological cascade are most likely molecules that lie downstream of Ca2+-signalling and are also present in excitatory synapses where Aβ oligomers likely initially bind. One candidate is the Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII), the major post-synaptic protein at excitatory synapses. This kinase is fundamentally important for synaptic plasticity and memory formation. Here we discuss evidence for the involvement of CaMKII in AD pathogenesis.
CaMKII: regulation and function
CaMKII is a holoenzyme of 12 subunits, each derived from one of four genes (α, β, γ and δ) [28]. In rat forebrain, αCaMKII and βCaMKII are the most abundant subunits, with the former expressed 3–4 times more than the latter [29], and can assemble into homo- or heteromeric holoenzymes [30]. The expression and function of α and βCaMKII differ; while α is expressed exclusively in glutamatergic neurons [31], the β subunit is also expressed in inhibitory interneurons [32]. Further, βCaMKII, but not α, binds to F-actin, which is relieved upon activation by Ca2+/CaM [33]. This dissociation is thought to regulate morphological changes at the synapse [34]. Functionally, αCaMKII activity is essential for synaptic plasticity and memory formation, as elegantly demonstrated in knock-in mutant mice [35]. It may also have a structural role as it can bind to various proteins at the synapse [36] and its expression is extremely abundant (about 1.4% of hippocampal protein) [29]. In contrast, βCaMKII activity is not required for synaptic plasticity and memory formation [37], indicating that the primary function of this subunit is structural.
CaMKII holoenzymes are activated by the binding of Ca2+/CaM, and also by NMDA receptors (NMDARs) and L-type voltage-gated Ca2+ channels (VGCCs) at the synapse [38]. An important aspect of αCaMKII activity is its autophosphorylation at threonine-286 (T286) (for review see [39]). This autophosphorylation results from an interaction between subunits within the holoenzyme and switches the subunit activity from a Ca2+/CaM-dependent to – independent state. This ‘autonomous’ activity persists at the synapse for about one minute after stimulation [38]. However, T286 autophosphorylation can last longer and the dissociation between prolonged autophosphorylation and autonomous activity is not understood [39, 40]. Studies with T286 autophosphorylation-deficient knock-in mutants have shown that this event is fundamentally important for NMDAR-dependent LTP at hippocampal CA1 synapses [41–43] but not at perforant path-granule cell synapses [44]. Furthermore, T286 autophosphorylation is essential for spatial memory formation [41, 45]. Besides T286 autophosphorylation, αCaMKII is also regulated by other autophosphorylation events, phosphatase activity and endogenous inhibitor proteins (for reviews see [36, 46]).
CaMKII abnormalities in AD
Expression analyses of post-mortem disease brain can be very informative, in that prominent disease-related dysfunction is detectable. In contrast, studies with AD models, in rodents or in vitro, suffer from inadequate modelling of disease cause. The limitation of post-mortem studies is that they may be confounded by post-mortem delay, which can range from several hours to one day, during which protein expression may decrease and, in particular, post-translational protein modifications such as phosphorylation may be compromised. Another limitation is that they offer only one time point for analysis; however the severity of disease at the time of death can be estimated [47].
Semi-quantitative western blot studies with post-mortem tissue have suggested that αCaMKII protein expression level is not altered in hippocampus, frontal cortex or other cortical areas in the severe stages of AD [48, 49]. However, immunohistochemical analyses have indicated that αCaMKII-expressing neurons, which are excitatory, are selectively lost in hippocampal area CA1 in severe AD [50, 51] (but see [52, 53]). The remaining excitatory neurons in CA1 appear to express increased levels of αCaMKII [50, 51]. Interestingly, increased αCaMKII expression is not found in hippocampal area CA3 in severe AD [51], a region which has almost no neuronal loss in the end stages of AD, in stark contrast to substantial neuronal loss in CA1 [54].
Changes in distribution of CaMKII mRNA in AD brain are more difficult to determine. One study finds reduced hybridisation of αCaMKII mRNA in CA1, but only when neuronal loss associated with severe NFT formation is observed [55], echoing the findings of Simonian et al. However another study finds an increase in hybridisation throughout the AD hippocampus, especially in the dentate gyrus (DG) and CA3 regions [56]. A more recent microarray analysis of several brain regions from AD patients discloses that alterations in the expression of CaMKII mRNA may be far more composite than previously thought, with genes encoding different subunits showing different directions in expression changes across brain regions [57].
