

Abstract
Differential natural selection acting on populations in contrasting environments often results in adaptive divergence in multivariate phenotypes. Multivariate trait divergence across populations could be caused by selection on pleiotropic alleles or through many independent loci with trait-specific effects. Here, we assess patterns of association between a suite of traits contributing to life history divergence in the common monkey flower, Mimulus guttatus, and examine the genetic architecture underlying these correlations. A common garden survey of 74 populations representing annual and perennial strategies from across the native range revealed strong correlations between vegetative and reproductive traits. To determine whether these multitrait patterns arise from pleiotropic or independent loci, we mapped QTLs using an approach combining high-throughput sequencing with bulk segregant analysis on a cross between populations with divergent life histories. We find extensive pleiotropy for QTLs related to flowering time and stolon production, a key feature of the perennial strategy. Candidate genes related to axillary meristem development colocalize with the QTLs in a manner consistent with either pleiotropic or independent QTL effects. Further, these results are analogous to previous work showing pleiotropy-mediated genetic correlations within a single population of M. guttatus experiencing heterogeneous selection. Our findings of strong multivariate trait associations and pleiotropic QTLs suggest that patterns of genetic variation may determine the trajectory of adaptive divergence.
Keywords: adaptive divergence, bulk segregant analysis, flowering time, QTL, stolons
Introduction
Divergent natural selection occurring between populations in different habitats is a fundamental cause of phenotypic variation in species (Endler 1986; Schluter 2000; Grant & Grant 2008). However, the efficacy of natural selection in driving evolutionary responses in suites of traits will be influenced by the genetic architecture underlying the traits. Depending on the genetic architecture (i.e. the number of loci as well as the magnitude, type and direction of their allelic effects), the evolutionary trajectories of multivariate trait responses to spatially heterogeneous selection pressures may result in concerted change along particular phenotypic axes or more flexible patterns of change in individual characters (Via & Hawthorne 2002; Rajon & Plotkin 2013). Genetic correlations can arise due to pleiotropy, where alleles at single loci have effects on more than one trait under selection. Alternatively, alleles at multiple independent loci could combine or interact to produce divergent multitrait phenotypes (Mitchell-Olds et al. 2007).
The genetic basis of diversity in multivariate traits has repercussions for adaptation to heterogeneous selection pressures. Although both pleiotropy at individual loci (or loci in tight linkage) and linkage disequilibrium (LD) among many separate loci can facilitate adaptive divergence, LD of unlinked loci is far less likely to persist under a regime of varying selection or upon admixture of subdivided populations. Moreover, loci in LD are likely to evolve as a by-product of divergence, whereas pleiotropy of individual loci or loci in tight linkage may be involved in early responses to selection or even constrain evolution by preventing traits from responding independently to selection (Lande 1979, 1980; Lynch & Walsh 1998).
The impacts of spatially variable selection on multitrait phenotypic evolution are readily apparent in the divergence of annual and perennial ecotypes of the common monkey flower (Mimulus guttatus, Phrymaceae). The annual populations typically thrive in a Mediterranean soil moisture regime characterized by a wet autumn, winter and spring followed by intense summer drought. As such, annual plants flower early in the season and complete their life cycle before summer desiccation. In contrast, perennial populations are protected from summer drought by growing in soils that remain wet year-round. Perennials invest heavily in vegetative growth early in the growing season, produce horizontal spreading stems (stolons) from basal nodes and delay flowering until later in the summer (Hall & Willis 2006; van Kleunen 2007). Because the life history strategies constitute alternative multivariate phenotypes, adaptation to contrasting moisture regimes may involve a shared genetic basis for traits related to growth and reproduction.
Past studies of life history differences in M. guttatus have largely focused on flowering time (Hall & Willis 2006; van Kleunen 2007; Lowry et al. 2008; Hall et al. 2010) and used perennial plants from coastal populations that may be morphologically and genetically distinct from inland perennials (Hitchcock & Cronquist 1973; A. D. Twyford & J. Friedman, unpublished data). We are interested in rigorously evaluating the extent of phenotypic differentiation between annuals and perennials throughout the geographic range. Furthermore, to better understand the key features of perenniality, we place greater emphasis on stolons. Stolons are key to the perennial strategy as the following season's flowering shoots come from rosettes formed at the stolon tip and along the nodes of the stolon. A comparison of shoot architecture determined that an M. guttatus perennial population produced more primary branches at the expense of flowering than an annual population (Baker et al. 2012), due to changes in both meristem outgrowth and meristem fate. Previous quantitative trait locus (QTL) mapping studies of trait differences between annual and perennial ecotypes of M. guttatus have typically identified a few large QTLs underlying flowering time, with pleiotropic effects on other floral and vegetative traits (Hall et al. 2006; Lowry et al. 2008). By applying new mapping approaches using next-generation sequencing, we can generate genetic maps with high marker densities increasing the potential for mapping with greater precision. Although these approaches remain recombination limited, it may be possible to distinguish among competing models of genetic architecture underlying multitrait divergence.
Here, we evaluate the phenotypic and genetic relationships between multiple traits in annual and perennial populations of M. guttatus. Our first aim is to establish the extent of phenotypic associations among reproductive and vegetative traits in a very broad sample of populations of M. guttatus, using wild-collected individuals grown under common greenhouse conditions. In the light of our findings, our second aim is to understand whether correlations among traits that differ between annuals and perennials are due to individual QTL (either individual genes, or loci in tight linkage), or unlinked genes. We focus on two key traits related to vegetative growth (stolon number) and reproduction (flowering time), and obtain high-resolution QTL maps. We use a bulk segregant approach with Illumina sequencing and develop a novel computational pipeline for data analysis. Overall, our joint observation of phenotypic correlations among life history traits across natural populations and the genetic architecture of these traits provides new evolutionary and mechanistic insight into how multivariate ecotypic differences are maintained by selection in heterogeneous environments.
Methods
Study system
Mimulus guttatus, the common monkey flower (section Simiolus, Phrymaceae), is a widespread species with a native range extending across western North America (Vickery 1978). The species is self-compatible and predominantly outcrossing (Ritland 1989), with populations that are fully interfertile. Populations demonstrate marked differences in many phenotypes, including branching pattern, hair type, flower size, time to flowering, leaf shape, anthocyanin spotting and anther-stigma separation (Carr et al. 1997; van Kleunen 2007; Lowry et al. 2012). The species is variable in the production of stolons, which emerge as vegetative stems from basal nodes and subsequently root and flower, providing a means of clonal growth. The presence or absence of stolons is a key trait used to distinguish annual and perennial M. guttatus populations (Lowry et al. 2008) and has led to uncertainty in species delimitation, with various authors recognizing distinct varieties or additional taxa within M. guttatus sensu lato (Hitchcock & Cronquist 1973; Nesom 2012).
