

Abstract
Abstract
Objective
Accurate prenatal diagnosis of genetic conditions can be challenging and usually requires invasive testing. Here, we demonstrate the potential of next-generation sequencing (NGS) for the analysis of cell-free DNA in maternal blood to transform prenatal diagnosis of monogenic disorders.
Methods
Analysis of cell-free DNA using a PCR and restriction enzyme digest (PCR–RED) was compared with a novel NGS assay in pregnancies at risk of achondroplasia and thanatophoric dysplasia.
Results
PCR–RED was performed in 72 cases and was correct in 88.6%, inconclusive in 7% with one false negative. NGS was performed in 47 cases and was accurate in 96.2% with no inconclusives. Both approaches were used in 27 cases, with NGS giving the correct result in the two cases inconclusive with PCR–RED.
Conclusion
NGS provides an accurate, flexible approach to non-invasive prenatal diagnosis of de novo and paternally inherited mutations. It is more sensitive than PCR–RED and is ideal when screening a gene with multiple potential pathogenic mutations. These findings highlight the value of NGS in the development of non-invasive prenatal diagnosis for other monogenic disorders. © 2015 The Authors. Prenatal Diagnosis published by John Wiley & Sons, Ltd.
What's already known about this topic?
Non-invasive prenatal diagnosis (NIPD) using PCR-based methods has been reported for the detection or exclusion of individual paternally inherited or de novo alleles in maternal plasma.
What does this study add?
NIPD using next generation sequencing provides an accurate, more sensitive approach which can be used to detect multiple mutations in a single assay and so is ideal when screening a gene with multiple potential pathogenic mutations. Next generation sequencing thus provides a flexible approach to non-invasive prenatal diagnosis ideal for use in a busy service laboratory.
Introduction
Prenatal diagnosis of monogenic disorders, including those presenting with ultrasound abnormalities, traditionally required analysis of fetal material obtained following invasive procedures such as amniocentesis and chorionic villus sampling, both of which carry a small but significant miscarriage risk.1 The discovery of cell-free fetal DNA (cffDNA) circulating in maternal blood2 is allowing the development of early non-invasive prenatal diagnosis (NIPD) based on analysis of maternal blood,3 eliminating the risk of directly causing fetal loss from invasive testing, which can only be performed after 11 weeks' gestation.1 Prenatal testing for Down syndrome and other aneuploidies using cell-free DNA (cfDNA) is now widely available in the USA, Asia, and Europe, implementation being based on next-generation DNA sequencing [next-generation sequencing (NGS)] technology4 and driven by commercial providers.5 However, in the UK during 2011–2012, more than 2000 invasive tests were performed because of a high risk for a monogenic disorder.6 While there are many single case reports and the occasional small series reporting NIPD for single gene disorders, there has been little by way of implementation into routine clinical practice.7 In our National Health Service (NHS) Regional Genetics Service, we have been offering fetal sex determination based on the analysis of cffDNA for several years8 and, in 2012, obtained UK Genetic Testing Network approvals to deliver a diagnostic service based on the analysis of cffDNA using a PCR-based method for achondroplasia9 and thanatophoric dysplasia (TD).10
Achondroplasia is the most common non-lethal skeletal dysplasia with an incidence of ∼5–15 per 100 000 live births. It is an autosomal dominant disorder, and ∼98% of cases are caused by a c.1138G>A mutation in the fibroblast growth factor receptor 3 (FGFR3) gene,11,12 with ∼1% caused by a c.1138G>C mutation.12 Most cases arise de novo with no prior familial history of skeletal dysplasia13 and present late in pregnancy when short limbs are detected by ultrasound scanning.9 TD is the most common lethal skeletal dysplasia, also resulting from FGFR3 mutations,15 but presents earlier in pregnancy with short limbs, a small chest, and other features.10 Unlike achondroplasia, TD arises from several FGFR3 mutations15 and can be separated radiologically into two forms, TD I and TD II with features that overlap. TD I has curved femora with infrequent craniosynostosis, whereas TD II is characterized by straight femora and a clover-shaped skull. A number of FGFR3 mutations cause TD I, with the c.742C>T mutation accounting for more than 50% of cases. All cases of TD II are caused by a single c.1948A>G FGFR3 mutation.15
The early PCR-based methods developed for NIPD of single gene disorders are suitable for the detection of paternally inherited alleles and can be useful in conditions such as achondroplasia where there is a single common causative mutation.7,11 In conditions such as TD, while this approach may be suitable to exclude recurrence, albeit the risk is very low, of a known mutation in families with a relevant family history, it is less useful for the diagnosis of ultrasound abnormalities in cases arising de novo when there are multiple potential causative mutations. Furthermore, as the majority of cfDNA is maternal in origin,16 simple molecular techniques such as PCR–RED may not be sufficiently sensitive for the delivery of accurate results in all cases.
