

Abstract
The development of effective and well-tolerated biologic therapies has advanced the management of psoriasis by enabling clinicians to treat underlying disease mechanisms. Biologics approved for the treatment of moderate-to-severe psoriasis include three tumor necrosis factor alpha inhibitors and an interleukin-12/interleukin-23 inhibitor. The establishment of the immunological basis of psoriasis has led to the development of biologic agents targeting specific downstream mediators in the psoriatic cascade. These drugs inhibit cytokines and cytokine signaling/transcription mediators like interleukin-17, which plays an important role in immunopathogenesis. Several interleukin-17 inhibitors are undergoing phase 3 clinical studies. In addition, biologics that selectively inhibit interleukin-23 have been assessed in phase 2 studies. This review describes how the dissection of pathways in the immunopathogenesis of psoriasis has led to the development of therapeutic agents and highlights the latest clinical efficacy, safety and tolerability data on new and emerging biologic therapies that selectively target interleukin-17 or interleukin-23.
Keywords: biologic, interleukin-17, interleukin-23, psoriasis
Introduction
The development of new biologic drugs that target specific mediators in the immunopathogenesis of plaque psoriasis has transformed management of the disease and has increased the range of options beyond phototherapies and traditional immunosuppressants. Physicians in the Europe Union and America can now choose among five approved biologic therapies that fall into three classes: the tumor necrosis factor alpha (TNF-α) inhibitors (etanercept, infliximab, adalimumab), the interleukin-12/interleukin-23 (IL-12/23) inhibitors (ustekinumab), and an IL-17 inhibitor (secukinumab). In the European Union, all five biologics, including two recent biosimilars of infliximab, are indicated in patients with moderate-to-severe plaque psoriasis, generally for second-line use after immunosuppressants (cyclosporine and methotrexate) or phototherapies. The TNF-α inhibitor etanercept is also approved for use in children and adolescents with chronic severe plaque psoriasis from the age of 6 years. In America, the situation is similar with biologics being approved for use in patients that are unresponsive to other systemic therapies, except secukinumab, which is indicated for patients who are candidates for systemic therapy or phototherapy. However, etanercept has no pediatric indication in America. Treatment guidelines developed by dermatology societies in the European Union (1–7) and North America (8,9) provide guidance to physicians on selection of approved biologics and treatment sequencing, as well as issues such as use in patients with comorbidities.
Although outcomes for patients with plaque psoriasis have improved since the introduction of biologic therapies at the end of the 1990s, safety concerns may limit their use, including the risk of serious infections (e.g., tuberculosis), and the potential for increased risk of malignancies or major adverse cardiovascular events (MACE). A greater understanding of the immune pathways involved in psoriasis has led to the identification of new therapeutic options (10). Here, we briefly review the immunopathogenesis of psoriasis, identify key mediators of psoriatic plaques that are being targeted by new and emerging biologic therapies, and highlight the latest efficacy and safety data from trials of these new agents.
Immunopathogenesis of psoriasis
Psoriasis is a disorder involving keratinocyte hyperproliferation, which interferes with the keratinocyte terminal differentiation program. This results in poorly adhesive stratum corneum and the formation of the characteristic plaques seen in psoriasis (11–13). The psoriatic keratinocyte is now known to be driven by an immune-mediated chronic inflammatory response. Although the exact cause is unknown, the development of psoriasis involves a complex interplay between genetic and environmental factors (e.g., skin injury, infection, stress, certain prescription drugs, smoking, alcohol, and obesity) (13,14). More than 20 different susceptibility genes associated with psoriasis have been identified through several genome-wide association studies, indicating the heterogeneous nature of the genetic susceptibility (15). Psoriasis genes fall into five main functional categories: those associated with acquired immunity (antigen presentation and helper T cell [TH] 17 activation) and those associated with innate immunity (nuclear factor kappaB pathway signaling, type 1 interferon induction, and skin barrier function) (16).
Initially, psoriasis was considered to be primarily a keratinization disorder (11,17–19). Since the 1970s, however, it has been established that immune cells (particularly T cells and dendritic cells) are present in psoriatic lesions and play a pathologic role. Later research established that the epidermal abnormalities are, in fact, T-cell driven (11–13). The role played by various cytokines in the immunopathogenesis of psoriasis is shown in FIG. 1 (12,13,20).
FIG. 1.
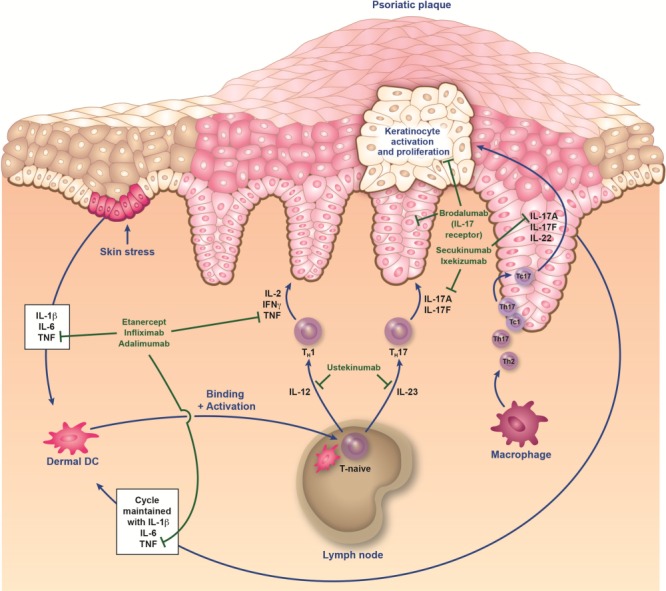
Immunopathogenesis of psoriasis and sites of action of biologics. It is proposed that an initial trigger (e.g., injury, infection, stress, etc.) precipitates a cascade of events that starts with the activation of innate immune cells (plasmacytoid dendritic cells, natural killer cells, and keratinocytes). These activated cells secrete cytokines (e.g., interferon [IFN] α, tumor necrosis factor [TNF] α, interleukin [IL]-1β, and IL-6), which in turn activate myeloid dendritic cells (DC). DC are central to the immune system, providing a link between innate and adaptive responses. The activated myeloid DC enter the draining lymph nodes, causing naive T cells (T-naive) to differentiate into helper type 17 (TH17) and type 1 (TH1) cells via the presentation of antigens and secretion of IL-12 and IL-23. These effector T cells then migrate into skin tissue, where they secrete mediators (eg, IL-17A, IL-17F, and IL-22 from TH17/cytotoxic T17 [TC17] cells, and IFNγ and TNFα from TH1/cytotoxic T1 [TC1] cells) that stimulate keratinocyte activation and proliferation, leading to plaque formation (12,13,20). Activated keratinocytes produce antimicrobial peptides, proinflammatory cytokines (IL-1β, TNF, and IL-6), and various chemokines that feedback into the proinflammatory cycle. This feedback loop, as well as others involving fibroblasts and endothelial cells, results in the continued immunopathologic progression of psoriasis.