Plant material and morphological variation
We used wild-collected and open-pollinated seed material from 74 annual and perennial M. guttatus populations from across the species’ native range (Fig.1, Table S1, Supporting Information: 32 annual and 42 perennial). Sampling extended from 31.2° to 53.5° in latitude to −111.1° to −132.2° in longitude. We planted five seeds per maternal family in 6.5-cm pots filled with moist compost (Fafard 4P), stratified at 4 °C for 1 week and grew plants under 16-h days (21 °C day/18 °C night) in the Syracuse University greenhouses. We thinned the pots to one individual per maternal family, for a total of 274 plants (mean 3.7 individuals from unique maternal families per population). To measure phenotypic associations between growth and reproduction, we phenotyped plants for nine traits. We scored flowering time as the number of days between germination and the day the first corolla was fully expanded. We measured all other traits on the day of first flowering. We measured second internode length, stem width at the midpoint between second and third nodes, and leaf length at the third node. We counted the number of stolons (any horizontal stem), recorded the length of the longest stolon, counted the number of nodes to first flower and measured the corolla length and width of the first flower. All measurements were made to the nearest millimetre, except stem width, which was measured to the nearest half millimetre.
Fig. 1.
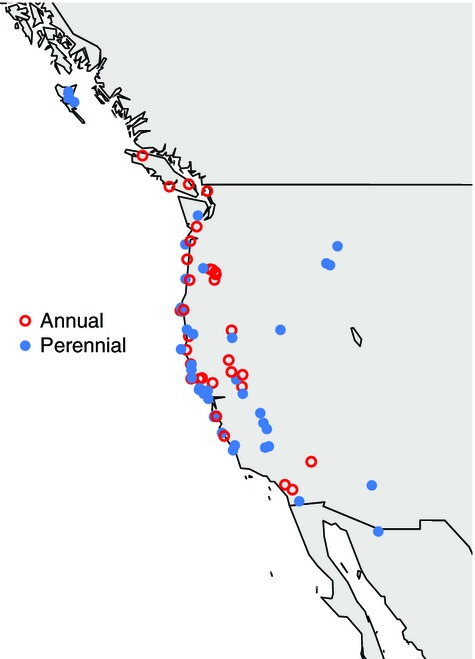
Map of Mimulus guttatus populations used for the analysis of phenotypic associations. The annual/perennial designation is based on field observations and confirmed in the greenhouse.
We calculated least-squares means (±SE) for each trait separately using a mixed model with life history as a fixed effect and population within life history as a random effect (proc mixed in sas, release 9.2, SAS Institute Inc., 2009, Cary, NC, USA). We transformed all variables to Z-scores, calculated Pearson pairwise correlation coefficients between all traits (PROC CORR) and performed a principal component analysis on this correlation matrix (PROC FACTOR). To determine whether there is overall multivariate morphological divergence between annual and perennial populations, we implemented a multivariate analysis of variance (manova) with all nine vegetative and flowering traits as the dependent variables and life history (annual or perennial) as the independent variable.
QTL mapping for flowering time and stolon number
To examine the genetic basis of flowering and vegetative strategies, we created a mapping population between two well-studied populations that differ in their timing and allocation to flowering and vegetative structures (Hall & Willis 2006; Hall et al. 2006; Lowry et al. 2008; Baker & Diggle 2011; Baker et al. 2012). We used the Iron Mountain (IM) population in Oregon's western cascades, where individuals are short-lived annuals that flower relatively early and rarely branch vegetatively, and the Oregon Dunes National Recreation Area (DUN) population, where individuals are perennials with numerous branches and flower relatively late (Hall & Willis 2006). We crossed one highly inbred individual from each population to create an F1 generation. A single F1 individual was selfed to create recombinant F2 progeny. Seeds from F2 DUNxIM individuals were sown in 2.5″ pots or 2.35″ × 1.45″ cell packs filled with moist Fafard 4P potting mix and stratified (4 °C, 1 week). Flats were then moved into an 18-h day greenhouse (21 °C day/18 °C night) at Duke University. Pots were thinned following germination to a single seedling per pot, so that 1280 individuals remained. Flowering time was scored as above. Stolon number was scored on a single day (once most plants had flowered) so that plants were at the same age.
Bulk segregant analysis
We performed a bulk segregant analysis (BSA; Michelmore et al. 1991; Magwene et al. 2011) to identify genomic regions containing loci affecting our traits of interest (flowering time and stolon number). BSA has long been used to quickly but crudely map major QTLs using traditional markers. High-throughput sequencing technologies provide genome-wide information at much reduced cost, allowing rapid quantification of allele frequencies at densely spaced markers and greater mapping precision (Magwene et al. 2011). For each trait, we selected the 48 individuals with the most extreme phenotypic values (i.e. earliest and latest flowering; and fewest and most stolons). Because many plants had zero stolons, we randomly selected 48 individuals for inclusion in this group. We collected a small fresh leaf per individual and pooled tissue from 12 individuals into a 50-mL tube (i.e. for each of the four phenotypic classes, we had four tubes with tissue from 12 individuals). Tissue was kept frozen at −80 °C until extraction. We ground tissue in liquid nitrogen and lysed cells in a urea-containing buffer (7 m urea, 0.35 m NaCl, 50 mm Tris, pH 8.0, 20 mm EDTA). We purified DNA by a series of phenol:chloroform extractions followed by isopropanol and ethanol precipitation. We pooled DNA for the four sets of 12 individuals from the same phenotype category by equal molarity, resulting in a single bulk each for early flowering, late flowering, few stolons and many stolons. Each genomic DNA pool was sequenced using single-end 75-bp reads in a single lane on an Illumina Genome Analyzer IIx by the University of North Carolina at Chapel Hill Genome Analysis Facility (Table S2, Supporting Information).
Statistical analysis
We developed a custom pipeline to perform SNP calling and read-based allele frequency analysis. We first aligned each FASTQ file to the M. guttatus reference genome (v 2.0, http://www.phytozome.net) using bwa (Li & Durbin 2010). The four alignments were then processed with samtools (Li et al. 2009) to generate an mpileup file from all the reads combined. In calling SNPs, we excluded positions with <4× or >40× coverage and positions where more than two alleles segregated or where either allele was observed in only one read. Following filtering (1 187 359 SNPs called), parental allele counts at each SNP position were counted separately for each alignment. Unmapped reads and reads with multiple equally likely best alignments were excluded, and reads overlapping multiple SNP positions were counted only once.