Early western blot studies suggest that autophosphorylation of αCaMKII at T286 is reduced in hippocampus and frontal cortex of the severe AD brain [48]. This is also reflected by the fact that cortical regions show a total loss of immunoreactivity for active conformations of CaM and reduced immunoreactivity for other forms [58]. However, this result has not been replicated [59]. Instead, it has emerged that in CA3 and the DG of AD brain, the subcellular localisation of αCaMKII autophosphorylation is altered [59]. p(T286)-αCaMKII is specifically decreased in dendrites and synapses, and increased in perikarya of CA3 neurons and granule cells of the DG. This altered distribution correlates with cognitive impairment both in patients with AD and its prodrome MCI [59]. Studies using cultured fibroblasts and lymphocytes from patients also suggest dysregulated CaMKII activity in AD [60, 61].
CaMKII dysregulation in AD models
The study of molecular dysfunction in AD has been greatly advanced by the development of transgenic mouse models that recapitulate some AD hallmarks. However, such models usually overexpress mutated forms of the human APP gene, and therefore are not fully representative of the causes underlying AD [62]. Additionally, they are confounded by artefacts due to increased transgene expression, and ageing, the main risk factor of AD, is not sufficiently addressed. Nonetheless, if a molecular dysregulation is similar in post-mortem AD brain and in AD models, it is very likely that it occurs in the disease.
Studies on AD models, like post-mortem analyses, suggest abnormalities in regulation of CaMKII. One of the most widely used AD models is the Tg2576 mouse, which carries the APPSwe mutation (K670N/M671L). While the total levels of α/βCaMKII are not altered in the frontal cortex of these mice, there is a significant alteration in their subcellular distribution, from synapse to cytosol. This change is not due to synaptic loss and is also seen in levels of active αCaMKII, suggesting a selective loss of synaptic CaMKII [63]. Another commonly used mouse model contains mutations in both APP and presenilin-1 (PS1), a component of the γ-secretase complex. Two studies find altered hippocampal expression of p(T286)-αCaMKII in these mice, and one additionally finds reduced levels of the CaMKII-binding VGCC Cav1.2 and elevated CaM [64, 65]. Altered αCaMKII distribution is also found in a mouse model of sporadic AD in which amyloid oligomers are injected into the ventricles. This acute treatment results in a shift of p(T286)-αCaMKII from apical dendrites/spines to the somata of CA3 pyramidal neurons and is blocked by inhibition of the phosphatase calcineurin, which augments phosphatase-1 activity [59].
A calcineurin-dependent redistribution of autophosphorylated αCaMKII also occurs in Aβ oligomer-treated primary neuronal cultures [59, 63]. Moreover, treating hippocampal neurons with Aβ oligomers impairs αCaMKII activation [64, 66]. In contrast to rodent models, there is no change in CaM levels and greater expression of Cav1.2 channels [64], a finding which is confirmed by an independent study [67]. This may be the result of cell cultures modelling earlier stages of the disease where there is no neuronal loss [64], or due to a lack of fully functional synapses.
Impact of dysregulated CaMKII in AD
Post-mortem analyses and studies with AD models indicate that T286-autophosphorylation of αCaMKII is impaired at synapses in the disease. Considering this autophosphorylation is essential for NMDAR-dependent LTP at CA1 synapses and spatial memory formation [41, 42, 45, 68, 69], the redistribution of p(T286)-αCaMKII could contribute to cognitive impairment in AD. Consistent with this, the reduction of T286-autophosphorylation in apical dendrites of granule cells of the DG in subjects with MCI and AD correlates with cognitive dysfunction as measured by MMSE scores [59]. Moreover, spatial training of Tg2576 mice increases T286-autophosphorylation of αCaMKII in the hippocampus and rescues deficits in contextual memory formation [70], suggesting deficits in T286 autophosphorylation are key to causing impairments in synaptic plasticity and memory formation in AD. This idea is confirmed in studies with Aβ-treated cultured primary neurons, which have reduced surface expression of AMPA receptor (AMPAR) subunit GluA1 and impaired AMPAR-mediated synaptic transmission. Knockdown of CaMKII mimics these effects and CaMKII overexpression rescues these [63]. An analogous observation is seen when treating rat hippocampal slices with Aβ1-42, where Aβ inhibits CaMKII activation and blocks the stimulation-dependent phosphorylation of a CaMKII-specific site on GluA1 [71]. Furthermore, it has been suggested that neurotrophin-induced enhancement of p(T286)-αCaMKII leads to rescue of Aβ-induced deficits in LTP at hippocampal synapses [72].