Emerging immunotherapeutic targets for psoriasis
Targeting IL-23 alone
Evidence is accumulating to suggest that IL-23 and its resulting TH17 pathway play a more important role in psoriasis than IL-12 (21,22). Thus, selective inhibition of IL-23 may be viewed as a refinement on inhibition of both IL-12 and IL-23 by ustekinumab. Both IL-12 and IL-23 are composed of 2 subunits: IL-12 has p40 and p35 subunits, whereas IL-23 shares the p40 subunit but has a distinct p19 subunit. Results of numerous studies have indicated that levels of p19 and p40 mRNA, but not p35 mRNA, are substantially increased in psoriatic lesional skin compared with psoriatic nonlesional skin (21,22). Immunohistochemical analyses show p40 and p19 protein expression in dermal dendritic cells and keratinocytes within psoriatic lesions. Moreover, correlations have been demonstrated between reductions in IL-23 levels and clinical improvements in psoriasis (21). Secretion of IL-23 by dermal dendritic cells is believed to activate TH17 cells to release proinflammatory cytokines such as IL-17A, which in turn, drives and sustains the psoriatic disease process (23).
The above findings, coupled with the role of IL-12 in the TH1 response and host defense against intracellular pathogens, suggest that preserving this cytokine may be beneficial and have led to the development of specific anti-p19 monoclonal antibodies (22). The most advanced of these agents are tildrakizumab and guselkumab, both of which have shown promising results in phase 2 trials (Table 1) (24,25).
Table 1.
Indirect comparison of key efficacy results from phase 2/3 studies of interleukin (IL)-23– and IL-17–targeted biologic therapies in patients with moderate-to-severe chronic plaque psoriasis
| IL-23–targeted therapies | |||||
|---|---|---|---|---|---|
| Active therapy (trial design) | Trial acronym/lead author (year) [primary outcome] | Comparators | n | Patients with a PASI 75 at week 16 (p value versus placebo) | Patients with a PGA score of 0 or 1 at week 16 (p value versus placebo) |
| Guselkumab subcutaneously every 8 weeks (q8w), or on weeks 0 and 4, then every 12 weeks (q12w) (52-week randomized, double-blind, dose-ranging phase 2 trial) | X-PLORE (2014) (24) | Guselkumab 5 mg q12w | 41 | 43.9% (<0.001) | 34.1% (=0.002) |
| [PGA score of 0 or 1 at week 16] | Guselkumab 15 mg q8w Guselkumab 50 mg q12w Guselkumab 100 mg q8w Guselkumab 200 mg q12w Adalimumaba Placebo | 41 42 42 42 43 42 | 75.6% (<0.001) 81.0% (<0.001) 78.6% (<0.001) 81.0% (<0.001) 69.8% (<0.001) 4.8% | 61.0% (<0.001) 78.6% (<0.001) 85.7% (<0.001) 83.3% (<0.001) 58.1% (<0.001) 7.1% | |
| Tildrakizumab subcutaneously on weeks 0 and 4, then every 12 weeks (52-week randomized, double-blind phase 2 trial) | Langley et al. (2014) (25) [PASI 75 at week 16] | Tildrakizumab 5 mg Tildrakizumab 25 mg Tildrakizumab 100 mg Tildrakizumab 200 mg Placebo | 42 92 89 86 46 | 33.3% (≤0.001) 64.4% (≤0.001) 66.3% (≤0.001) 74.4% (≤0.001) 4.4% | NRb NRb NRb NRb NRb |
| IL-17–targeted therapies | |||||||
|---|---|---|---|---|---|---|---|
| Patients with improvement in PASI score at week 12 (p value versus comparator) |
Patients with a PGA/IGAd score of 0 or 1 at week 12 (p value versus comparator) | ||||||
| Active therapy (trial design) | Trial acronym/lead author (year) [primary outcome] | Comparators | nc | PASI 75 | PASI 90 | PASI 100 | |
| Phase 3 trials | |||||||
| Secukinumab subcutaneously once weekly for 5 weeks, then every 4 weekse | FIXTURE (2014) (26) | Secukinumab 300 mg | 323 | 77.1%(<0.001)f | 54.2%(<0.001)f | 24.1%(<0.001)g | 62.5%(<0.001)f |
| (5 randomized, double-blind, phase 3 trials) | [Coprimary: PASI 75/IGA score of 0 or 1 at week 12] | ||||||
| Secukinumab 150 mg | 327 | 67.0%(<0.001)f | 41.9%(<0.001)f | 14.4%(<0.001)g | 51.1%(<0.001)f | ||
| Etanercept SC 50 mg | 323 | 44.0% | 20.7% | 4.3% | 27.2% | ||
| Placebo | 324 | 4.9% | 1.5% | 0% | 2.8% | ||
| ERASURE (2014) (26) | Secukinumab 300 mg | 245 | 81.6%(<0.001) | 59.2%(<0.001) | 28.6%(<0.001) | 65.3%(<0.001) | |
| [Coprimary: PASI 75/IGA score of 0 or 1 at week 12] | |||||||
| Secukinumab 150 mg | 243 | 71.6%(<0.001) | 39.1%(<0.001) | 12.8%(<0.001) | 51.2%(<0.001) | ||
| Placebo | 246 | 4.5% | 1.2% | 0.8% | 2.4% | ||
| FEATURE (2014) (27) | Secukinumab 300 mg | 58 | 75.9%(<0.0001) | 60.3%(<0.0001) | 43.1%(<0.0001) | 69.0%(<0.0001) | |
| [Coprimary: PASI 75/IGA score of 0 or 1 at week 12] | |||||||
| Secukinumab 150 mg | 59 | 69.5%(<0.0001) | 45.8%(<0.0001) | 8.5% (=0.057) | 52.5%(<0.0001) | ||
| Placebo | 59 | 0% | 0% | 0% | 0% | ||
| JUNCTURE (2014) (28) | Secukinumab 300 mg | 60 | 86.7%(<0.0001) | 55.0%(<0.0001) | 26.7%(<0.0001) | 73.3%(<0.001) | |
| [Coprimary: PASI 75/IGA score of 0 or 1 at week 12] | |||||||
| Secukinumab 150 mg | 60 | 71.7%(<0.0001) | 40.0%(<0.0001) | 16.7%(=0.0006) | 53.3%(<0.001) | ||
| Placebo | 61 | 3.3% | 0% | 0% | 0% | ||
| SCULPTUREe (2013) (29) | Secukinumab 300 mg | 483 | 90.1%(NA) | 64.2%(NA) | 25.7%(NA) | 76.0%(NA) | |
| [PASI 75 at week 12] | Secukinumab 150 mg | 482 | 84.4%(NA) | 49.3%(NA) | 16.2%(NA) | 62.8%(NA) | |
| Phase 2 trials | |||||||
| Secukinumab subcutaneously 150 mg at 1 of 3 induction regimens and 2 maintenance regimensh | Rich et al. (2013) (30) [PASI 75 at week 12] | Secukinumab 150 mg, single dose | 66 | 10.6%(NS) | 3.0%(NS) | NA | 4.5%(NS) |
| Secukinumab 150 mg, monthly dosing at weeks 0, 4, and 8 | 138 | 42.0%(<0.001) | 17.4%(<0.001) | NA | 22.6%(= 0.003) | ||
| (32-week randomized, double-blind, phase 2 trial) | Secukinumab 150 mg, early dosing at weeks 0, 1, 2, 4, and 8 | 133 | 54.5%(<0.001) | 31.8%(<0.001) | NA | 37.1%(<0.001) | |
| Placebo | 67 | 1.5% | 1.5% | NA | 1.5% | ||
| Ixekizumab subcutaneously every other week for 4 weeks, then every 4 weeks | Leonardi et al. (2012) (31) | Ixekizumab 10 mg | 28 | 28.6%(NS) | 18.0%(NS) | 0%(NS) | 25.0%(NS) |
| [PASI 75 at week 12] | Ixekizumab 25 mg | 30 | 76.7%(≤0.001) | 50.0%(≤0.001) | 16.7%(NS) | 70.0%(NS) | |
| (20-week randomized, double-blind, phase 2 trial) | Ixekizumab 75 mg | 29 | 83.0%(≤0.001) | 58.6%(≤0.001) | 37.9%(≤0.001) | 72.4%(≤0.001) | |
| Ixekizumab 150 mg | 28 | 82.0%(≤0.001) | 71.4%(≤0.001) | 39.3%(≤0.001) | 71.4%(≤0.001) | ||
| Placebo | 26 | 7.7% | 0% | 0% | 7.7% | ||
| Brodalumab subcutaneously every week for 2 weeks, then every 2 weeks for the 70-mg, 140-mg, or 210-mg doses, or every 4 weeks for the 280-mg dose | Papp et al. (2012) (32) | Brodalumab 70 mg | 39 | 33.3%(<0.001) | 18.0%(<0.01) | 10.3%(<0.05) | 26.0%(<0.0.1) |
| [Percentage change in PASI score from baseline to week 12] | |||||||
| Brodalumab 140 mg | 39 | 77.0%(<0.001) | 71.8%(<0.001) | 38.5%(<0.001) | 84.6%(<0.001) | ||
| Brodalumab 210 mg | 40 | 82.5%(<0.001) | 75.0%(<0.001) | 62.5%(<0.001) | 80.0%(<0.001) | ||
| (12-week randomized, double-blind, phase 2 trial) | |||||||
| Brodalumab 280 mg | 42 | 66.7%(<0.001) | 57.1%(<0.001) | 28.6%(<0.001) | 69.1%(<0.001) | ||
| Placebo | 38 | 0% | 0% | 0% | 2.6% | ||
Eighty milligram at week 0, 40 mg at week 1, then 40 mg once every 2 weeks.