We compared allele frequencies between each pair of bulk DNAs (early vs. late flowering; few vs. many stolons). We used a sliding window approach to accommodate for low overall coverage and low read counts for any given SNP. Consistent parental genotype assignment within windows was facilitated because the IM parent inbred line is the genome reference line (IM62). SNPs were divided into intervals containing a minimum number of counted reads in each bulk. Different minimum counts per interval were used for each comparison (flowering bulks: 250; stolon bulks: 125) to account for differences in the overall read number and to yield equivalent numbers of intervals across comparisons (Table S2, Supporting Information). Allele frequencies were then calculated for sliding windows consisting of 10 intervals each (Table S2).
Differences in allele frequencies between two bulks are expected to be close to zero at markers unlinked to loci contributing to phenotypic divergence, while allele frequency differences will increase at markers closely linked to the underlying QTLs. A nonparametric test of allele frequency divergence between bulks that accounts for sampling effects of read coverage and bulk size was applied (Magwene et al. 2011). Briefly, we calculated a modified G-statistic for each interval, which was then smoothed by a weighted average procedure (G’) to yield a statistic for a sliding window. We then calculated Z-scores and P-values based upon an empirical estimate of an underlying log-normal null distribution derived from the observed data (Magwene et al. 2011). P-values were corrected for spatial autocorrelation (3-Mb windows, 3 kb step size) and multiple testing at a false discovery rate of 0.05 using comb-p (Pedersen et al. 2012).
Although BSA highlights the locations of QTLs, it does not provide information about the phenotypic effects of these regions. To determine the magnitude, type and direction of genotypic effects at each QTL, we genotyped 384 random F2s for known markers located within the QTL intervals. Because differences in power resulting from read coverage differences may have caused small effect QTLs detected in the flowering comparison to go undetected in the stolon comparison, we considered an allele frequency difference between bulks exceeding ±0.2 and a raw P-value below 0.05 in either bulk comparison as threshold criteria to select QTLs for this analysis. We first screened the inbred parental lines for length polymorphism using exon-primed intron-crossing (EPIC) markers. The development of these markers is outlined elsewhere (Fishman et al. 2002), and primers can be found at http://www.mimulusevolution.org. The PCR products were analysed on an ABI 3730xl DNA Analyser (Applied Biosystems, Foster City, CA, USA) and scored with GeneMarker (SoftGenetics, State College, PA, USA). We estimated relative effect size using general linear models in sas (proc mixed), with block included as a random factor. Marker effects on stolon number and flowering time were analysed separately, and we present the mean genotypic effect for one representative marker of three markers screened per interval.
Results
Phenotypic variation
Allocation to vegetative growth, flowers, and the timing of flowering vary tremendously between the two life histories (Table 1). We detect significant positive correlations between days to first flower and number (and length) of stolons, stem width and leaf size (i.e. plants that flower later have more, longer stolons, larger stems and larger leaves; Table 2). There is a very strong positive correlation between days to first flower and node of first flower, with later flowering individuals flowering at a more advanced node (Table 2). Furthermore, days to first flower are positively associated with both corolla length and width; individuals that flower later have larger flowers (Table 2).
Table 1.
Summary of the least-squares mean values for each phenotypic trait by life history
| Trait | Annual Mean ± SE | Perennial Mean ± SE |
|---|---|---|
| Days to flower | 31.37 ± 1.46 | 42.37 ± 1.27 |
| Nodes to flower | 3.25 ± 0.22 | 5.19 ± 0.19 |
| Corolla length | 20.99 ± 0.97 | 27.65 ± 0.84 |
| Corolla width | 23.77 ± 1.04 | 30.71 ± 0.90 |
| Internode length | 48.36 ± 4.99 | 29.11 ± 4.26 |
| Number of stolons | 0.77 ± 0.26 | 2.95 ± 0.22 |
| Stolon length | 34.55 ± 6.43 | 67.17 ± 3.77 |
| Stem thickness | 2.21 ± 0.19 | 4.18 ± 0.16 |
| Leaf length | 33.10 ± 1.79 | 39.50 ± 1.52 |
Table 2.
Pearson correlation coefficients for nine phenotypic traits in 74 populations of M. guttatus grown in the greenhouse under 16-h day lengths
| Days to flower | Nodes to flower | Corolla length | Corolla width | Internode length | Number of stolons | Stolon length | Stem thickness | Leaf length | |
|---|---|---|---|---|---|---|---|---|---|
| Days to flower | 0.846*** | 0.656*** | 0.679*** | −0.564*** | 0.686*** | 0.630*** | 0.763*** | 0.426*** | |
| Nodes to flower | 0.698*** | 0.675*** | −0.517*** | 0.820*** | 0.670*** | 0.832*** | 0.431*** | ||
| Corolla length | 0.955*** | −0.326* | 0.565*** | 0.496*** | 0.807*** | 0.589*** | |||
| Corolla width | −0.321* | 0.548*** | 0.516*** | 0.821*** | 0.598*** | ||||
| Internode length | −0.562*** | −0.502*** | −0.333* | −0.023 | |||||
| Number of stolons | 0.611*** | 0.662*** | 0.413*** | ||||||
| Stolon length | 0.621*** | 0.589*** | |||||||
| Stem thickness | 0.654*** | ||||||||
| Leaf length |
P < 0.05,
P < 0.001.
When we perform a multivariate principal component analysis, populations are largely distinguished along PC1, which accounts for 64% of the variation (Fig.2). The highest loading variables on PC1 include node of first flower (0.91), stem thickness (0.91), flowering time (0.88), corolla width (0.86), corolla length (0.85) and stolon number (0.81). PC2 accounts for 14.5% of variation and is largely comprised of internode distance (0.72) and leaf length (0.65). Although annual and perennial populations overlap considerably, there is a large space that is exclusively composed of perennials and a smaller space that is mostly comprised of annuals (Fig.2). Life history explains a significant proportion of the variation in PC1 (F1,271 = 168.43, P < 0.0001), but not in PC2 (F1,271 = 0.08, P > 0.5). Joint analysis of the nine flowering and vegetative traits showed a significant effect of life history (manova, F9,232 = 22.82, P < 0.0001). Considering days to flowering and stolon number as representative traits, it is clear that the distributions for annual and perennial populations are different, although there is considerable overlap (Fig.3).
Fig. 2.