At the neuropathological level, the finding that APP can be phosphorylated in vitro by several kinases including CaMKII [73], puts forward the hypothesis that there could be a possible link between CaMKII and Aβ production. Both McKee and Wang remark on some co-localisation of αCaMKII with SPs, with differences in the deposition pattern around diffuse and neuritic plaques [50, 51]. It has been found that phosphorylation on T668 of APP is elevated in AD brain and can regulate its cleavage by β-secretases [74], but this is not known to be a CaMKII site of phosphorylation. It has also been suggested that phosphorylation of CaMKII sites (T654/S655) can alter the conformation of APP [75] and regulate its trafficking [76], but direct evidence that CaMKII is involved is lacking.
The correlation between CaMKII and tau phosphorylation is much stronger. Increased αCaMKII expression in CA1 neurons [50, 51] and increased αCaMKII autophosphorylation in cell bodies of CA3 neurons and granule cells in the DG [59, 63] suggest that outside of synapses, αCaMKII is hyperactive. Being a tau kinase, this hyperactivity could contribute to NFT formation. NFTs are made of paired helical filaments (PHFs) which contain tau protein hyperphosphorylated at many sites [77]. Several analyses of AD brain find that αCaMKII expression in cell bodies frequently co-localises with NFTs or tau mRNA [50, 51, 53, 55, 78, 79]. Mass spectrometry has also revealed that AD brain tau is phosphorylated by CaMKII at several different sites [80]. CaMKII phosphorylation of tau alters its electrophoretic mobility and structure, in a manner specific to PHF-tau [81–83]. Additionally, isolation of PHFs from AD brains results in co-purification with αCaMKII, 4–7 times more than is observed in controls [78]. The difficulty in analysing the importance of CaMKII in tau hyperphosphorylation arises from the fact that tau can be phosphorylated by several other kinases at CaMKII sites. It has been found that phosphorylation by CaMKII alone only partially inhibits binding of tau to microtubules [84]. Additionally several post-mortem studies note that not all αCaMKII-expressing neurons develop NFTs [51, 53, 55], suggesting that other tau kinases/phosphatases are involved. A likely scenario is one where αCaMKII phosphorylation of tau can prime its phosphorylation by other kinases such as cdk5 and GSK3-β [85, 86]. Collectively, it is conceivable that CaMKII can contribute to NFT formation in AD.
The loss of synaptic proteins in AD, combined with dysregulated CaMKII, may also lead to neuronal death. It has been suggested that αCaMKII and the post-synaptic protein PSD-95 can compete for binding to the C-terminus of the NMDAR subunit NR2A upon physiological stimulus [87]. Treating hippocampal neurons with antisense oligonucleotides to PSD-95 leads to increased association of both total and p(T286)-αCaMKII with NR2A/B subunits, although the total levels of αCaMKII are unaltered [88]. This is paralleled by an increase in cell death which can be rescued by pharmacological inhibition of CaMKII. Interestingly, hippocampal neurons are more susceptible to this type of injury than cortical neurons, and in organotypic hippocampal slices, CA1 neurons show greater susceptibility than CA3 or DG neurons. This reflects the hierarchical decline of brain areas during disease progression [54], further suggesting that αCaMKII/NR2A co-expression may be a causal factor for cell death in AD. Additionally, selectively inhibiting CaMKII in Aβ-treated primary cortical cultures reduces amyloid-induced activity of caspases-2 and -3 as well as tau phosphorylation [89]. It is conceivable that the upregulation of αCaMKII in CA1 may be directly responsible for the severe atrophy seen in this region. CaMKII may also be involved in other signalling cascades related to neuronal decline [90–93].