Assessed as 1 of 2 secondary outcome measures, but not reported.
Efficacy evaluable population.
In the two trials of secukinumab, a more rigorous version of the PGA scale was used, which also required an a reduction in score from baseline of at least 2 points.
In the SCULPTURE trial, the treatment regimen changed after week 12; PASI 75 responders were rerandomized to receive secukinumab at the same dose, but with fixed-interval, monthly dosing, or with retreatment as needed (dosing only at the start of a relapse) up to week 52.
Versus etanercept and versus placebo.
Versus etanercept; no comparison versus placebo was performed because no patient in the placebo group responded.
Results for the maintenance regimens are not shown in the table because this was assessed after week 12.
IGA: investigator global assessment; NA: not assessed; NR: not reported; NS: not significant; PASI: Psoriasis Area-and-Severity Index; PGA: Physician's Global Assessment.
Tildrakizumab
Tildrakizumab is a humanized immunoglobulin (Ig)G1k monoclonal anti-IL-23p19 antibody that does not bind to IL-12 or p40 (25). Phase 3 studies in moderate-to-severe chronic plaque psoriasis are ongoing, and initial results of a 52-week randomized, double-blind, dose-finding, phase 2 study were reported recently (25).
Efficacy
In the phase 2 study, the efficacy and safety of four doses of tildrakizumab (5, 25, 100, and 200 mg) administered by subcutaneous injection at weeks 0 and 4 were compared with placebo (25). The proportion of patients with a 75% improvement in psoriasis area-and-severity index score (PASI 75) at week 16 (primary endpoint) was significantly higher in all four active treatment groups compared with placebo (Table 1). Patients who achieved a PASI 75 response at week 16 were eligible to receive treatment every 12 weeks for a further 40 weeks. Results have not been reported in patients who continued to receive the tildrakizumab 5-mg or 25-mg doses, but the number of patients continuing to achieve PASI 75 in the 100-mg and 200-mg groups appeared to remain relatively constant during weeks 16–52 and at a follow-up assessment 20 weeks after treatment discontinuation. In patients who were rerandomized from 100 to 25 mg at week 16, there was a significant reduction on the proportion of patients achieving PASI 75 by week 52 (p = 0.005) and Physician's Global Assessment (PGA) scores of 0 or 1 (p = 0.02) compared with those who continued to receive the 100-mg dose (25). Together, these results indicate that the two highest doses (100 and 200 mg) have promising efficacy and that a strategy of reducing the dose below 100 mg may be associated with deterioration in clinical response.
Safety and tolerability
The overall incidence of adverse events (AEs) and serious AEs (SAEs) during the 52-week treatment phase of this study have not been reported. However, the most frequent AEs across the tildrakizumab groups were nasopharyngitis, headache, hypertension, and diarrhea (25). The SAEs that were considered to be possibly related to tildrakizumab included bacterial arthritis, lymphedema, melanoma, stroke, epiglottitis, and knee infection. One death of undetermined cause was reported (treatment group unspecified), and malignancies (rectal cancer, malignant melanoma and malignant melanoma in situ), serious infections (sinusitis, epiglottitis, and cellulitis), and ischemic stroke were reported in one patient each. In the 20-week posttreatment follow-up period, three patients had serious infections (mycoplasma pneumonia, pneumonia, and soft tissue infection) and one major cardiovascular event was reported (thrombotic cerebral infarction) (25). At present, it is unclear whether there was a relationship between the dose of tildrakizumab and the incidence of AEs.
Guselkumab
Guselkumab is a human IgG1 monoclonal anti-IL-23 antibody (33,34). It is in a similar stage of development as tildrakizumab: phase 3 studies are ongoing and initial results of a phase 2, dose-ranging study are available (24).
Efficacy
In the phase 2, double-blind study, patients were randomized to receive subcutaneous injections of guselkumab 5, 50, or 200 mg (at weeks 0 and 4, then every 12 weeks), guselkumab 15 or 100 mg (at weeks 0 and 8, then every 8 weeks), adalimumab (as indicated in the label), or placebo for up to 52 weeks (24). At week 16, proportionately more patients in all five guselkumab groups achieved PGA scores of 0 or 1 (primary endpoint) and PASI 75 (secondary endpoint) than in the placebo group (Table 1). The change in mean dermatology life quality index (DLQI) scores from baseline to week 16 (secondary endpoint) also significantly favored guselkumab over placebo (p ≤ 0.008, all comparisons) (24). A post hoc analysis indicated that the proportions of patients achieving a response at week 40 were higher with the guselkumab 50-mg, 100-mg, and 200-mg dose groups than with adalimumab (24).
Safety and tolerability
Safety findings have not been reported in detail. However, it has been reported that AEs and SAEs at week 16 were experienced by 49 and 1.4% of patients, respectively, in the guselkumab groups compared with 53.5 and 2.3% in the adalimumab group, and 50.0 and 2.4% in the placebo group (35). At week 52, the incidence of AEs and SAEs was 63.4 and 2.8%, respectively, in the guselkumab groups and 72.1 and 4.7% in the adalimumab group (24). The most frequent AE was infection (36.6% of patients in the guselkumab groups versus 41.9% in the adalimumab group) of which three were serious (lung abscess and appendicitis in the guselkumab 50-mg group and pneumonia in adalimumab group). MACE were reported in one patient receiving guselkumab 5 mg (fatal myocardial infarction) and two patients receiving the 100-mg dose (nonfatal myocardial infarction and stroke). A grade III cervical intraepithelial neoplasia was reported in one patient who received guselkumab 200 mg.