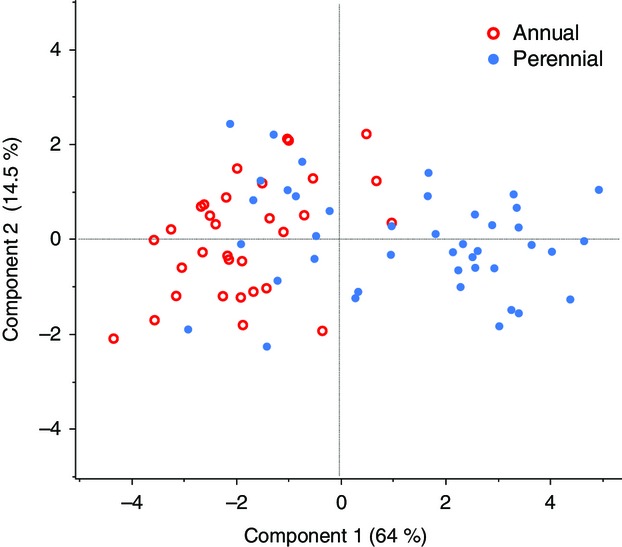
Principal component analysis on nine phenotypic traits in 74 populations of M. guttatus grown in the greenhouse under 16-h day lengths.
Fig. 3.
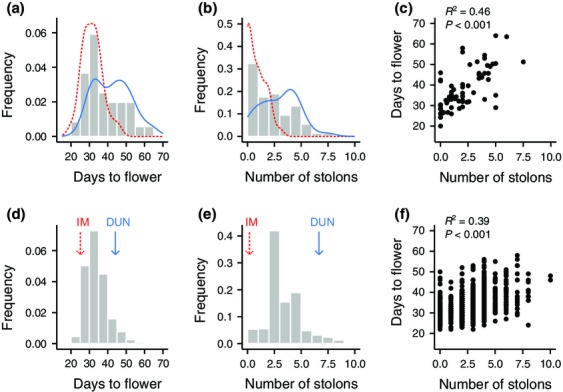
Distributions of days to flower (a) and stolons (b) in 74 populations of M. guttatus, measured in the greenhouse under 16-h day lengths. The lines depict the density curves for the annual (red-dashed) and perennial (blue-solid) populations separately. (c) depicts the relation between number of stolons and flowering time. (d, e, and f) illustrate the same variables in a mapping population of 1280 recombinant F2 plants from parents IM (annual) and DUN (perennial). The arrows in (d) and (e) represent the mean phenotypic trait for the annual IM parent (red-dashed) and perennial DUN (blue-solid) parent.
Genetic correlations and quantitative trait locus analyses
To investigate the genetic architecture underlying the phenotypic correlations between traits across populations, we scored 1280 recombinant DUNxIM F2 individuals for flowering time and stolon number. As observed for the natural accessions, days to flowering and stolon number are strongly positively correlated with the F2 population (R2 = 0.39, P < 0.001; Fig.3f), indicating that single, pleiotropic loci or groups of tightly linked loci with effects on both traits may be segregating in the population.
To formally assess this prediction, we mapped QTL for each trait using a bulk segregant analysis coupled with next-generation sequencing (Magwene et al. 2011). Many regions of the genome exhibited significant differences in allele frequency between bulks in each comparison, demonstrating that the genetic architecture of divergence in both traits is polygenic (Fig.4, Table S3, Supporting Information). We find remarkable similarity between the QTL maps constructed for flowering time and number of stolons. The observation of multiple overlapping QTL intervals is consistent with a shared genetic basis for variation in these traits (Fig.4). Correction for spatial autocorrelation and multiple testing confirms this pattern (Table S3, Supporting Information), but the QTL peaks span greater physical distances and are consequently less informative.
Fig. 4.

Shared genetic architecture for flowering and stolon number revealed through bulk segregant analysis. Allele frequency divergence (a) between early- vs. late-flowering (orange) or few vs. many stolon (green) DUNxIM F2 plants grown in a common 18-h greenhouse. The y-axis shows the allele frequency difference between the highest 5% and the lowest 5% pools, with positive values indicating a preponderance of alleles from the annual IM parent in the early-flowering or few stolon pools, and negative values indicating an excess of alleles from the perennial DUN parent in the early-flowering or few stolon pools. Dashed lines indicate allele frequency differences of ±0.2. G'-statistics for (b) flowering and (c) stolon number identify regions of high allele frequency divergence; solid and dashed lines indicate raw P-values of 0.05 and 0.01, respectively.
Single-marker analyses, which provide estimates of individual QTL effects and have greater power to test specific hypotheses as correction for genome-wide testing is not needed, confirmed that the QTL regions on linkage groups (LGs) 6, 7, 8 and 9 are significantly associated with both flowering time and number of stolons, with individual markers explaining 3–12% of the variation (Table 3). Another unique QTL region was found for flowering time (on LG 10), and two other regions (LG 13 and 14) were significantly associated with stolon production. Curiously, more than half of these QTLs have effects in the opposite direction to expectation given the difference in phenotype observed between the parents (Table 3). This may implicate complex patterns of epistasis between loci. However, no significant epistatic interactions among QTLs were detectable, even after genotyping an additional 384 individuals for several QTLs to gain statistical power (data not shown). Alternatively, for some QTL regions, our parental lines may have sampled alleles maintained by balancing selection as polymorphisms in one or both natural populations (Mojica et al. 2012). Epistasis or limited power to detect loci of small effect may explain why a few QTLs identified by BSA (e.g. LG2 for stolon number, LG13 for flowering time) did not have significant effects in the single-marker analysis. Several QTLs also overlap flowering-related QTLs previously identified in crosses within M. guttatus or between M. guttatus and M. nasutus (Zuellig et al. 2014).
Table 3.