Conclusions
It has been established that CaMKII is dysregulated in AD hippocampus (Fig. 1). We suggest that this dysregulation is a key contributor to synaptic degeneration, NFT formation and memory deficits. However, the nature of CaMKII dysregulation is undoubtedly complex and several questions remain unanswered. One key question is ‘how’ this dysregulation can occur. So far, the focus has been on levels of total or T286 autophosphorylated αCaMKII. Other aspects of CaMKII regulation and activity need to be addressed, such as distribution of unphosphorylated CaMKII in AD brain, and other important sites of autophosphorylation such as T305/6. Other subunits such as β and γ may also be integral to CaMKII dysregulation. For example, βCaMKII autophosphorylation can regulate its dissociation from F-actin, thereby allowing cytoskeletal remodelling in glutamatergic excitatory synapses, a necessary occurrence for LTP induction [94]. Impaired Ca2+ signalling could therefore impact upon both this dissociation and the reassociation between βCaMKII and F-actin, an event crucial for stabilisation of newly remodelled actin and LTP maintenance. Additionally, γCaMKII can act as a Ca2+/CaM shuttle to the nucleus to alter gene expression (for review see [95]). Another fundamental issue is how CaMKII is dysregulated specifically in the CA1 region, an area showing devastating neuronal loss in AD compared to normal ageing. Is increased αCaMKII expression in remaining CA1 neurons a compensatory effect or a precursor to neurotoxicity? How does this relate to the subcellular distribution of total and p(T286) αCaMKII in CA1, and are these changes also calcineurin-dependent? Finally, it remains to be determined whether CaMKII is essential for synaptic dysfunction, cognitive impairment and NFT formation in AD. Can restoring synaptic activity of CaMKII in models of AD prevent cognitive dysfunction? Can reducing somatic CaMKII in an in vivo model of tau pathology prevent or abolish tangle formation? Elucidating these questions will investigate the hypothesis that dysregulated CaMKII is a key contributor to synaptic dysfunction, neurodegeneration and memory impairment in AD, and may point to novel treatment routes.
Fig. 1.
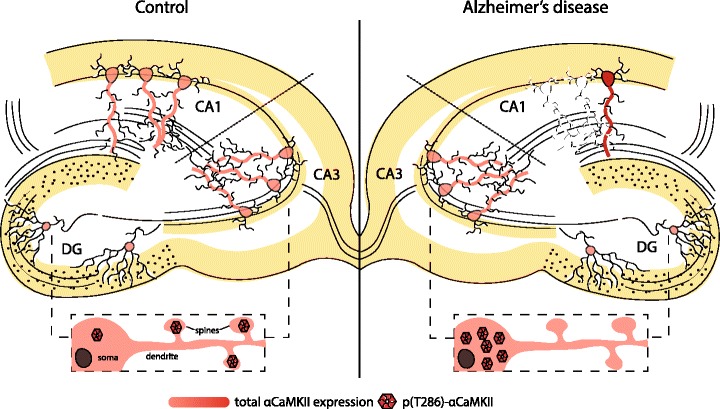
αCaMKII is dysregulated in the Alzheimer’s disease hippocampus. αCaMKII-expressing neurons are selectively lost in the hippocampal CA1 subfield in AD [50, 51], a region that shows devastating atrophy when compared to age-matched controls [54]. Remaining pyramidal neurons of this region show increased expression of αCaMKII. This increased expression may critically contribute to tau hyperphosphorylation and other neurodegenerative processes, such as caspase-3 overactivation, in CA1 pyramidal neurons [for references, see main text]. On the other hand, CA3 pyramidal neurons and granule cells of the DG do not develop these changes in total αCaMKII. They do however show a change in subcellular distribution of T286-autophosphorylated αCaMKII (inset) [59]. This change is suggested to shift CaMKII activity from the synapse to soma leading to synaptic deficits, neurodegenerative processes, and impaired memory formation. AD, Alzheimer’s disease; CA1/3, Cornu Amonis areas 1/3; αCaMKII, α subunit of calcium/calmodulin-dependent protein kinase II; DG, dentate gyrus
Acknowledgements
This work was supported by the Medical Research Council. We would like to thank Dr. Wendy Noble for comments on the final manuscript.
This article is a contribution to a series of invited reviews on Molecules of Neural Disorders.