BI 655066
BI 655066 is a human IgG1 monoclonal anti-IL-23A antibody (36). Phase 2 studies in patients with moderate-to-severe chronic plaque psoriasis are ongoing and results from a phase 1 single-rising-dose trial of 39 patients were recently reported (36).
Efficacy
In the phase 1 study, the efficacy and safety of a single dose of BI 655066 administered intravenously (0.01, 0.05, 0.25, 1, 3, or 5 mg/kg) or subcutaneously (0.25 or 1 mg/kg) was compared with placebo (36). At week 12, PASI 75 was achieved by 87% of patients receiving any dose of BI 655066 (p <0.001 compared with placebo). Similarly, 87% of patients treated with any dose of BI 655066 achieved static physician global assessment (sPGA) values of 0 or 1 at week 12.
Safety and tolerability
AEs were reported in 65% of patients receiving any dose of BI 655066 over the 24 weeks of the study; with the most common AEs being nasopharyngitis (13%), headache (10%), and upper respiratory tract infection (10%)(36). SAEs were reported in 13% of patients treated with BI 655066 and consisted of a single event of alcoholic pancreatitis, ischemic thalamic stroke, transient ischemic attack, and polymyositis. None of the SAEs were considered related to study treatment by the investigators. There were no deaths during the study.
Targeting IL-17
Evidence is now emerging that IL-17 is a key cytokine in the immunopathogenesis of psoriasis at the keratinocyte level (37,38). IL-17 consists of a family of six members (IL-17A–F), with the role of IL-17A in psoriasis being the most extensively researched. Because of new evidence supporting a central role of IL-17A (and thus the IL-17A–producing helper [TH17] and cytotoxic T cells [TC17]), investigators have questioned the importance of T cells producing interferon γ and TNF-α in the pathogenesis of psoriasis (38). IL-17A has proinflammatory effects on keratinocytes, macrophages, and endothelial cells and is responsible for increased downstream production of cytokines (20,39,40). IL-17A binds to receptors on keratinocytes and induces expression of neutrophil, T-cell, and dendritic-cell chemokines that lead to the migration of neutrophils, memory T cells, and dendritic cells to psoriatic lesions (37). Neutrophils attracted to the skin by chemokines released from IL-17A-stimulated keratinocytes can affect the growth and differentiation of keratinocytes, in addition to causing local tissue damage by producing reactive oxygen species and proteolytic enzymes (37).
Secukinumab, a monoclonal antibody that targets IL-17A, has been approved for the treatment of moderate-to-severe psoriasis, and one other monoclonal antibody that inhibits IL-17A (ixekizumab) and one that blocks the IL-17 receptor (brodalumab) are being researched for the treatment of psoriasis (FIG. 2; 38,40–42). Key findings from phase 2 and 3 trials of these agents in patients with moderate-to-severe plaque psoriasis are summarized in Tables 1 and 2 (26,29–32,43,44). RG7624, an antibody that targets both IL-17A and IL-17F, is also being investigated for the treatment of psoriasis but no clinical trial results have been reported for this agent (45).
FIG. 2.
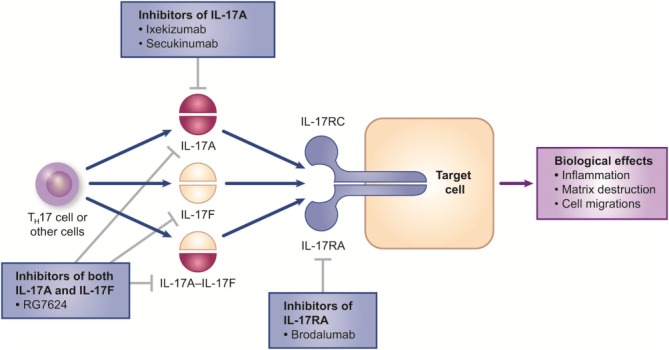
Targeting the interleukin (IL)-17 signaling pathway. IL-17RC and IL-17RA are subunits of the IL-17 receptor, which is present on various cell types including keratinocytes, dendritic cells, dermal fibroblasts, and endothelial cells. Secukinumab, a fully human immunoglobulin (Ig)G1κ monoclonal antibody, and ixekizumab, a humanized IgG4 monoclonal antibody, target the cytokine IL-17A. These agents prevent inflammatory-mediated effects by neutralizing IL-17 and therefore inhibit the binding of IL-17A to its receptor. Brodalumab is a human IgG2 monoclonal antibody that binds to IL-17RA, thereby blocking signaling of IL-17 via its receptor. Another agent, RG7624, is a fully human IgG1 monoclonal antibody against both IL-17A and IL-17F. All four agents are in clinical development for the treatment of psoriasis, with secukinumab and ixekizumab being the furthest along (40–42). Reprinted by permission from Macmillan Publishers Ltd: Nat Rev Drug Discov 12: 815–816, © 2013.
Table 2.
Indirect comparison of the incidences of adverse events during the induction period (first 12–20 weeks) in phase 2/3 trials of interleukin (IL)-17–targeted biologic therapies; values are number of patients (%)
| Safety and tolerability over 12 weeks | |||
|---|---|---|---|
|
Secukinumaba | |||
| Phase 3 ERASURE study (26) | |||
| Secukinumab 300 mg (n = 245) | Secukinumab 150 mg (n = 245) | Placebo (n = 247) | |
| Any AE | 135 (55.1) | 148 (60.4) | 116 (47.0) |
| Any SAE | 6 (2.4) | 4 (1.6) | 4 (1.6) |
| Death | 0 | 0 | 0 |
| Treatment discontinuation due to AE | 3 (1.2) | 5 (2.0) | 4 (1.6) |
| Most common AEsb | |||
| Nasopharyngitis | 22 (9.0) | 23 (9.4) | 19 (7.7) |
| Headache | 12 (4.9) | 13 (5.3) | 7 (2.8) |
| Infection or infestation | 72 (29.4) | 66 (26.9) | 40 (16.2) |
| Other safety findings | No cases of grade 3 or 4 neutropenia reported for the 12-week induction period | ||
| Phase 3 FIXTURE study (26) | ||||
|---|---|---|---|---|
| Secukinumab 300 mg (n = 326) | Secukinumab 150 mg (n = 327) | Etanercept (n = 323) | Placebo (n = 327) | |
| Any AE | 181 (55.5) | 191 (58.4) | 186 (57.6) | 163 (49.8) |
| Any SAE | 4 (1.2) | 7 (2.1) | 3 (0.9) | 6 (1.8) |
| Death | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AE | 4 (1.2) | 2 (0.6) | 6 (1.9) | 3 (0.9) |
| Most common AEsb | ||||
| Nasopharyngitis | 35 (10.7) | 45 (13.8) | 36 (11.1) | 26 (8.0) |