Marker details and effect sizes for QTLs in Mimulus guttatus flowering time and stolon number mapping experiments. For each QTL identified by BSA, the marker that explained the largest proportion of variation is listed. QTLs that are significant based on single-marker analysis are in bold, note that the QTL on linkage groups 6, 7, 8 and 9 are significant for both flowering time and stolons. In every case, the same marker for that QTL (of the 3 tested) explained the largest proportion of variation for both traits. Superscripts following the mean phenotypes denote significant differences between the three genotypes following Dunn–Sidak correction, with different letters indicating P < 0.05
| Trait | Linkage group | Marker | Position (bp) | Proportion of variation explained | Test of association | Mean phenotype for G11 (IM allele) | Mean phenotype for G12 | Mean phenotype for G22 (DUN allele) |
|---|---|---|---|---|---|---|---|---|
| Flowering time | 2 | MgSTS617 | 2 505 570 | F2,252.9 = 0.02 | 33.96 | 33.81 | 33.82 | |
| 2 | MgSTS589 | 14 990 762 | F2,330 = 0.87 | 33.57 | 34.06 | 34.79 | ||
| 6 | MgSTS323 | 17 853 596 | 0.08 | F2,319.3 = 7.75*** | 36.15A | 34.10B | 32.65C | |
| 7 | MgSTS331 | 8 480 344 | 0.10 | F2,317.9 = 9.76*** | 35.45A | 34.32A | 31.59B | |
| 8 | MgSTS675 | 2 044 482 | 0.08 | F2,297.9 = 5.31** | 32.68A | 34.15B | 35.38B | |
| 9 | MgSTS481 | 8 149 927 | 0.12 | F2,336.8 = 14.35*** | 31.75A | 33.72A | 36.34B | |
| 10 | MgSTS109 | 1 819 700 | 0.09 | F2,233.3 = 4.73** | 35.33A | 34.65A | 32.43B | |
| 13 | MgSTS281 | 16 049 688 | F2,334.6 = 1.19 | 34.95 | 33.74 | 34.04 | ||
| 14 | MgSTS17 | 21 295 067 | F2,335.7 = 0.21 | 34.41 | 34.03 | 33.89 | ||
| Stolons | 2 | MgSTS617 | 2 505 570 | F2,269.1 = 0.14 | 2.74 | 2.66 | 2.62 | |
| 2 | MgSTS589 | 14 990 762 | F2,356 = 1.57 | 3.07 | 2.76 | 2.59 | ||
| 6 | MgSTS323 | 17 853 596 | 0.07 | F2,343 = 11.04*** | 3.52A | 2.68B | 2.34B | |
| 7 | MgSTS331 | 8 480 344 | 0.03 | F2,344.1 = 5.42** | 3.05A | 2.87A | 2.18B | |
| 8 | MgSTS675 | 2 044 482 | 0.05 | F2,321.6 = 5.80** | 2.24A | 3.01B | 2.90B | |
| 9 | MgSTS481 | 8 149 927 | 0.07 | F2,363 = 11.25*** | 1.94A | 2.88B | 3.20B | |
| 10 | MgSTS109 | 1 819 700 | F2,256 = 1.95 | 3.22 | 2.73 | 2.69 | ||
| 13 | MgSTS281 | 16 049 688 | 0.02 | F2,361.2 = 3.52* | 3.12A | 2.82AB | 2.43B | |
| 14 | MgSTS17 | 21 295 067 | 0.05 | F2,363.4 = 7.19** | 3.33A | 2.76B | 2.37B |
P < 0.05,
P < 0.01,
P < 0.001.
Discussion
We have explored the range-wide phenotypic differences, and the underlying genetic architecture of trait differences, for two contrasting life history strategies in Mimulus guttatus. We find a strong association between flowering time and investment in vegetative structures. Specifically, M. guttatus populations that flower early tend to make fewer or no stolons compared to populations that take more time to reach reproductive maturity. Early-flowering populations also invest less in other aspects of vegetative growth and make smaller flowers. We performed QTL mapping to determine whether our broad population-wide patterns could derive from a history of selection on unlinked loci individually affecting flowering time and vegetative traits in different populations, or arise from shared QTLs that have produced joint responses to divergent selection. Both bulk segregant and single-marker analyses reveal that genomic intervals with pleiotropic effects on flowering time and stolons are common, with at least four genomic regions affecting both traits. Here, we discuss our results in the context of the implications for adaptive divergence between populations, and the developmental and genetic causes of pleiotropy.
Environmental heterogeneity and divergent selection maintain ecotypic differentiation
In our study of 74 populations, we find strong positive correlations between flowering time and flower size, and also between flowering time and stolons, with plants that produce more stolons flowering later. These associations between growth and reproductive timing among populations are comparable to a genetic trade-off between flowering time and flower size detected within the annual M. guttatus population at Iron Mountain, Oregon. Mojica et al. (2012) showed that near-isogenic lines (NILs) possessing alleles for small flowers typically finish flowering early in the season before the onset of summer drought. However, they set fewer seed than NILs possessing alleles that increase flower size and delay flowering, which in turn risk not reproducing before summer drought. They show that the genetic trade-off for survival to flowering and fecundity is a direct consequence of differences among genotypes in the transition from vegetative growth to flowering.
Our findings suggest the same type of association in the transition from vegetative growth to flowering across populations. Although we could not assess fitness in our greenhouse experiment, previous reciprocal transplant experiments using IM, DUN and other annual/perennial population pairs have repeatedly found a strong fitness advantage for early flowering in the annual habitat and weak selection for later flowering in the perennial habitat (Hall et al. 2006, 2010; Lowry et al. 2008). Moreover, transplant experiments using NILs for a large inversion on LG8 that distinguishes annuals and perennials confirm this fitness trade-off occurs at the level of individual QTLs (Lowry & Willis 2010). Thus, we believe that early-flowering minimally vegetative plants and late-flowering large vegetative plants represent alternate strategies that have been selected for in environments that differ in the risk of summer drought.
To more fully understand the fitness consequences of the correlation between vegetative growth and flowering, it will be necessary to study how stolons and vegetative size contribute to fitness. Stolons facilitate the perennial lifestyle as the following season's flowering shoots come from rosettes formed at the stolon tip and at nodes along the stolon and thus are key to the lifetime fitness of the plant. Additionally, within a single growing season, the total reproductive output of perennials may be higher than annuals in M. guttatus, given that perennials produce larger flowers and can potentially flower from the main rosette and the stolons. This difference may be even more pronounced in natural conditions, where resources may be limited, than in our greenhouse experiment. If so, this would contradict the expectation than annual plants should have higher reproductive effort in a given season than perennial plants (Harper 1967; Hirshfield & Tinkle 1975; Primack 1979).
Proximal causes of association between growth and reproduction in M. guttatus
Trade-offs between growth and reproduction are generally attributed to resource limitation. However, in plants, a trade-off between growth and reproduction may arise via the pattern of meristem commitment during development. Watson (1984) first proposed a meristem cost as distinct from a resource cost, by showing that the cost of reproductive structures could not account for difference in size between flowering clones and branching clones. The observed association between stolon production and flowering in M. guttatus may partly involve differential allocation of meristems to vegetative and floral competency (Moody et al. 1999), which would result in a direct trade-off due to individual meristem fate (Geber 1990). The developmental pattern of meristem commitment to vegetative growth vs. reproduction is analogous to the regulation of bud development in Arabidopsis thaliana. During vegetative growth, buds are activated in a bottom-up gradient. After the floral transition, the gradient reverses and the most apical buds activate first, producing a top-down pattern of floral growth (McSteen & Leyser 2005). Similarly, reduced apical dominance prior to flowering has been shown to favour vegetative lateral shoots in other perennial plants (Kim & Donohue 2011). Thus, our discovery of QTLs with congruent effects on stolon number and flowering time may represent alternative outcomes for differences in the transition between vegetative and reproductive states. This result implies that a plant cannot simultaneously produce many early stolons and flower rapidly, regardless of available resources.