Abbreviations
- Aβ
Amyloid-β
- AD
Alzheimer’s disease
- Aη
Amyloid-η
- AMPAR
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor
- APP
Amyloid precursor protein
- CA1/3
Cornu Ammonis areas 1/3
- CaMKII
Calcium/calmodulin binding protein kinase II
- cdk5
cyclin-dependent kinase 5
- DG
Dentate gyrus
- GluA1
AMPAR subunit
- GSK3-β
Glycogen synthase kinase 3-β
- LTD/LTP
Long-term depression/potentiation
- MCI
Mild cognitive impairment
- MMSE
Mini-mental state examination
- NFT
Neurofibrillary tangle
- NMDAR
N-methyl-D-aspartic acid receptor
- NR2A/B
NMDAR subunits
- PHF
Paired helical filament
- PS1
Presenilin-1
- PSD-95
Post-synaptic density protein 95
- SP
Senile plaque
- VGCC
Voltage-gated calcium channel
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Both AG and KPG wrote, read and approved the final manuscript.
Contributor Information
Anshua Ghosh, Email: anshua.ghosh@kcl.ac.uk.
Karl Peter Giese, Email: karl.giese@kcl.ac.uk.
References
- 1.Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8(6):429–31. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 2.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–890. doi: 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer‘s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122(3):1131–1135. doi: 10.1016/0006-291X(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 4.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82(12):4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85(11):4051–5. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83(13):4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem. 1986;99(6):1807–10. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- 8.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83(11):4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. doi: 10.1016/0165-6147(91)90609-V. [DOI] [PubMed] [Google Scholar]
- 10.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 11.Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S et al. Eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature. 2015;526(7573):443–7. doi:doi:10.1038/nature14864. [DOI] [PMC free article] [PubMed]
- 12.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 13.Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990;11(1):29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 14.Scheff SW, Price DA, Schmitt FA, DeKosky ST. Mufson EJ Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68(18):1501–8. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 15.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 16.Masliah E, Mallory M, Hansen L, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43(1):192–7. doi: 10.1212/WNL.43.1_Part_1.192. [DOI] [PubMed] [Google Scholar]
- 17.Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33(3):377–87. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- 18.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24(45):10191–200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 21.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marcello E, Epis R, Di Luca M. Amyloid flirting with synaptic failure: towards a comprehensive view of Alzheimer’s disease pathogenesis. Eur J Pharmacol. 2008;585(1):109–118. doi: 10.1016/j.ejphar.2007.11.083. [DOI] [PubMed] [Google Scholar]
- 24.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3(11):862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 25.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31(9):454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I. Rydel RE Beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12(2):376–89. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285(17):12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123(5):849–60. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 29.Hanson PI, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- 30.Bronstein J, Nishimura R, Lasher R, Cole R, de Vellis J, Farber D, et al. Calmodulin kinase II in pure cultured astrocytes. J Neurochem. 1988;50(1):45–9. doi: 10.1111/j.1471-4159.1988.tb13227.x. [DOI] [PubMed] [Google Scholar]
- 31.Liu XB, Jones EG. Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc Natl Acad Sci U S A. 1996;93(14):7332–7336. doi: 10.1073/pnas.93.14.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamsa K, Irvine EE, Giese KP, Kullmann DM. NMDA receptor-dependent long-term potentiation in mouse hippocampal interneurons shows a unique dependence on Ca(2+)/calmodulin-dependent kinases. J Physiol. 2007;584(Pt 3):885–94. doi: 10.1113/jphysiol.2007.137380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39(2):283–97. doi: 10.1016/S0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto K, Bosch M, Hayashi Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag? Physiology. 2009;24:357–366. doi: 10.1152/physiol.00029.2009. [DOI] [PubMed] [Google Scholar]