| Headache | 30 (9.2) | 16 (4.9) | 23 (7.1) | 23 (7.0) |
| Diarrhea | 17 (5.2) | 12 (3.7) | 11 (3.4) | 6 (1.8) |
| Infection or infestation | 87 (26.7) | 101 (30.9) | 79 (24.5) | 63 (19.3) |
| Phase 3 FEATURE study (27) | |||
|---|---|---|---|
| Secukinumab 300 mg (n = 59) | Secukinumab 150 mg (n = 59) | Placebo (n = 59) | |
| Any AE | 30 (50.8) | 34 (57.6) | 28 (47.5) |
| Any SAE | 3 (5.1) | 0 | 1 (1.7) |
| Death | 0 | 0 | 0 |
| Treatment discontinuation due to AE | 1 (1.7) | 0 | 1 (1.7) |
| Most common AEsb | |||
| Diarrhea | 5 (8.5) | 2 (3.4) | 1 (1.7) |
| Nasopharyngitis | 3 (5.1) | 3 (5.1) | 5 (8.5) |
| Headache | 0 | 4 (6.8) | 3 (5.1) |
| Back pain | 3 (5.1) | 0 | 0 |
| Nausea | 3 (5.1) | 0 | 1 (1.7) |
| Infection or infestation | NR | NR | NR |
| Phase 3 JUNCTURE study (28) | |||
|---|---|---|---|
| Secukinumab 300 mg (n = 60) | Secukinumab 150 mg (n = 61) | Placebo (n = 61) | |
| Any AE | 42 (70.0) | 39 (63.9) | 33 (54.1) |
| Any SAE | 1 (1.7) | 3 (4.9) | 1 (1.6) |
| Death | 0 | 0 | 0 |
| Treatment discontinuation due to AE | 0 | 0 | 1 (1.6) |
| Most common AEsb | |||
| Nasopharyngitis | 19 (31.7) | 14 (23.0) | 10 (16.4) |
| Headache | 3 (5.0) | 5 (8.2) | 3 (4.9) |
| Pruritus | 5 (8.3) | 1 (1.6) | 2 (3.3) |
| Sinusitis | 3 (5.0) | 1 (1.6) | 0 |
| Cough | 3 (5.0) | 0 | 2 (3.3) |
| Infection or infestation | NR | NR | NR |
| Phase 2 study (30) | ||||
|---|---|---|---|---|
| Single dose Secukinumab 150 mg (n = 66) | Monthly dosing Secukinumab 150 mg (n = 138) | Early dosing Secukinumab 150 mg (n = 133) | Placebo (n = 67) | |
| Any AE | 41 (62.1) | 91 (65.9) | 89 (66.9) | 47 (70.1) |
| Any SAE | 3 (4.5) | 3 (2.2) | 6 (4.5) | 1 (1.5) |
| Death | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AE | 1 (1.5) | 0 | 3 (2.3) | 1 (1.5) |
| Most common AEsb | ||||
| Nasopharyngitis | 8 (12.1) | 31 (22.5) | 30 (22.6) | 12 (17.9) |
| Headache | 6 (9.1) | 8 (5.8) | 11 (8.3) | 3 (4.5) |
| Psoriasisc | 6 (9.1) | 8 (5.8) | 4 (3.0) | 7 (10.4) |
| Infection or infestation | 14 (21.2) | 56 (40.6) | 45 (33.8) | 26 (38.8) |
| Other safety findings | No injection-site reactions or antibodies to secukinumab detected | |||
| No cases of grade 3 or 4 neutropenia | ||||
|
Brodalumab | |||||
|---|---|---|---|---|---|
| Phase 2 study (32) | |||||
| Brodalumab 70 mg (n = 38) | Brodalumab 140 mg (n = 39) | Brodalumab 210 mg (n = 40) | Brodalumab 280 mg (n = 41) | Placebo (n = 37) | |
| Any AE | 26 (68) | 27 (69) | 33 (82) | 30 (73) | 23 (62) |
| Any SAE | 1 (3) | 0 | 1 (2) | 0 | 1 (3) |
| Death | 0 | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AE | 0 | 0 | 2 (5) | 1 (2) | 1 (3) |
| Most common AEsb | |||||
| Nasopharyngitis | 6 (16) | 1 (3) | 4 (10) | 2 (5) | 3 (8) |
| Upper respiratory tract infection | 3 (8) | 3 (8) | 2 (5) | 5 (12) | 2 (5) |
| Arthralgia | 1 (3) | 2 (5) | 0 | 4 (10) | 1 (3) |
| Injection site erythema | 1 (3) | 1 (3) | 3 (8) | 4 (10) | 1 (3) |
| Pain in extremity | 1 (3) | 0 | 3 (8) | 4 (10) | 0 |
| Nausea | 4 (11) | 1 (3) | 1 (2) | 0 | 1 (3) |
| Infection or infestation | NR | NR | NR | NR | NR |
| Other safety findings | Approximately 6% of patients had injection-site erythema 5% to 10% of patients developed antibodies to brodalumab (none neutralizing) 2 Cases of grade 3 neutropenia (210-mg group) | ||||
| Safety and tolerability over 20 weeks | |||||
|---|---|---|---|---|---|
|
Ixekizumab | |||||
| Phase 2 study (31) | |||||
| Ixekizumab 10 mg (n = 28) | Ixekizumab 25 mg (n = 30) | Ixekizumab 75 mg (n = 29) | Ixekizumab 150 mg (n = 28) | Placebo (n = 27) | |
| Any AE | 21 (75) | 21 (70) | 17 (59) | 13 (46) | 17 (63) |
| Any SAE | 0 | 0 | 0 | 0 | 0 |
| Death | 0 | 0 | 0 | 0 | 0 |
| Treatment discontinuation due to AE | NR | NR | NR | NR | NR |
| Most common AEsb | |||||
| Nasopharyngitis | 3 (11) | 3 (10) | 3 (10) | 4 (14) | 5 (19) |
| Upper respiratory tract infection | 1 (4) | 3 (10) | 1 (3) | 1 (4) | 1 (4) |
| Injection-site reaction | 0 | 3 (10) | 1 (3) | 2 (7) | 0 |
| Headache | 4 (14) | 4 (13) | 1 (3) | 1 (4) | 1 (4) |
| Allergy or hypersensitivity | 1 (4) | 1 (3) | 2 (7) | 1 (4) | 2 (7) |
| Infection or infestation | 12 (43) | 9 (30) | 9 (31) | 8 (29) | 7 (26) |
| Other safety findings | 0% to 10% of patients had injection-site reactions Incidence of antibodies to ixekizumab not reported No cases of grade 3 or 4 neutropenia | ||||
The phase 3 SCULPTURE study of secukinumab has not been included because results at week 12 have not yet been reported.
Reported in more than 5% of patients in any active treatment group.
Indicates worsening of psoriasis; the protocol suggested that psoriasis, being the studied indication, was not to be reported as an AE by the investigators.
AE: adverse event; NR: not reported; SAE: serious adverse event.
Secukinumab
Secukinumab is a fully human IgG1κ monoclonal anti–IL-17A antibody (38). Results are available from five phase 3 studies: the Full year Investigative eXamination of secukinumab versus eTanercept Using 2 dosing Regimens to determine Efficacy in psoriasis (FIXTURE; 26), Efficacy of Response And Safety of two fixed secUkinumab REgimens in psoriasis (ERASURE; 26), Study Comparing secukinumab Use in Long-term Psoriasis maintenance therapy: fixed regimens versus retreatment Upon start of Relapse (SCULPTURE; 29), First study of sEcukinumAb in pre-filled syringes in subjecTs with chronic plaqUe-type psoriasis: REsponse at 12 weeks (FEATURE; 27), and Judging the efficacy of secUkinumab in patients with psoriasis using autoiNjector: a Clinical Trial evalUating treatment REsults (JUNCTURE; 28).