Here, we describe the use of NGS to improve the accuracy and scope of NIPD of monogenic disorders using skeletal dysplasias as an example. We compare the results obtained using NGS with PCR–RED and demonstrate for the first time how, in clinical practice, NGS has the potential to transform prenatal diagnosis for some families at high risk for genetic disorders.
Methods
Patient recruitment
The study included a mix of retrospective cases, ascertained by searching our nationally accredited Regional Genetics Laboratory records to identify all cases where cfDNA in maternal blood had been analyzed for an FGFR3 mutation and cases presenting prospectively. In all cases, blood samples were collected from women at risk of carrying a fetus with achondroplasia or TD either because of sonographic findings or because of a relevant past family history. Where appropriate, written informed consent was obtained prior to venepuncture, and the study was approved by the University College London Hospital Ethics Committee A (reference 01/0095). In all cases, confirmation of prenatal testing result was sought from postnatal molecular testing, radiology, or pathology.
Testing using PCR–RED was available in our laboratory from 2007. Where there was sufficient DNA or frozen maternal plasma, available tests were repeated using the NGS panel. For cases tested prospectively, if sufficient plasma was available, PCR–RED and NGS analyses were run concurrently. Registered clinical scientists with at least 3 years' experience performed the tests and were blinded to clinical information and previous laboratory results.
Sample processing and DNA extraction
Plasma was separated from 20 mL of blood within 48 h of blood draw and cfDNA extracted using the QIAamp MinElute Virus Spin Kit (Qiagen) or QIAamp Circulating Nucleic Acid kit (Qiagen).8–10
PCR–RED for FGFR3 mutations
PCR–RED for the diagnosis of achondroplasia and TD was carried out as previously described.9,10 Assays were designed for the common c.1138G>A achondroplasia mutation and two of the TD mutations, the 1948A>G mutation that causes TD II and the most common TD I mutation, c.742C>T.
FGFR3 – next-generation DNA sequencing assay
Five amplicons covering 29 known disease-causing mutations in the FGFR3 gene (Table1) were designed using Primer 3 software.17 Our custom design for the PCR primers included the adapters and index sequences. The panel was validated using normal and positive control genomic DNA samples obtained by chorionic villus sampling to ensure that the panel was able to detect the expected mutations.
Table 1.
FGFR3 details of the mutations included in the next-generation sequencing panel
| FGFR3 exon | Mutation | Amino acid change | Associated skeletal disorder |
|---|---|---|---|
| 6 | c.742C>T | p.Arg248Cys | TD I |
| 6 | c.746C>G | p.Ser249Cys | TD I |
| 6 | c.749C>G | p.Pro250Arg | Muenke syndrome19 |
| 6 | c.749C>T | p.Pro250Leu | Craniosynostosis20 |
| 8 | c.1108G>T | p.Gly370Cys | TD I |
| 8 | c.1111A>T | p.Ser371Cys | TD I |
| 8 | c.1118A>G | p.Tyr373Cys | TD I |
| 8 | c.1123G>T | p.Gly375Cys | Achondroplasia |
| 8 | c.1130T>G | p.Leu377Arg | Achondroplasia |
| 8 | c.1138G>C | p.Gly380Arg | Achondroplasia |
| 8 | c.1138G>A | p.Gly380Arg | Achondroplasia |
| 8 | c.1142T>A | p.Val381Glu | Hypochondroplasia |
| 11 | c.1619A>C | p.Asn540Thr | Hypochondroplasia |
| 11 | c.1619A>G | p.Asn540Ser | Hypochondroplasia |
| 11 | c.1620C>G | p.Asn540Lys | Hypochondroplasia |
| 11 | c.1620C>A | p.Asn540Lys | Hypochondroplasia |
| 13 | c.1948A>C | p.Lys650Gln | Hypochondroplasia |
| 13 | c.1948A>G | p.Lys650Glu | TD II |
| 13 | c.1949A>T | p.Lys650Met | TD I/SADDAN21 |
| 13 | c.1949A>C | p.Lys650Thr | Familial acanthosis22 nigricans/hypochondroplasia + acanthosis nigricans |
| 13 | c.1950G>C | p.Lys650Asn | Hypochondroplasia |
| 13 | c.1950G>T | p.Lys650Asn | Hypochondroplasia |
| 17 | c.2419T>G | p.*807Glyext*102 | TD I |
| 17 | c.2419T>A | p.*807Argext*102 | TD I |
| 17 | c.2420G>C | p.*807Serext*102 | TD I |
| 17 | c.2420G>T | p.*807Leuext*102 | TD I |
| 17 | c.2421A>T | p.*807Cysext*102 | TD I |
| 17 | c.2421A>C | p.*807Cysext*102 | TD I |
| 17 | c.2421A>G | p.*807Trpext*102 | TD I |
DNA sequencing library preparation