The genetic architectures of both stolon production and days to flowering are polygenic, similar to previous QTL mapping studies between annual and perennial M. guttatus, although we identify several new QTL regions relative to prior findings (Hall et al. 2006, 2010). Although there are some QTLs with trait-specific effects, most contribute to variation in both flowering time and stolon production. Similarly, although separate QTLs affect flowering time and vegetative growth in crosses between geographically disparate populations of the perennial species Arabidopsis lyrata, multiple QTLs had consistent effects on vegetative and reproductive allocation due to differences in early vegetative development (Remington et al. 2013). Thus, shared genetic architecture may reflect alternate developmental trajectories that have emerged due to spatially heterogeneous selection favouring divergent life history strategies. The joint QTL effects on flowering and stolon production in our study could result from a single-pleiotropic locus, or from selection on two or more loci that are maintained in linkage disequilibrium (Sinervo & Svensson 2002). If pleiotropic alleles are responsible for the different developmental strategies, then genetic correlations are much more likely to persist over time and to have played a major role in shaping the overall evolutionary trajectory (Lande 1979, 1980; Lynch & Walsh 1998).
Because the shared QTLs span large physical distances relative to the genome-wide recombination rate (∼3.9 cM/Mb; Holeski et al. 2014), further fine mapping is necessary to determine whether any impact on evolutionary trajectories would persist (i.e. single-pleiotropic allele) or could be eventually eliminated through recombination (i.e. trait-specific alleles at linked loci). Notably in this respect, the significant pleiotropic QTL on LG8 corresponds to a known recombination-suppressing inversion polymorphism (Lowry & Willis 2010). However, unlike in previous studies, this LG8 QTL does not explain the largest proportion of variation for either flowering time or stolon production. Various factors might play into this discrepancy. For one, previous efforts have used recombinant inbred line populations instead of F2s and therefore largely excluded heterozygous genotypes or backgrounds. In addition, our BSA approach reduces the number of genotypes sampled overall and thus may have greater power to pull out combinations of loci that have small or recessive individual effects or epistatic effects.
Candidate mechanisms for trait-specific and pleiotropic QTLs
Previous developmental studies have predicted that alleles responsible for differences in shoot architecture between annual and perennial M. guttatus will occur in genes that impact axillary meristem outgrowth or fate (Baker & Diggle 2011; Baker et al. 2012; Jorgensen & Preston 2014). Consistent with this prediction, clear candidate genes related to these processes colocalize with our QTLs in a manner consistent with QTL effects on stolon number and/or flowering time (Table S4, Supporting Information). For instance, MgMAX3, a homologue of carotenoid cleavage dioxygenase MORE AXILLARY GROWTH 3, colocalizes with the LG2 QTL identified only for stolon number. MAX3 homologues contribute to the production of strigolactones that inhibit branch outgrowth (Zou et al. 2006; Vogel et al. 2010; Janssen et al. 2014) and have been associated with dosage-dependent variation in branching (Booker et al. 2005; Ehrenreich et al. 2007). Most pertinently, differences in MgMAX3 expression between annual IM plants and perennial DUN plants correlate with differences in branching (Baker et al. 2012).
In contrast, regulators of axillary meristem fate are the strongest candidates located within QTLs with pleiotropic effects. A striking pattern is the frequent colocalization of FLOWERING LOCUS T/TERMINAL FLOWER 1 (FT/TFL1) homologues with QTLs associated with both traits (LG6, LG7, LG9, LG13) or with flowering time alone (LG10). Although best known for their role as systemic regulators of flowering time, members of this family interact antagonistically to more broadly set the balance between vegetative and floral developmental programmes, both at the shoot apex and in axillary buds (McGarry & Ayre 2012). Roles for FT/TFL1 homologues in modulating vegetative development have been demonstrated for shoot architecture in tomato (Shalit et al. 2009; Jiang et al. 2013), tuberization in potato (Navarro et al. 2011) and branching in A. thaliana (Niwa et al. 2013). Variation in FT/TFL1 homologues is often associated with natural variation in flowering time (e.g. Schwartz et al. 2009; Takahashi et al. 2009; Blackman et al. 2011). Additive or compensatory modulation of competing FT/TFL1 homologue activities on meristem fate provides a plausible evolutionary mechanism to explain our QTL observations.
Another well-known group of flowering time genes also regulates the fates of axillary meristems (Park et al. 2014; Yamaguchi et al. 2014). Active gibberellins, which promote inflorescence initiation and branch extension but inhibit floral fate, upregulate LEAFY (LFY) expression, but this then promotes expression of enzymes that catabolize these hormones. Consequently, DELLA proteins accumulate and interact with SQUAMOSA PROMOTER-BINDING PROTEIN-LIKE (SPL) transcription factors to promote floral fate. Genes homologous to regulators of gibberellin synthesis (LG8) and catabolism (LG8, LG13), a DELLA protein (LG8), LFY (LG9) and SPL-gene family members (LG13) are all found within pleiotropic QTLs, thus constituting a suite of mechanistically related candidates for divergence in flowering time and stolon number. Notably, the SPL3 homologue, MgSBP2, which colocalizes with the LG13 QTL, was recently implicated in differences in flowering and shoot architecture between annual and perennial M. guttatus (Jorgensen & Preston 2014).
Although we highlight candidate genes located in our QTLs homologous to genes with pleiotropic effects on reproductive timing and shoot architecture in other species, we recognize that our QTL regions are physically broad and contain many additional homologues of genes known to affect flowering time and/or axillary meristem outgrowth (Table S4, Supporting Information). Thus, although gene family and network relationships among certain candidates suggest plausible mechanisms for how single, pleiotropic alleles may affect flowering time and stolon number, further fine mapping and functional studies will be necessary to substantiate these possibilities and exclude control by tightly linked sets of genes with trait-specific effects.
Despite well over half a century of research, our understanding of the genetic architecture of quantitative traits and how they evolve under selection is tenuous; especially if we account for realistic scenarios including spatially fluctuating selection coupled with recurrent migration between environments. Furthermore, evidence is accumulating that pleiotropy is generally more pervasive than previously believed, and the challenge will be to catalogue the range of pleiotropic effects of individual alleles (Mackay et al. 2009). Empirical studies that demonstrate numerous pleiotropic QTL underlying fitness-related traits, as well as heterogeneous selection that favours divergent strategies across a species range, suggest that the mechanisms maintaining quantitative genetic variation are complex and varied.
Acknowledgments
We thank the numerous people who generously provided seed for wild populations (listed in Table S1, Supporting Information), and David Lowry for the F2 DUNxIM seed. We thank Lex Flagel for providing valuable input into the computational strategy for QTL mapping, Peter Nakagami for assistance with phenotyping and DNA extraction, John Kelly for helpful discussions, and Jeff Conner for comments on an earlier version of the manuscript. Funding for this project was provided by Syracuse University (JF), Natural Science and Engineering Research Council of Canada post-doctoral fellowship (JF), University of Virginia (BKB), a National Science Foundation Postdoctoral Fellowship in Biology to BKB (DBI-0905958), and National Science Foundation grant (IOS-1024966) to JHW. The article is funded by NERC (grant number: NE/L011336/1).