Efficacy
FIXTURE and ERASURE were both 1-year randomized, double-blind studies that had the same coprimary endpoint and assessed secukinumab at doses of 150 and 300 mg. Secukinumab was compared with placebo (FIXTURE and ERASURE) and with etanercept (FIXTURE only). A nonresponder imputation analysis was used to evaluate both trials. Both studies met the coprimary endpoint at week 12, with significantly higher proportions of secukinumab-treated patients achieving PASI 75 and scores of 0 (clear skin) or 1 (almost clear skin) with an improvement from baseline of at least two points on the static Investigator's Global Assessment (IGA modified 2011) of overall psoriasis severity than those receiving placebo or etanercept (p < 0.001, all comparisons; Table 1). All secondary endpoints were also met in both studies. Significantly higher proportions of patients in the secukinumab groups achieved PASI score improvements of 90% (PASI 90; almost clear skin) and 100% (PASI 100; completely clear skin) than those receiving either etanercept or placebo (p < 0.001, all comparisons; Table 1). Improvements in PASI score became evident as early as week 2, with a significantly shorter median time to 50% reduction in mean PASI score from baseline with secukinumab 300 mg (3.0 weeks) and 150 mg (3.9 weeks) than with etanercept in the FIXTURE study (7.0 weeks; p < 0.001, both comparisons)(26). Importantly, the improvements were also maintained at 1 year in significantly higher proportions of secukinumab-treated patients than those receiving etanercept; 82–84% of patients receiving secukinumab maintained a PASI 75 response between weeks 12 and 52 compared with 73% in the etanercept group (p < 0.01, both comparisons) and a similar trend was observed for maintenance of a PASI 90 response between weeks 16 and 52 (26). In ERASURE, the benefits were similar, with further increases in the response rates for PASI 75, PASI 90, PASI 100, and modified IGA scores of 0 or 1 from week 12 to week 16 in both secukinumab groups, which were then maintained to 52 weeks (26). In both studies, a significantly higher proportion of patients reported no impact of disease on their quality of life (as indicated by a DLQI score of 0 or 1) in both secukinumab groups (300 mg, 57–59%; 150 mg, 46–51%) than in the placebo groups (7–10%; p < 0.001, all comparisons); in the FIXTURE study the difference was also significant versus etanercept (35%; p < 0.001, both doses). These DLQI response rates were maintained at 1 year with both doses of secukinumab (300 mg, 66–70%; 150 mg, 49–56%).
Initial results have also been reported for the 1-year SCULPTURE study, which assessed the maintenance of efficacy of secukinumab (300 and 150 mg) when administered at fixed, monthly intervals versus retreatment as needed (i.e., only at the start of a relapse) after an initial induction period of 12 weeks (29). At the end of the induction period, the proportions of patients achieving PASI 75, PASI 90, PASI 100, or modified IGA scores of 0 or 1 were numerically similar to those reported in FIXTURE and ERASURE (Table 1). Patients who initially achieved PASI 75 or PASI 90 at week 12 were more likely to maintain their response at 1 year if they received secukinumab at fixed, monthly intervals compared with treatment only at start of relapse. The secukinumab 300-mg dose was considered to be more effective than the 150-mg dose (29). Similar efficacy was observed at week 12 in the FEATURE and JUNCTURE trials which evaluated self-administration of secukinumab by pre-filled syringe or autoinjector, respectively (27,28). In both trials, patients reported high levels of device usability.
Safety and tolerability
In the FIXTURE study, the overall exposure-adjusted incidence rates over 52 weeks for AEs and SAEs were similar with secukinumab 300 mg (252 and 6.8 events per 100 patient-years, respectively), secukinumab 150 mg (236 and 6.0 events per 100 patient-years) and etanercept (243 and 7.0 events per 100 patient-years)(26). In the placebo group, the equivalent rates were slightly higher (330 and 8.3 events per 100 patient-years). The incidence rates of AEs and SAEs in ERASURE were consistent with those of FIXTURE (26). There were no clinically apparent differences in the types of SAEs among the study groups in either study and no deaths occurred during the treatment period in either study. In both studies, the most common AEs in the secukinumab groups (reported in more than 5% of patients) during the induction period and the entire treatment period were nasopharyngitis and headache; in FIXTURE, diarrhea was also reported in more than 5% of patients. Across both studies, there were no apparent dose-related effects in AEs between the secukinumab groups, with the exception of nonserious Candida infections, which were reported in a higher proportion of patients in the 300-mg groups (FIXTURE, 4.7%; ERASURE, 2.0%) compared with the 150-mg groups (FIXTURE, 2.3%; ERASURE, 0.8%); all Candida infections were mild or moderate in severity, localized, and resolved spontaneously or with standard therapy, without the need to discontinue secukinumab. AEs in the system organ class infections and infestations were more frequent in both studies in the secukinumab groups than in the placebo groups during the entire 52-week treatment period (secukinumab, 92–105 events per 100 patient-years; placebo 84–90 events per 100 patient-years). Across both studies and over 52 weeks, serious infections (n = 13), malignant or unspecified tumors (n = 12) and MACE (n = 6) were reported in < 1% of patients with either secukinumab dose. Two patients in 300-mg group had MACE (one patient had two myocardial infarction events and one patient had a cerebrovascular infarction); the remaining four MACE occurred in the secukinumab 150-mg group (ischemic stroke, moyamoya disease, cerebrovascular accident, and myocardial infarction). Evaluation by a blinded adjudication committee determined that the moyamoya disease event did not meet MACE adjudication criteria. Grade 3 neutropenia was reported in 10 patients treated with secukinumab in FIXTURE or ERASURE; grade 4 neutropenia was not reported in any patient receiving secukinumab across both studies. In FIXTURE, injection-site reactions were 15-fold less frequent with secukinumab (0.7%) than with etanercept (11.1%). Development of antibodies to secukinumab was uncommon: 0.4% of patients in the FIXTURE study (none were neutralizing) and 0.3% in ERASURE (classified as neutralizing in 1 patient; 26).
Ixekizumab
Ixekizumab is a humanized IgG4 monoclonal antibody that neutralizes IL-17A. Phase 3 studies, including head-to-head comparisons with etanercept and adalimumab, are ongoing and the results of a phase 2, double-blind, placebo-controlled trial have been reported (31).
Efficacy
In the phase 2 study, patients were randomized to receive ixekizumab 10, 25, 75, or 150 mg, or placebo (31). At 12 weeks, significantly higher proportions of patients in the ixekizumab 25-mg, 75-mg, and 150-mg groups, but not the 10-mg group, achieved PASI 75 (primary endpoint) or PASI 90 (secondary endpoint) than in the placebo group (p ≤ 0.001, all comparisons; Table 1). Differences in the proportions of patients achieving PASI 100 (secondary endpoint) were significant versus placebo for the ixekizumab 75-mg and 150-mg groups only (p ≤ 0.001, both comparisons). Static PGA scores of 0 or 1 were achieved by proportionately more patients at week 12 in the three highest dose groups (25, 75, and 150 mg) compared with placebo (Table 1). With the two highest doses, the differences in response rates for PASI 75 and PGA scores of 0 or 1 were observed at week 1 (p values not reported) and were maintained through week 20 (p < 0.05 at all assessments from week 6 onwards; 31). Changes in score from baseline to week 12 favored the highest doses of ixekizumab over placebo for Nail Psoriasis Severity Index (75-mg and 150-mg doses), Psoriasis Scalp Severity Index (25-mg, 75-mg, and 150-mg doses), and a joint pain visual analogue scale (150-mg dose; p ≤ 0.05, all comparisons). In addition, changes in mean DLQI scores and an itch severity score at weeks 8 and 16 favored the 25-mg, 75-mg, and 150-mg doses of ixekizumab over placebo (p < 0.001, all comparisons).