PCR was carried out on plasma cfDNA samples using 10 μL of Phusion High-Fidelity PCR Master Mix (NEB), 500 nM of each primer, and 4 μL plasma cfDNA, in a final reaction volume of 20 μL. Cycling conditions were: 98 °C for 1 min, followed by 42 cycles of 98 °C for 10 s, 64 °C for 30 s, and 72 °C for 30 s, followed by 72 °C for 10 min. All amplicons for all cases are pooled into a single pool, and individual samples are tagged with Illumina indexes and purified using a MinElute PCR Purification Kit (Qiagen), quantified using a Qubit dsDNA BR Assay Kit (Invitrogen), and amplicon quality was assessed using a Bioanalyzer (Agilent). Purified PCR products were diluted to 2 nM in Elution Buffer (Qiagen), and equal amounts of all samples were pooled to give a single 2-nM library, which was denatured using sodium hydroxide according to MiSeq sample preparation instructions (Illumina) and diluted to a final concentration of 8 pM and mixed with an 8-pM PhiX control to give a 20% PhiX spike, providing sequence diversity. The library was loaded onto the Illumina MiSeq for initiation of cluster generation and a single-end 100 cycle sequencing protocol.
Confirmation of the presence of cffDNA
Where there was sufficient DNA available, the presence of cffDNA in the sample was confirmed using assays for SRY, ZFY, or RASSAF1A.10,23 Quantification of cffDNA was not undertaken.
Statistical analysis
Sensitivities and specificities with 95% confidence intervals for both approaches were calculated and 95% confidence intervals for the difference between detection rates calculated using the exact method.
Results
A total of 108 cases were ascertained. Of these, 68 were referred because of a risk of achondroplasia, 53 of the 68 after 20 weeks' gestation (mean 29, range of 20–37 weeks) because of ultrasound findings suggestive of a skeletal problem. A further 13 were referred between 9 and 14 weeks (mean 10.3 weeks) because a previous affected pregnancy conferred a small recurrence risk due to germline mosaicism, that is, a mutation present in some germline cells but not in somatic cells, so the individual parent shows no signs of the condition but is at a very small increased risk of having an affected child. The remaining two cases were referred because of raised paternal age or because the father was affected. There were 40 cases at risk of TD, nine with a germline mosaicism recurrence risk tested between 9 and 16 (mean 12.7) weeks' gestation and the other 31 with ultrasound findings detected between 12 and 36 (mean 18.6) weeks' gestation. Three cases have been lost to follow-up, and in 11 the pregnancies are ongoing. These have all been excluded from further analysis and will not be discussed further. Not all cases were tested using both approaches, leaving 72 cases tested with PCR–RED and 47 with the NGS panel (Table2). Twenty-seven cases, where there was sufficient DNA or plasma available, were tested using both platforms.
Table 2.
The performance of the PCR–RED and NGS panel for the detection of FGFR3 mutations causing achondroplasia and thanatophoric dysplasia with confirmed postnatal diagnoses
| Confirmed postnatal diagnosis | Mutation | Prenatal test result | |||||
|---|---|---|---|---|---|---|---|
| PCR–RED | NGS panel | ||||||
| True positive | False negative | Inconclusive | True positive | False negative | Inconclusive | ||
| Achondroplasia | c.1138G>A (p.Gly380Arg) | 14 | 0 | 1 | 8 | 0 | 0 |
| c.835A>T (p.Ser279Cys) | ND | ND | ND | 0 | 1a | 0 | |
| Thanatophoric dysplasia | c.742C>CT (p.Arg248Cys) | 6b | 1c | 1b | 9b | 0 | 0 |
| c.1948A>AT (p.Lys650Glu) | 3 | 0 | 0 | 3 | 0 | 0 | |
| c.1118A>G (p.Tyr373Cys) | ND | ND | ND | 2 | 0 | 0 | |
| c.746C>G (p.Ser249Cys) | ND | ND | ND | 2 | 0 | 0 | |
| c.2419T>A (p.*807Argext*101) | ND | ND | ND | 1 | 0 | 0 | |
| bDoes not carry FGFR3 mutation | No mutant allele detected | 43b | 0 | 3 | 20b | 0 | 0 |
| Total | 66 | 1 | 5 | 46 | 1 | 0 | |
| Sensitivity | 88.6% (95% CI 71–96) taking inconclusive that were positive as false negative | 96.2% (81–99) | |||||
| Specificity | 100% (92–100) | 100% (85–100) | |||||
| Positive predictive value | 100% (86–100) | 100% (87–100) | |||||
| Negative predictive value | 93.9 (83.5–97.9) taking inconclusive that were positive as false negative | 95.5% (78.2–99.2) | |||||
| Inconclusive rate | 7% | 0 | |||||
ND, not done; PCR–RED, PCR–restriction enzyme digest; NGS, next-generation sequencing; FGFR3, fibroblast growth factor receptor 3.