- 35.Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, et al. Kinase-dead knock-in mouse reveals an essential role of kinase activity of Ca2+/calmodulin-dependent protein kinase IIalpha in dendritic spine enlargement, long-term potentiation, and learning. J Neurosci. 2009;29(23):7607–18. doi: 10.1523/JNEUROSCI.0707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hell JW. CaMKII: claiming center stage in postsynaptic function and organization. Neuron. 2014;81(2):249–265. doi: 10.1016/j.neuron.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borgesius NZ, van Woerden GM, Buitendijk GH, Keijzer N, Jaarsma D, Hoogenraad CC, et al. BetaCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting alphaCaMKII to synapses. J Neurosci. 2011;31(28):10141–8. doi: 10.1523/JNEUROSCI.5105-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee SJ, Escobedo-Lozoya Y, Szatmari EM. Yasuda R Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458(7236):299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irvine EE, von Hertzen LS, Plattner F. Giese KP AlphaCaMKII autophosphorylation: a fast track to memory. Trends Neurosci. 2006;29(8):459–65. doi: 10.1016/j.tins.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 40.Lengyel I, Voss K, Cammarota M, Bradshaw K, Brent V, Murphy KP, et al. Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. Eur J Neurosci. 2004;20(11):3063–72. doi: 10.1111/j.1460-9568.2004.03748.x. [DOI] [PubMed] [Google Scholar]
- 41.Giese KP, Fedorov NB, Filipkowski RK. Silva AJ Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279(5352):870–3. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 42.Radwanska K, Medvedev NI, Pereira GS, Engmann O, Thiede N, Moraes MF, et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proc Natl Acad Sci U S A. 2011;108(45):18471–5. doi: 10.1073/pnas.1109680108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Irvine EE, Danhiez A, Radwanska K, Nassim C, Lucchesi W, Godaux E, et al. Properties of contextual memory formed in the absence of alphaCaMKII autophosphorylation. Mol Brain. 2011;4:8. doi: 10.1186/1756-6606-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooke SF, Wu J, Plattner F, Errington M, Rowan M, Peters M, et al. Autophosphorylation of alphaCaMKII is not a general requirement for NMDA receptor-dependent LTP in the adult mouse. J Physiol. 2006;574(Pt 3):805–18. doi: 10.1113/jphysiol.2006.111559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Need AC, Giese KP. Handling and environmental enrichment do not rescue learning and memory impairments in alphaCamKII(T286A) mutant mice. Genes Brain Behav. 2003;2(3):132–139. doi: 10.1034/j.1601-183X.2003.00020.x. [DOI] [PubMed] [Google Scholar]
- 46.Lucchesi W, Mizuno K, Giese KP. Novel insights into CaMKII function and regulation during memory formation. Brain Res Bull. 2011;85(1-2):2–8. doi: 10.1016/j.brainresbull.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 47.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 48.Amada N, Aihara K, Ravid R. Horie M Reduction of NR1 and phosphorylated Ca2+/calmodulin-dependent protein kinase II levels in Alzheimer’s disease. Neuroreport. 2005;16(16):1809–13. doi: 10.1097/01.wnr.0000185015.44563.5d. [DOI] [PubMed] [Google Scholar]
- 49.Tannenberg RK, Scott HL, Tannenberg AE. Dodd PR Selective loss of synaptic proteins in Alzheimer’s disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49(7):631–9. doi: 10.1016/j.neuint.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 50.McKee AC, Kosik KS, Kennedy MB. Kowall NW Hippocampal neurons predisposed to neurofibrillary tangle formation are enriched in type II calcium/calmodulin-dependent protein kinase. J Neuropathol Exp Neurol. 1990;49(1):49–63. doi: 10.1097/00005072-199001000-00006. [DOI] [PubMed] [Google Scholar]
- 51.Wang YJ, Chen GH, Hu XY, Lu YP, Zhou JN, Liu RY. The expression of calcium/calmodulin-dependent protein kinase II-alpha in the hippocampus of patients with Alzheimer’s disease and its links with AD-related pathology. Brain Res. 2005;1031(1):101–8. doi: 10.1016/j.brainres.2004.10.061. [DOI] [PubMed] [Google Scholar]
- 52.Ferrer I, Blanco R, Carmona M, Puig B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J Neural Transm. 2001;108(12):1397–415. doi: 10.1007/s007020100016. [DOI] [PubMed] [Google Scholar]
- 53.Simonian NA, Elvhage T, Czernik AJ, Greengard P. Hyman BT Calcium/calmodulin-dependent protein kinase II immunostaining is preserved in Alzheimer’s disease hippocampal neurons. Brain Res. 1994;657(1-2):294–9. doi: 10.1016/0006-8993(94)90979-2. [DOI] [PubMed] [Google Scholar]