Safety and tolerability
Over 20 weeks, the overall incidence of AEs was similar to placebo when all doses of ixekizumab were combined (Table 2)(31). No SAEs or deaths were reported. Infection or infestation AEs, including nasopharyngitis and upper respiratory infections, were the most common AEs. No grades 3 or 4 neutropenia was reported. Injection-site reactions were reported in 5.2% of patients receiving ixekizumab.
Brodalumab
Brodalumab is a human IgG2 monoclonal antibody that binds to and blocks IL-17R, the receptor subunit shared by IL-17A, IL-17F, and IL-17A/F heterodimer ligands (38). Phase 3 studies in plaque psoriasis (140-mg or 210-mg doses) are ongoing and the results of a phase 2, double-blind, placebo-controlled, dose-ranging study have been reported (32).
Efficacy
In the phase 2 study, patients were randomized to receive subcutaneous brodalumab 70, 140, 210 (at day 1 and weeks 1, 2, 4, 6, 8, and 10), or 280 mg (once monthly), or placebo (32). Mean percentage improvement in PASI score from baseline to week 12 (primary endpoint) favored brodalumab over placebo (p < 0.001, all comparisons). PASI 50, 75, 90, and 100, and PGA scores of 0 or 1 at week 12 (secondary endpoints) were also reached in proportionately more patients in the brodalumab groups than in the placebo group (p ≤ 0.05, all comparisons; Table 1). At week 12, brodalumab also reduced the mean body surface area affected and was associated with lower mean DLQI scores than placebo (p ≤ 0.01, all comparisons). The brodalumab 280-mg regimen did not appear to confer any additional benefits over the 210-mg or 140-mg doses (32).
Safety and tolerability
The overall incidence of AEs was higher with the 210-mg dose of brodalumab (82%) than with the other three doses (68–73%; 32). Although the overall incidence of infections and infestations was not reported, nasopharyngitis and upper respiratory tract infections were the most frequent AEs, followed by arthralgia, injection-site erythema, extremity pain, and nausea (all reported in more than 5% of patients in any group). In the brodalumab groups, one case of renal colic (in the 70-mg group) and one case of grade 3 neutropenia (in the 210-mg group) were reported as SAEs. Grade 3 neutropenia was reported in two patients in the brodalumab 210-mg group; in one of these patients who re-initiated brodalumab after normalization of neutrophil counts, neutropenia returned, and treatment was discontinued. Antibodies to brodalumab were detected in 5–10% of patients (none were classified as neutralizing antibodies)(32).
Summary
Based on evidence from clinical studies, IL-23 and IL-17 appear to be suitable targets for moderate-to-severe psoriasis. Emerging biologic therapies that selectively inhibit either IL-23 or IL-17 significantly reduced the severity of moderate-to-severe plaque psoriasis during 12–16 weeks of treatment. Initial results from the phase 2 studies of the selective IL-23 inhibitors indicate that this efficacy may be maintained for up to 40 weeks (guselkumab) or 52 weeks (tildrakizumab). However, full results from these studies and further phase 3 studies are awaited. The three selective IL-17 inhibitors have also all shown convincing efficacy over 12 weeks, although the 1-year results of large-scale phase 3 studies have only been reported for secukinumab. In these studies, the efficacy of secukinumab was not only superior to placebo, but also to etanercept. In addition, the PASI and IGA modified 2011 response rates were maintained up to 52 weeks, with numerically higher rates than in patients treated with etanercept.
Whether selective inhibition of IL-23 or IL-17 will result in similar or improved safety profiles compared with TNF-α or IL-12/23 inhibition, remains to be established from long-term studies and real-world evidence because, although pivotal trials are powered to evaluate efficacy and common AEs, AEs of special interest require broader patient populations and longer time periods to detect. However, these new biologic therapies show great promise and, if approved, would increase the range of options for patients with plaque psoriasis.
Acknowledgments
Technical assistance with editing and styling of the manuscript for submission was provided by Oxford PharmaGenesis Inc, Newtown, PA. Funding sources: Novartis Pharmaceuticals Corporation (East Hanover, NJ, USA) provided funding for editing and styling of the manuscript. The authors were fully responsible for all content and editorial decisions and received no financial support or other form of compensation related to the development of this manuscript. Dr Gaspari has participated in Data Monitoring Committee activities for Johnson & Johnson (New Brunswick, NJ) and has served on the Novartis (East Hanover, NJ) Advisory Board for secukinumab. Dr Tyring's institution has received grants from the manufacturers of etanercept, adalimumab, secukinumab, guselkumab, ixekizumab, brodalumab, and tildrakizumab for clinical studies of these drugs in the treatment of psoriasis. In addition, Dr Tyring is a speaker for the manufacturers of etanercept and adalimumab and is a consultant for the manufacturers of adalimumab and secukinumab.
REFERENCES
- 1.Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303:1–10. doi: 10.1007/s00403-010-1080-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nast A, Boehncke WH, Mrowietz U, et al. S3—Guidelines on the treatment of psoriasis vulgaris (English version). Update. J Dtsch Dermatol Ges. 2012;10(Suppl 2):S1–S95. doi: 10.1111/j.1610-0387.2012.07919.x. [DOI] [PubMed] [Google Scholar]
- 3.National Institute for Health and Clinical Excellence. 2012. Psoriasis: The assessment and management of psoriasis. NICE clinical guideline 153. Available at: http://www.nice.org.uk/guidance/cg153/resources/guidance-psoriasis-pdf. Accessed February 13, 2015.