False negative for rare achondroplasia mutation c.835A>T (p.Ser279Cys). Panel redesigned to incorporate this mutation.
Includes one twin pregnancy, one twin normal, and one with a skeletal dysplasia. The case that was inconclusive using PCR–RED for c.742C>CT (p.Arg248Cys) was positive when tested with NGS.
False negative for c.742C>CT (p.Arg248Cys) due to low fetal fraction. There was insufficient sample available to analyze this sample using NGS.
Includes normal, growth retarded fetuses and other skeletal dysplasias.
An example of the results obtained in the PCR–RED assay is shown in Figure1. The sensitivity and specificity (with 95% confidence intervals) of the PCR–RED assay if taking inconclusive results as negative were 88.6% (71–96%) and 100% (92–100%), while for the NGS panel, the sensitivity was 96.2% (81–99.3%) and the specificity, 100% (85–100%) (Table2). Comparison of the sensitivities for each approach, as well as the comparison of the specificities, showed no significance [−7.7 (−25.4–9.1) and 0, respectively], but as the confidence intervals are wide, differences that may be of clinical significance cannot be excluded. Furthermore, the NGS assay yielded no inconclusive results and indeed gave a result in both cases where PCR–RED was inconclusive. Overall, PCR–RED was inconclusive in five cases (7%), all tested after 20 weeks' gestation with one being a twin pregnancy. In one case, testing for an independent fetal marker was also inconclusive, and, as the sample was very small, repeat testing could not be performed. In two further cases, SRY assays confirmed the presence of cffDNA, but the FGFR3 PCR–RED assay was inconclusive. In the last two, including the twin pregnancy, there was sufficient sample to run the NGS panel as well, and this detected the wild-type or mutant FGFR3 allele in both cases. Both the PCR–RED and NGS assays have high positive predictive values of 100% (86–100%) and 100% (87–100%), respectively, with negative predictive values of 93.9 (83.5–97.9%) and 95.5 (78.2–99.2%), respectively (Table2). There was one false negative result using PCR–RED where a c.742C>CT (p.Arg248Cys) TD-causing mutation was not detected at 21 weeks' gestation although the RASSAF1A assay had confirmed the presence of cffDNA. With NGS, one case was reported as ‘no mutation detected’ but postnatally found to be heterozygous for a very rare achondroplasia-causing mutation [c.835A>T (p.Ser279Cys)],18 which was not included in the panel at that time. The panel has since been modified to cover this mutation.
Figure 1.