Data accessibility
Data uploaded to Dryad: doi:10.5061/dryad.76f45.
Trait measurements for wild populations grown in a greenhouse: traits_pops.xls.
Flowering time and stolon number for F2 mapping population: DUN_IM_F2.xls.
Genotype for single-marker analysis for 384 F2 individuals: F2_markers.xls.
Illumina sequence data from the bulk segregant analyses have been deposited in the NCBI Sequence Read Archive (Accession nos. SRX658835, SRX658836, SRX658838, SRX658839).
Supporting information
Additional supporting information may be found in the online version of this article.
Table S1 Location information for Mimulus guttatus populations used for the analysis of phenotypic trade-offs.
Table S2 Summary statistics for bulk segregant sequencing and analysis.
Table S3 QTL regions defined by comb-p following correction of P-values for spatial autocorrelation and multiple testing at a false discovery rate of 0.05.
Table S4 Candidate genes in QTL intervals.
References
- Baker RL, Diggle PK. Node-specific branching and heterochronic changes underlie population-level differences in Mimulus guttatus (Phrymaceae) shoot architecture. American Journal of Botany. 2011;98:1924–1934. doi: 10.3732/ajb.1100098. [DOI] [PubMed] [Google Scholar]
- Baker RL, Hileman LC, Diggle PK. Patterns of shoot architecture in locally adapted populations are linked to intraspecific differences in gene regulation. New Phytologist. 2012;196:271–281. doi: 10.1111/j.1469-8137.2012.04245.x. [DOI] [PubMed] [Google Scholar]
- Blackman BK, Rasmussen DA, Strasburg JL, et al. Contributions of flowering time genes to sunflower domestication and improvement. Genetics. 2011;187:271–287. doi: 10.1534/genetics.110.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker J, Sieberer T, Wright W, et al. MAX1 Encodes a cytochrome P450 family member that acts downstream of MAX3/4 to produce a carotenoid-derived branch-inhibiting hormone. Developmental Cell. 2005;8:443–449. doi: 10.1016/j.devcel.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Carr DE, Fenster C, Dudash MR. The relationship between mating-system characters and inbreeding depression in Mimulus guttatus. Evolution. 1997;51:363–372. doi: 10.1111/j.1558-5646.1997.tb02423.x. [DOI] [PubMed] [Google Scholar]
- Ehrenreich IM, Stafford PA, Purugganan MD. The genetic architecture of shoot branching in Arabidopsis thaliana: a comparative assessment of candidate gene associations vs. quantitative trait locus mapping. Genetics. 2007;176:1223–1236. doi: 10.1534/genetics.107.071928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endler JA. Natural Selection in the Wild. Princeton, New Jersey: Princeton University Press; 1986. [Google Scholar]
- Fishman L, Kelly AJ, Willis JH. Minor quantitative trait loci underlie floral traits associated with mating system divergence in Mimulus. Evolution. 2002;56:2138–2155. doi: 10.1111/j.0014-3820.2002.tb00139.x. [DOI] [PubMed] [Google Scholar]
- Geber MA. The cost of meristem limitation in Polygonum arenastrum: negative genetic correlations between fecundity and growth. Evolution. 1990;44:799–819. doi: 10.1111/j.1558-5646.1990.tb03806.x. [DOI] [PubMed] [Google Scholar]
- Grant PR, Grant BR. How and Why Species Multiply: The Radiation of Darwin's Finches. Princeton, New Jersey: Princeton University Press; 2008. [Google Scholar]
- Hall MC, Willis JH. Divergent selection on flowering time contributes to local adaptation in Mimulus guttatus populations. Evolution. 2006;60:2466–2477. [PubMed] [Google Scholar]
- Hall MC, Basten C, Willis JH. Pleiotropic quantitative trait loci contribute to population divergence in traits associated with life-history variation in Mimulus guttatus. Genetics. 2006;172:1829–1844. doi: 10.1534/genetics.105.051227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Lowry DB, Willis JH. Is local adaptation in Mimulus guttatus caused by trade-offs at individual loci? Molecular Ecology. 2010;19:2739–2753. doi: 10.1111/j.1365-294X.2010.04680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JL. A Darwinian approach to plant ecology. Journal of Applied Ecology. 1967;4:267–290. [Google Scholar]
- Hirshfield MF, Tinkle DW. Natural selection and the evolution of reproductive effort. Proceedings of the National Academy of Sciences, USA. 1975;72:2227–2231. doi: 10.1073/pnas.72.6.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock CL, Cronquist A. Flora of the Pacific Northwest. Seattle, Washington: University of Washington Press; 1973. [Google Scholar]
- Holeski LM, Monnhan P, Koseva B, et al. A high-resolution genetic map of yellow monkeyflower identifies chemical defense QTLs and recombination rate variation. Genes | Genomes | Genetics. 2014;4:813–821. doi: 10.1534/g3.113.010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen BJ, Drummond RSM, Snowden KC. Regulation of axillary shoot development. Current Opinion in Plant Biology. 2014;17:28–35. doi: 10.1016/j.pbi.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Jiang K, Liberatore KL, Park SJ, Alvarez JP, Lippman ZB. Tomato yield heterosis is triggered by a dosage sensitivity of the florigen pathway that fine-tunes shoot architecture. PLoS Genetics. 2013;9:e1004043. doi: 10.1371/journal.pgen.1004043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SA, Preston JC. Differential SPL gene expression patterns reveal candidate genes underlying flowering time and architectural differences in Mimulus and Arabidopsis. Molecular Phylogenetics and Evolution. 2014;73:129–139. doi: 10.1016/j.ympev.2014.01.029. [DOI] [PubMed] [Google Scholar]
- Kim E, Donohue K. Population differentiation and plasticity in vegetative ontogeny: effects on life-history expression in Erysimum capitatum (Brassicaceae) American Journal of Botany. 2011;98:1752–1761. doi: 10.3732/ajb.1100194. [DOI] [PubMed] [Google Scholar]
- van Kleunen M. Adaptive genetic differentiation in life-history traits between populations of Mimulus guttatus with annual and perennial life-cycles. Evolutionary Ecology. 2007;21:185–199. [Google Scholar]
- Lande R. Quantitative genetic analysis of multivariate evolution, applied to brain-body size allometry. Evolution. 1979;33:402–416. doi: 10.1111/j.1558-5646.1979.tb04694.x. [DOI] [PubMed] [Google Scholar]