- 4.Pathirana D, Ormerod AD, Saiag P, et al. European S3-guidelines on the systemic treatment of psoriasis vulgaris. J Eur Acad Dermatol Venereol. 2009;23(Suppl 2):1–70. doi: 10.1111/j.1468-3083.2009.03389.x. [DOI] [PubMed] [Google Scholar]
- 5.Puig L, Carrascosa JM, Daudén E, et al. [Spanish evidence-based guidelines on the treatment of moderate-to-severe psoriasis with biologic agents] Actas Dermosifiliogr. 2009;100:386–413. [PubMed] [Google Scholar]
- 6.Smith CH, Anstey AV, Barker JN, et al. British Association of Dermatologists' guidelines for biologic interventions for psoriasis 2009. Br J Dermatol. 2009;161:987–1019. doi: 10.1111/j.1365-2133.2009.09505.x. [DOI] [PubMed] [Google Scholar]
- 7.Zweegers J, de Jong EM, Nijsten TE, et al. Summary of the Dutch S3-guidelines on the treatment of psoriasis 2011. Dermatol Online J. 2014;20 Mar 17. [PubMed] [Google Scholar]
- 8.Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826–850. doi: 10.1016/j.jaad.2008.02.039. [DOI] [PubMed] [Google Scholar]
- 9.Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 6. Guidelines of care for the treatment of psoriasis and psoriatic arthritis: case-based presentations and evidence-based conclusions. J Am Acad Dermatol. 2011;65:137–174. doi: 10.1016/j.jaad.2010.11.055. [DOI] [PubMed] [Google Scholar]
- 10.Levy LL, Solomon SM, Emer JJ. Biologics in the treatment of psoriasis and emerging new therapies in the pipeline. Psoriasis: Targets Therapy. 2012;2:29–43. [Google Scholar]
- 11.Bos JD, de Rie MA, Teunissen MB, Piskin G. Psoriasis: dysregulation of innate immunity. Br J Dermatol. 2005;152:1098–1107. doi: 10.1111/j.1365-2133.2005.06645.x. [DOI] [PubMed] [Google Scholar]
- 12.Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis. 2005;64 Suppl 2:ii30–ii36. doi: 10.1136/ard.2004.031120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villaseñor-Park J, Wheeler D, Grandinetti L. Psoriasis: evolving treatment for a complex disease. Cleve Clin J Med. 2012;79:413–423. doi: 10.3949/ccjm.79a.11133. [DOI] [PubMed] [Google Scholar]
- 15.Capon F, Barker JN. The quest for psoriasis susceptibility genes in the postgenome-wide association studies era: charting the road ahead. Br J Dermatol. 2012;166:1173–1175. doi: 10.1111/j.1365-2133.2012.10895.x. [DOI] [PubMed] [Google Scholar]
- 16.Blauvelt A, Brown M, Gordon KB, et al. Updates on psoriasis and cutaneous oncology: proceedings from the 2013 MauiDerm meeting. J Clin Aesthet Dermatol. 2013;6(9 Suppl):S2–S20. [PMC free article] [PubMed] [Google Scholar]
- 17.Voorhees JJ, Duell EA. Psoriasis as a possible defect of the adenyl cyclase-cyclic AMP cascade. A defective chalone mechanism? Arch Dermatol. 1971;104:352–358. [PubMed] [Google Scholar]
- 18.Voorhees JJ, Marcelo CL, Duell EA. Cyclic AMP, cyclic GMP, and glucocorticoids as potential metabolic regulators of epidermal proliferation and differentiation. J Invest Dermatol. 1975;65:179–190. doi: 10.1111/1523-1747.ep12598125. [DOI] [PubMed] [Google Scholar]
- 19.Weinstein GD, McCullough JL, Ross PA. Cell kinetic basis for pathophysiology of psoriasis. J Invest Dermatol. 1985;85:579–583. doi: 10.1111/1523-1747.ep12283594. [DOI] [PubMed] [Google Scholar]
- 20.Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–1211. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 21.Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: recent advances on IL-23 and Th17 cytokines. Curr Rheumatol Rep. 2007;9:461–467. doi: 10.1007/s11926-007-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin AA, Gottlieb AB. Specific targeting of interleukin-23p19 as effective treatment for psoriasis. J Am Acad Dermatol. 2014;70:555–561. doi: 10.1016/j.jaad.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 23.Di Meglio P, Nestle FO. The role of IL-23 in the immunopathogenesis of psoriasis. F1000 Biol Rep. 2010;2:40. doi: 10.3410/B2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Callis-Duffin K, Wasfi Y, Shen YK, Gordon K, Bissonnette R, Prinz J. 2014. A phase 2 multicenter, randomized, placebo- and active-controlled, dose-ranging trial to evaluate guselkumab for the treatment of patients with moderate to severe plaque-type psoriasis (X-PLORE). Oral presentation at: American Academy of Dermatology, March 21–25, Denver, CO.
- 25.Langley RG, Papp KA, Reich A, et al. 2014. MK-3222, an anti-IL-23p19 humanized monoclonal antibody, provide significant improvement in psoriasis over 52 weeks of treatment that is maintained after discontinuation of dosing. Oral presentation at: American Academy of Dermatology, March 21–25, Denver, CO.
- 26.Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326–338. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 27.Blauvelt A, Prinz JC, Gottlieb AB, et al. Secukinumab administration by pre-filled syringe: efficacy, safety, and usability results from a randomized controlled trial in psoriasis (FEATURE) Br J Dermatol. 2014;172:484–493. doi: 10.1111/bjd.13348. [DOI] [PubMed] [Google Scholar]
- 28.Paul C, Lacour JP, Tedremets L, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE) J Eur Acad Dermatol Venereol. 2014 doi: 10.1111/jdv.12751. doi: 10.1111/jdv.12751. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29.Mrowietz U, Leonardi C, Girolomoni G, et al. Secukinumab 'fixed-interval' vs. 'retreatment-as-needed' regimen for moderate-to-severe plaque psoriasis: a study comparing secukinumab use in long-term psoriasis maintenance therapy (SCULPTURE). Oral presentation at: 22nd Annual Congress of the European Academy of Dermatology and Venereology, 2–6 October 2013, Istanbul, Turkey.
- 30.Rich P, Sigurgeirsson B, Thaci D, et al. Secukinumab induction and maintenance therapy in moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled, phase II regimen-finding study. Br J Dermatol. 2013;168:402–411. doi: 10.1111/bjd.12112. [DOI] [PubMed] [Google Scholar]
- 31.Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190–1199. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 32.Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–1189. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- 33.Hu C, Wasfi Y, Zhuang Y, Zhou H. Information contributed by meta-analysis in exposure-response modeling: application to phase 2 dose selection of guselkumab in patients with moderate-to-severe psoriasis. J Pharmacokinet Pharmacodyn. 2014;41:239–250. doi: 10.1007/s10928-014-9360-6. [DOI] [PubMed] [Google Scholar]
- 34.Sofen H, Smith S, Matheson RT, et al. Guselkumab (an IL-23-specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J Allergy Clin Immunol. 2014;133:1032–1040. doi: 10.1016/j.jaci.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 35.Callis-Duffin K, Gordon K, Wasfi Y, Shen YK. A phase 2 multicenter, randomized, placebo- and active-comparator-controlled, dose-ranging trial to evaluate guselkumab for the treatment of patients with moderater to severe plaque-type psoriasis (X-PLORE) J Am Acad Dermatol. 2014;70(5 Suppl 1):AB162. Abstract P8353. [Google Scholar]
- 36.Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: Safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015 doi: 10.1016/j.jaci.2015.01.018. pii: S0091-6749(15)00108-6. doi: 10.1016/j.jaci.2015.01.018. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 37.Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159:1092–1102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiricozzi A, Krueger JG. IL-17 targeted therapies for psoriasis. Expert Opin Investig Drugs. 2013;22:993–1005. doi: 10.1517/13543784.2013.806483. [DOI] [PubMed] [Google Scholar]
- 39.Gaffen SL. Recent advances in the IL-17 cytokine family. Curr Opin Immunol. 2011;23:613–619. doi: 10.1016/j.coi.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii116–ii123. doi: 10.1136/annrheumdis-2012-202371. [DOI] [PubMed] [Google Scholar]
- 41.Tse MT. IL-17 antibodies gain momentum. Nat Rev Drug Discov. 2013;12:815–816. doi: 10.1038/nrd4152. [DOI] [PubMed] [Google Scholar]
- 42.Lynde CW, Poulin Y, Vender R, Bourcier M, Khalil S. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141–150. doi: 10.1016/j.jaad.2013.12.036. [DOI] [PubMed] [Google Scholar]
- 43.Papp KA, Langley RG, Sigurgeirsson B, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol. 2013;168:412–421. doi: 10.1111/bjd.12110. [DOI] [PubMed] [Google Scholar]
- 44.Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2:52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- 45.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]