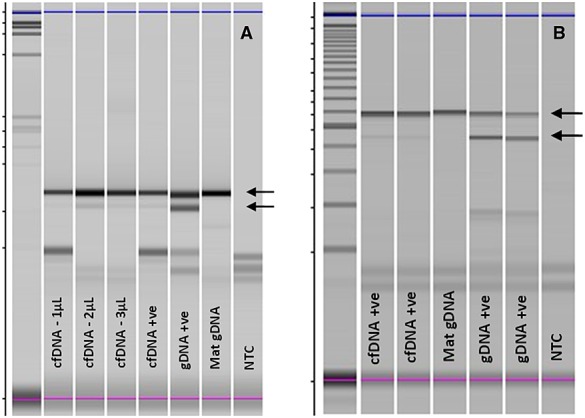
PCR-restriction enzyme digest results for (a) achondroplasia [fibroblast growth factor receptor 3 (FGFR3): c.1138G>A p.(Gly380Arg)/restriction enzyme BsrG1] and (b) thanatophoric dysplasia [FGFR3: c.742C>T p.(Arg248Cys)/restriction enzyme DraIII]. Upper arrows indicate wild-type (normal) allele that is strongly present in all samples. Bottom arrows indicate the mutant alleles that are faint in affected cell-free DNA (cfDNA+) and stronger mutation positive control genomic DNA (gDNA+ve). There is no mutant band present in the unaffected maternal genomic DNA (Mat gDNA). The figure shows uncropped images visualized on Shimadzu Multi-NA microchip electrophoresis system. NTC, blank, no template control
Discussion
This study represents the largest reported data set for the NIPD of de novo or paternally inherited autosomal dominant monogenic disorders and confirms that analysis of cfDNA in maternal plasma offers a safe alternative approach to invasive prenatal diagnosis. These data clearly demonstrate that NGS provides a more sensitive approach than PCR–RED, as it detected mutations in cases that were reported as inconclusive using a PCR–RED assay while yielding no inconclusive results. While PCR–RED is a very cost-effective technique, which is widely used in laboratories that do not have access to NGS platforms, mutations have to be screened for individually, interpretation of agarose gel electrophoresis can be very subjective (Figure1a and b), and it is only possible to test for mutations where a restriction enzyme cleavage site is created or destroyed by the mutation and therefore cannot be applied to all mutations. Thus, while this approach may be of use when testing for the recurrence of a known mutation for which an assay can be designed, when attempting diagnosis in cases presenting de novo, it would be very time consuming to use PCR–RED to individually test for all possible disease-causing mutations.
To date, there has been relatively little NIPD available clinically for families at high risk of single gene disorders, with most reported cases being performed on a research basis.9 Fetal sex determination in pregnancies at risk of sex-linked disorders to enable targeting of invasive testing in male-bearing pregnancies is in use in many parts of Europe and has been the standard of care in the UK NHS since 2011 (http://ukgtn.nhs.uk/news-events/article/non-invasive-prenatal-diagnosis-nipd-ukgtn-approval-of-gene-dossiers-for-fetal-sex-determination-for-congenital-adrenal-hyperplasia-cah-and-x-linked-conditions-excluding-hemophilia-7/).24 This non-invasive approach is welcomed by women and health professionals alike,25 as it avoids the risk of miscarriage and can be carried out earlier in pregnancy. The PCR–RED approach to NIPD for achondroplasia and TD was approved for use in the UK NHS in 2012. Although there are relatively few reports of NIPD for single gene disorders, a recent survey of women who had undergone this testing reported that they too valued the safety, ease of access, and early testing available.26 Health professionals, who have also been found to view the introduction of NIPD for single gene disorders positively, however, emphasized the need for new tests to be highly accurate and thoroughly validated.27
Non-invasive prenatal diagnosis can be a very useful aid to clinical management. TD is a condition that is increasingly detected by ultrasound early in pregnancy,12 but the differential diagnosis includes the short-ribbed polydactyly syndromes and other autosomal recessively inherited conditions associated with a high recurrence risk, while TD is a new dominant mutation with a low recurrence risk. If positive for TD, NIPD can allow for a definitive diagnosis without recourse to invasive testing and allows the option of a surgical termination, as a post-mortem will not be required. NIPD can also significantly alter clinical management in multiple pregnancies where the fetuses are discordant for abnormalities, as in three of the cases reported here. In this situation, definitive diagnosis using NIPD avoids the risk of miscarriage for the normal fetus. In addition, in lethal conditions, such as TD, it allows for conservative management of pregnancy with no requirement for feticide. In cases presenting late in pregnancy at risk of achondroplasia, NIPD allows for definitive diagnosis