- 54.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344(8925):769–72. doi: 10.1016/S0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 55.Mah VH, Eskin TA, Kazee AM, Lapham L, Higgins GA. In situ hybridization of calcium/calmodulin dependent protein kinase II and tau mRNAs; species differences and relative preservation in Alzheimer’s disease. Brain Res Mol Brain Res. 1992;12(1-3):85–94. doi: 10.1016/0169-328X(92)90071-I. [DOI] [PubMed] [Google Scholar]
- 56.Murray KD, Gall CM, Jones EG, Isackson PJ. Differential regulation of brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase messenger RNA expression in Alzheimer’s disease. Neuroscience. 1994;60(1):37–48. doi: 10.1016/0306-4522(94)90202-X. [DOI] [PubMed] [Google Scholar]
- 57.Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, et al. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics. 2008;33(2):240–56. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solomon B, Koppel R, Jossiphov J. Immunostaining of calmodulin and aluminium in Alzheimer’s disease-affected brains. Brain Res Bull. 2001;55(2):253–256. doi: 10.1016/S0361-9230(01)00466-X. [DOI] [PubMed] [Google Scholar]
- 59.Reese LC, Laezza F, Woltjer R, Taglialatela G. Dysregulated phosphorylation of Ca(2+) /calmodulin-dependent protein kinase II-alpha in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J Neurochem. 2011;119(4):791–804. doi: 10.1111/j.1471-4159.2011.07447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cavazzin C, Bonvicini C, Nocera A, Racchi M, Kasahara J, Tardito D, et al. Expression and phosphorylation of delta-CaM kinase II in cultured Alzheimer fibroblasts. Neurobiol Aging. 2004;25(9):1187–96. doi: 10.1016/j.neurobiolaging.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 61.Esteras N, Munoz U, Alquezar C, Bartolome F, Bermejo-Pareja F. Martin-Requero A Altered calmodulin degradation and signaling in non-neuronal cells from Alzheimer’s disease patients. Curr Alzheimer Res. 2012;9(3):267–77. doi: 10.2174/156720512800107564. [DOI] [PubMed] [Google Scholar]
- 62.Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17(5):661–3. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- 63.Gu Z, Liu W, Yan Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. 2009;284(16):10639–10649. doi: 10.1074/jbc.M806508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang DM, Yang YJ, Zhang L, Zhang X, Guan FF, Zhang LF, et al. The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer’s disease and vascular dementia. Neurosci Lett. 2013;538:60–5. doi: 10.1016/j.neulet.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 65.Wang DM, Yang YJ, Zhang L, Zhang X, Guan FF. Zhang LF Naringin Enhances CaMKII Activity and Improves Long-Term Memory in a Mouse Model of Alzheimer’s Disease. Int J Mol Sci. 2013;14(3):5576–86. doi: 10.3390/ijms14035576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282(46):33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- 67.Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, Woltjer RL. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol Dis. 2011;41(1):62–70. doi: 10.1016/j.nbd.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6(1):15–6. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- 69.Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129(Pt 7):1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- 70.Jiang X, Chai GS, Wang ZH, Hu Y, Li XG, Ma ZW, et al. Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci Rep. 2015;5:9488. doi: 10.1038/srep09488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao D, Watson JB, Xie CW. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004;92(5):2853–2858. doi: 10.1152/jn.00485.2004. [DOI] [PubMed] [Google Scholar]
- 72.Zeng Y, Zhao D, Xie CW. Neurotrophins enhance CaMKII activity and rescue amyloid-beta-induced deficits in hippocampal synaptic plasticity. J Alzheimers Dis. 2010;21(3):823–831. doi: 10.3233/JAD-2010-100264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci U S A. 1988;85(16):6218–6221. doi: 10.1073/pnas.85.16.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163(1):83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramelot TA, Nicholson LK. Phosphorylation-induced structural changes in the amyloid precursor protein cytoplasmic tail detected by NMR. J Mol Biol. 2001;307(3):871–884. doi: 10.1006/jmbi.2001.4535. [DOI] [PubMed] [Google Scholar]