and accurate parental counseling without risk of precipitating preterm labor. In all cases, it also allows for a safe, non-invasive test early in future pregnancies to exclude a recurrence or inheritance of a paternal mutant allele. This can be carried out from 9 weeks' gestation, earlier than invasive testing, which cannot safely be performed until 11 weeks1 and much earlier than an ultrasound scan for those women not wanting to put the pregnancy at risk with an invasive test. Early access to NIPD in these circumstances has been welcomed by parents and has improved access to prenatal diagnosis for families where moral or religious reasons necessitate testing very early in pregnancy.25,26
Overall, our data demonstrate that NGS will be the best approach for NIPD for many monogenic conditions. Unlike PCR–RED, it is possible to screen for all mutations in a single assay, making it appropriate both for the exclusion of a recurrence and for diagnosis in cases arising de novo. The digital output from NGS is easy to interpret although in view of the low level of background counts in normal samples, maternal genomic DNA should be analyzed alongside the cfDNA sample (Table3). NGS offers optimal turnaround times as the entire procedure can be carried out in 24 h, which allows timely reporting for a prenatal test result. Furthermore, multiple patients with different conditions can be analyzed in a single run, allowing optimization of the workflow in a routine NHS diagnostic laboratory. Finally, the use of NGS may allow definitive NIPD in autosomal recessive conditions or X-linked where there is a high background of mutant allele in maternal plasma as she is a carrier herself. In this situation, NIPD cannot depend on detecting the presence or absence of an allele not present in the mother but requires an assessment of the relative proportion of mutant allele because, if the fetus is affected, there will be more mutant than wild-type allele present in maternal plasma. However, the amount of cffDNA (the fetal fraction) in maternal plasma varies between and within women and will directly affect the total amount of mutant allele in plasma if the fetus is affected. Estimation of the fetal fraction requires measurement of a fetal DNA marker present in the fetus but not in the mother.28 While this is possible in male-bearing pregnancies using SRY or DYS14 alleles, there is no universal marker for female fetuses. If using the NGS approach, panels of single nucleotide polymorphisms with high degrees of heterogeneity can be incorporated into the panel to facilitate estimation of fetal fraction. Furthermore, NGS offers the potential for a haplotyping approach using single nucleotide polymorphisms closely linked to the disease-causing gene in conditions where the mutations are not amenable to direct detection.29
Table 3.
Mutation counts
| Target | ACH Case 9 | ACH Case 10 | TD Case 17 | TD Case 21 | TD Case 22 | TD Case 23 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sequence | cfDNA | Mat gDNA | cfDNA | Mat gDNA | cfDNA | cfDNA | Mat gDNA | cfDNA | Mat gDNA | cfDNA | Mat gDNA |
| Exon 6 – wt | 21 386 | 7046 | 24 897 | 11 975 | 24 071 | 65 527 | 90 155 | ||||
| c.742C>T | 703 | 1 | 7 | 1 | 2 | 12 | 18 | ||||
| c.749C>G | 0 | 180 | 2 | 2 | 2 | 4 | 1 | ||||
| c.749C>G | 2 | 0 | 5 | 3 | 7 | 2 | 3 | ||||
| c.749C>T | 1 | 3 | 8 | 2 | 4 | 14 | 18 | ||||
| Exon 8 – wt 1 | 59 065 | 163 007 | 103 269 | 48 748 | 44 888 | 223 530 | 228 583 | ||||
| c.1108G>T | 10 | 18 | 9 | 6 | 1 | 30 | 27 | ||||
| c.1111A>T | 18 | 41 | 39 | 23 | 15 | 33 | 35 | ||||
| c.1118A>G | 8 | 22 | 16 | 5 | 13 | 8487 | 26 | ||||
| Exon 8 – wt 2 | 5183 | 13 112 | 3794 | 1737 | 12 584 | 6999 | 5753 | ||||
| c.1123G>T | 15 | 43 | 13 | 3 | 8 | 9 | 3 | ||||
| c.1130T>G | 12 | 35 | 7 | 4 | 2 | 98 | 99 | ||||
| c.1138G>C | 60 | 95 | 39 | 16 | 66 | 10 | 4 | ||||
| c.1138G>A | 515 | 19 | 207 | 0 | 11 | 1 | 4 | ||||
| Exon 8 – wt 3 | 114 | 297 | 178 | 97 | 208 | 119 | 159 | ||||
| c.1142T>A | 2 | 16 | 5 | 9 | 3 | 21 | 38 | ||||
| Exon 11 – wt | 0 | 43 894 | 94 651 | 56 888 | 63 064 | 166 808 | 235 016 | ||||
| c.1619A>C | 0 | 10 | 37 | 15 | 14 | 27 | 47 | ||||
| c.1619A>G | 0 | 4 | 2 | 5 | 6 | 20 | 23 | ||||
| c.1620C>G | 0 | 1 | 0 | 2 | 1 | 6 | 9 | ||||
| c.1620C>A | 0 | 10 | 24 | 16 | 17 | 40 | 53 | ||||
| Exon 13 – wt | 111 829 | 84 573 | 127 642 | 70 368 | 91 694 | 264 715 | 247 971 | ||||
| c.1948A>C | 14 | 7 | 15 | 11 | 6 | 29 | 31 | ||||
| c.1948A>G | 104 | 9 | 19 | 4139 | 2 | 39 | 29 | ||||
| c.1949A>T | 10 | 4 | 9 | 4 | 9 | 36 | 41 | ||||
| c.1949A>C | 62 | 25 | 43 | 18 | 28 | 39 | 60 | ||||
| c.1950G>C | 5 | 2 | 6 | 6 | 0 | 21 | 10 | ||||
| c.1950G>T | 82 | 21 | 46 | 28 | 32 | 52 | 42 | ||||
| Exon 17 – wt | 35 976 | 37 747 | 105 463 | 48 311 | 53 519 | 121 602 | 229 702 | ||||
| c.2419T>G | 287 | 67 | 278 | 103 | 133 | 858 | 1662 | ||||
| c.2419T>A | 6 | 12 | 30 | 8 | 11 | 15 | 22 | ||||
| c.2420G>T | 15 | 41 | 93 | 52 | 53 | 46 | 94 | ||||
| c.2420G>C | 3 | 1 | 8 | 3 | 3 | 17 | 18 | ||||
| c.2421A>T | 0 | 6 | 5 | 5 | 1 | 12 | 22 | ||||
| c.2421A>C | 20 | 32 | 105 | 55 | 61 | 25 | 62 | ||||
| c.2421A>G | 10 | 3 | 13 | 7 | 7 | 15 | 41 | ||||
ACH, achondroplasia; TD, thanatophoric dysplasia; wt, wild type or non-diseased causing; cfDNA, cell-free DNA; Mat gDNA, maternal genomic DNA.
Case 17 was analyzed early in the NGS series, before we routinely ran the maternal gDNA in parallel to facilitate interpretation in view of the low background counts in normal samples.
Disease-causing mutations are shown in bold, highlighted, and underlined. Note the difference between fetal and maternal counts.
Conclusion
The development of cfDNA aneuploidy testing has been driven by the commercial sector,3–5 as there is a large market opportunity. Implementation of NIPD for monogenic disorders has not attracted such attention, largely because development must be on a patient or disease-specific basis and demand is less. Here, we show how NGS can offer a comprehensive and speedy approach to NIPD for de novo and paternally inherited autosomal dominant conditions, which is welcomed by patients and health professionals alike and which can significantly improve clinical management. With increasing availability of sequencing platforms in public sector laboratories, it is hoped that there will be developments in NIPD for a wider range of monogenic disorders for the benefit of these families who are at high risk of genetic disease.
What's Already Known About this Topic?
Non-invasive prenatal diagnosis (NIPD) using PCR-based methods has been reported for the detection or exclusion of individual paternally inherited or de novo alleles in maternal plasma.
What Does this Study Add?
NIPD using next generation sequencing provides an accurate, more sensitive approach which can be used to detect multiple mutations in a single assay and so is ideal when screening a gene with multiple potential pathogenic mutations. Next generation sequencing thus provides a flexible approach to non-invasive prenatal diagnosis ideal for use in a busy service laboratory.
References
- Tabor A, Alfirevic Z. Update on procedure-related risks for prenatal diagnosis techniques. Fetal Diagn Ther. 2010;271:1–7. doi: 10.1159/000271995. [DOI] [PubMed] [Google Scholar]
- Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–7. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- Chitty LS, Bianchi DW. Non-invasive prenatal testing: the paradigm is shifting rapidly. Prenat Diagn. 2013;33:511–3. doi: 10.1002/pd.4136. [DOI] [PubMed] [Google Scholar]
- Boon EM, Faas BH. Benefits and limitations of whole genome versus targeted approaches for non-invasive prenatal testing for fetal aneuploidies. Prenat Diagn. 2013;33:563–8. doi: 10.1002/pd.4111. [DOI] [PubMed] [Google Scholar]
- Agarwal A, Sayres LC, Cho MK, et al. Commercial landscape of non-invasive prenatal testing in the United States. Prenat Diagn. 2013;33:521–31. doi: 10.1002/pd.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CMGS. 2012. Audit report. Available at: http://cmgsweb.shared.hosting.zen.co.uk/CMGS%20audit/2012%20audit/CMGSAudit11_12_FINAL.pdf [accessed on 27 November 2014]
- Lench N, Barrett A, Fielding S, et al. The clinical implementation of non-invasive prenatal diagnosis for single gene disorders: challenges and progress made. Prenat Diagn. 2013;33:555–62. doi: 10.1002/pd.4124. [DOI] [PubMed] [Google Scholar]
- Hill M, Finning K, Martin P, et al. Non-invasive prenatal determination of fetal sex: translating research into clinical practice. Clin Genet. 2011;80:68–75. doi: 10.1111/j.1399-0004.2010.01533.x. [DOI] [PubMed] [Google Scholar]
- Chitty LS, Griffin DR, Meaney C, et al. New aids for the non-invasive prenatal diagnosis of achondroplasia: dysmorphic features, charts of fetal size and molecular confirmation using cell-free fetal DNA in maternal plasma. Ultrasound Obstet Gynecol. 2011;37:283–9. doi: 10.1002/uog.8893. [DOI] [PubMed] [Google Scholar]