- 76.Vieira SI, Rebelo S, Esselmann H, Wiltfang J, Lah J, Lane R, et al. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol Neurodegener. 2010;5:40. doi: 10.1186/1750-1326-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71(6):2465–76. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 78.Xiao J, Perry G, Troncoso J, Monteiro MJ. Alpha-calcium-calmodulin-dependent kinase II is associated with paired helical filaments of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55(9):954–63. doi: 10.1097/00005072-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 79.Yamamoto H, Hiragami Y, Murayama M, Ishizuka K, Kawahara M, Takashima A. Phosphorylation of tau at serine 416 by Ca2+/calmodulin-dependent protein kinase II in neuronal soma in brain. J Neurochem. 2005;94(5):1438–47. doi: 10.1111/j.1471-4159.2005.03307.x. [DOI] [PubMed] [Google Scholar]
- 80.Yoshimura Y, Ichinose T, Yamauchi T. Phosphorylation of tau protein to sites found in Alzheimer’s disease brain is catalyzed by Ca2+/calmodulin-dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci Lett. 2003;353(3):185–188. doi: 10.1016/j.neulet.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 81.Baudier J, Cole RD. Phosphorylation of tau proteins to a state like that in Alzheimer’s brain is catalyzed by a calcium/calmodulin-dependent kinase and modulated by phospholipids. J Biol Chem. 1987;262(36):17577–17583. [PubMed] [Google Scholar]
- 82.Steiner B, Mandelkow EM, Biernat J, Gustke N, Meyer HE, Schmidt B, et al. Phosphorylation of microtubule-associated protein tau: identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. 1990;9(11):3539–44. doi: 10.1002/j.1460-2075.1990.tb07563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagestedt T, Lichtenberg B, Wille H, Mandelkow EM, Mandelkow E. Tau protein becomes long and stiff upon phosphorylation: correlation between paracrystalline structure and degree of phosphorylation. J Cell Biol. 1989;109(4 Pt 1):1643–51. doi: 10.1083/jcb.109.4.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh TJ, Wang JZ, Novak M, Kontzekova E, Grundke-Iqbal I, Iqbal K. Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett. 1996;387(2-3):145–8. doi: 10.1016/0014-5793(96)00485-1. [DOI] [PubMed] [Google Scholar]
- 85.Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357(2):299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- 86.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25(1):59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gardoni F, Schrama LH, Kamal A, Gispen WH, Cattabeni F, Di Luca M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci. 2001;21(5):1501–9. doi: 10.1523/JNEUROSCI.21-05-01501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gardoni F, Bellone C, Viviani B, Marinovich M, Meli E, Pellegrini-Giampietro DE, et al. Lack of PSD-95 drives hippocampal neuronal cell death through activation of an alpha CaMKII transduction pathway. Eur J Neurosci. 2002;16(5):777–86. doi: 10.1046/j.1460-9568.2002.02141.x. [DOI] [PubMed] [Google Scholar]
- 89.Ashpole NM, Song W, Brustovetsky T, Engleman EA, Brustovetsky N, Cummins TR, et al. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J Biol Chem. 2012;287(11):8495–506. doi: 10.1074/jbc.M111.323915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bok J, Wang Q, Huang J. Green SH CaMKII and CaMKIV mediate distinct prosurvival signaling pathways in response to depolarization in neurons. Mol Cell Neurosci. 2007;36(1):13–26. doi: 10.1016/j.mcn.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Churn SB, Limbrick D, Sombati S, DeLorenzo RJ. Excitotoxic activation of the NMDA receptor results in inhibition of calcium/calmodulin kinase II activity in cultured hippocampal neurons. J Neurosci. 1995;15(4):3200–14. doi: 10.1523/JNEUROSCI.15-04-03200.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin KF, Chang RC, Suen KC, So KF. Hugon J Modulation of calcium/calmodulin kinase-II provides partial neuroprotection against beta-amyloid peptide toxicity. Eur J Neurosci. 2004;19(8):2047–55. doi: 10.1111/j.0953-816X.2004.03245.x. [DOI] [PubMed] [Google Scholar]
- 93.Pike CJ, Balazs R, Cotman CW. Attenuation of beta-amyloid neurotoxicity in vitro by potassium-induced depolarization. J Neurochem. 1996;67(4):1774–1777. doi: 10.1046/j.1471-4159.1996.67041774.x. [DOI] [PubMed] [Google Scholar]
- 94.Kim K, Lakhanpal G, Lu HE, Khan M, Suzuki A, Kato-Hayashi M, et al. A Temporary Gating of Actin Remodeling during Synaptic Plasticity Consists of the Interplay between the Kinase and Structural Functions of CaMKII. Neuron. 2015;87(4):813–26. doi: 10.1016/j.neuron.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ma H, Li B, Tsien RW. Distinct roles of multiple isoforms of CaMKII in signaling to the nucleus. Biochim Biophys Acta. 2015;1853(9):1953–1957. doi: 10.1016/j.bbamcr.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]