- Chitty LS, Khalil A, Barrett AN, et al. Safe, accurate, prenatal diagnosis of thanatophoric dysplasia using ultrasound and free fetal DNA. Prenat Diagn. 2013;33:416–23. doi: 10.1002/pd.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang R, Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–42. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Hefferon TW, Ortiz de Luna RI, et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368–73. [PMC free article] [PubMed] [Google Scholar]
- Wilkin DJ, Szabo JK, Cameron R, et al. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am J Hum Genet. 1998;63:711–6. doi: 10.1086/302000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavormina PL, Shiang R, Thompson LM, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321–8. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- Passos-Bueno MR, Wilcox WR, Jabs EW, et al. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–25. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Lo YM, Tein MS, Lau TK, et al. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for non-invasive prenatal diagnosis. Am J Hum Genet. 1998;62:768–75. doi: 10.1086/301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. New Jersey, USA: Humana Press; 2000. pp. 365–86. [DOI] [PubMed] [Google Scholar]
- Heuertz S, Le Merrer M, Zabel B, et al. Novel FGFR3 mutations creating cysteine residues in the extracellular domain of the receptor cause achondroplasia or severe forms of hypochondroplasia. Eur J Hum Genet. 2006;14:1240–7. doi: 10.1038/sj.ejhg.5201700. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Spector EB, Speiser PW, et al. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am J Hum Genet. 2000;67:1411–21. doi: 10.1086/316892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenke M, Gripp KW, McDonald-McGinn DM, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997;60:555–64. [PMC free article] [PubMed] [Google Scholar]
- Schindler S, Friedrich M, Wagener H, et al. Heterozygous P250L mutation of fibroblast growth factor receptor 3 in a case of isolated craniosynostosis. J Med Genet. 2002;39:764–6. doi: 10.1136/jmg.39.10.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Feijoo L, Loidi L, Vidal A, et al. Hypochondroplasia and acanthosis nigricans: a new syndrome due to the p.Lys650Thr mutation in the fibroblast growth factor receptor 3 gene? Eur J Endocrinol. 2008;159:243–9. doi: 10.1530/EJE-08-0393. [DOI] [PubMed] [Google Scholar]
- White HE, Dent CL, Hall VJ, et al. Evaluation of a novel assay for detection of the fetal marker RASSF1A: facilitating improved diagnostic reliability of non-invasive prenatal diagnosis. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045073. :e45073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Non-invasive prenatal diagnosis (NIPD) – UK Genetic Testing Network 2010. Available at: http://ukgtn.nhs.uk/news-events/article/non-invasive-prenatal-diagnosis-nipd-ukgtn-approval-of-gene-dossiers-for-fetal-sex-determination-for-congenital-adrenal-hyperplasia-cah-and-x-linked-conditions-excluding-haemophilia-7/. [Accessed 27 November 2014]
- Lewis C, Hill M, Skirton H, Chitty LS. Non-invasive prenatal diagnosis for fetal sex determination – benefits and disadvantages from the service users' perspective. Eur J Hum Genet. 2012;20:1127–33. doi: 10.1038/ejhg.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis C, Hill M, Chitty LS. Non-invasive prenatal diagnosis for single gene disorders: experience of patients. Clin Genet. 2014;85:336–42. doi: 10.1111/cge.12179. [DOI] [PubMed] [Google Scholar]
- Hill M, Karunaratna M, Lewis C, et al. Views and preferences for the implementation of non-invasive prenatal diagnosis for single gene disorders from health professionals in the United Kingdom. Am J Med Genet A. 2013;161:1612–8. doi: 10.1002/ajmg.a.35972. [DOI] [PubMed] [Google Scholar]
- Lun FM, Tsui NB, Chan KC, et al. Non-invasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci U S A. 2008;105:19920–5. doi: 10.1073/pnas.0810373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New MI, Tong YK, Yuen T, et al. Non-invasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014;99:E1022–30. doi: 10.1210/jc.2014-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]