- Lande R. The genetic covariance between characters maintained by pleiotropic mutations. Genetics. 1980;94:203–215. doi: 10.1093/genetics/94.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Willis JH. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biology. 2010;8:e1000500. doi: 10.1371/journal.pbio.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Rockwood RC, Willis JH. Ecological reproductive isolation of coast and inland races of Mimulus guttatus. Evolution. 2008;62:2196–2214. doi: 10.1111/j.1558-5646.2008.00457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Sheng CC, Lasky JR, Willis JH. Five anthocyanin polymorphisms are associated with an R2R3-MYB cluster in Mimulus guttatus (Phrymaceae) American Journal of Botany. 2012;99:82–91. doi: 10.3732/ajb.1100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sunderland, Massachusetts: Sinauer Associates Inc; 1998. [Google Scholar]
- Mackay TFC, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nature Reviews Genetics. 2009;10:565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- Magwene PM, Willis JH, Kelly JK. The statistics of bulk segregant analysis using next generation sequencing. PLoS Computational Biology. 2011;7:e1002255. doi: 10.1371/journal.pcbi.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry RC, Ayre BG. Manipulating plant architecture with members of the CETS gene family. Plant Science. 2012;188–189:71–81. doi: 10.1016/j.plantsci.2012.03.002. [DOI] [PubMed] [Google Scholar]
- McSteen P, Leyser O. Shoot branching. Annual Review of Plant Biology. 2005;56:353–374. doi: 10.1146/annurev.arplant.56.032604.144122. [DOI] [PubMed] [Google Scholar]
- Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proceedings of the National Academy of Sciences, USA. 1991;88:9828–9832. doi: 10.1073/pnas.88.21.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell-Olds T, Willis JH, Goldstein DB. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nature Reviews Genetics. 2007;8:845–856. doi: 10.1038/nrg2207. [DOI] [PubMed] [Google Scholar]
- Mojica JP, Lee Y-W, Willis JH, Kelly JK. Spatially and temporally varying selection on intrapopulation quantitative trait loci for a life history trade-off in Mimulus guttatus. Molecular Ecology. 2012;21:3718–3728. doi: 10.1111/j.1365-294X.2012.05662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody A, Diggle P, Steingraeber D. Developmental analysis of the evolutionary origin of vegetative propagules in Mimulus gemmiparus (Scrophulariaceae) American Journal of Botany. 1999;86:1512–1522. [PubMed] [Google Scholar]
- Navarro C, Abelenda JA, Cruz-Oró E, et al. Control of flowering and storage organ formation in potato by FLOWERING LOCUS T. Nature. 2011;478:119–122. doi: 10.1038/nature10431. [DOI] [PubMed] [Google Scholar]
- Nesom GL. Taxonomy of Erythranthe sect. Simiola (Phrymaceae) in the USA and Mexico. Phytoneuron. 2012;40:1–123. [Google Scholar]
- Niwa M, Daimon Y, Kurotani K-I, et al. BRANCHED1 interacts with FLOWERING LOCUS T to repress the floral transition of the axillary meristems in Arabidopsis. The Plant Cell. 2013;25:1228–1242. doi: 10.1105/tpc.112.109090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Eshed Y, Lippman ZB. Meristem maturation and inflorescence architecture—lessons from the Solanaceae. Current Opinion in Plant Biology. 2014;17:70–77. doi: 10.1016/j.pbi.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–2988. doi: 10.1093/bioinformatics/bts545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primack R. Reproductive effort in annual and perennial species of Plantago (Plantaginaceae) American Naturalist. 1979;114:51–62. [Google Scholar]
- Rajon E, Plotkin JB. The evolution of genetic architectures underlying quantitative traits. Proceedings of the Royal Society of London, Series B: Biological Sciences. 2013;280:20131552. doi: 10.1098/rspb.2013.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington DL, Leinonen PH, Leppälä J, Savolainen O. Complex genetic effects on early vegetative development shape resource allocation differences between Arabidopsis lyrata populations. Genetics. 2013;195:1087–1102. doi: 10.1534/genetics.113.151803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritland K. Correlated matings in the partial selfer Mimulus guttatus. Evolution. 1989;43:848–859. doi: 10.1111/j.1558-5646.1989.tb05182.x. [DOI] [PubMed] [Google Scholar]
- Schluter D. The Ecology of Adaptive Radiation. New York, New York: Oxford University Press; 2000. [Google Scholar]
- Schwartz C, Balasubramanian S, Warthmann N, et al. Cis-regulatory changes at FLOWERING LOCUS T mediate natural variation in flowering responses of Arabidopsis thaliana. Genetics. 2009;183:723–732. doi: 10.1534/genetics.109.104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalit A, Rozman A, Goldshmidt A, et al. The flowering hormone florigen functions as a general systemic regulator of growth and termination. Proceedings of the National Academy of Sciences, USA. 2009;106:8392–8397. doi: 10.1073/pnas.0810810106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinervo B, Svensson E. Correlational selection and the evolution of genomic architecture. Heredity. 2002;89:329–338. doi: 10.1038/sj.hdy.6800148. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Teshima KM, Yokoi S, Innan H, Shimamoto K. Variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proceedings of the National Academy of Sciences, USA. 2009;106:4555–4560. doi: 10.1073/pnas.0812092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via S, Hawthorne DJ. The genetic architecture of ecological specialization: correlated gene effects on host use and habitat choice in pea aphids. American Naturalist. 2002;159(Suppl 3):S76–S88. doi: 10.1086/338374. [DOI] [PubMed] [Google Scholar]
- Vickery RK. Case studies in the evolution of species complexes in Mimulus. Evolutionary Biology. 1978;11:405–507. [Google Scholar]
- Vogel JT, Walter MH, Giavalisco P, et al. SlCCD7 controls strigolactone biosynthesis, shoot branching and mycorrhiza-induced apocarotenoid formation in tomato. Plant Journal. 2010;61:300–311. doi: 10.1111/j.1365-313X.2009.04056.x. [DOI] [PubMed] [Google Scholar]
- Watson MA. Developmental constraints: effect on population growth and patterns of resource allocation in a clonal plant. American Naturalist. 1984;123:411–426. [Google Scholar]
- Yamaguchi N, Winter CM, Wu M-F, et al. Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis. Science. 2014;344:638–641. doi: 10.1126/science.1250498. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhang S, Zhang W, et al. The rice HIGH-TILLERING DWARF1 encoding an ortholog of Arabidopsis MAX3 is required for negative regulation of the outgrowth of axillary buds. Plant Journal. 2006;48:687–698. doi: 10.1111/j.1365-313X.2006.02916.x. [DOI] [PubMed] [Google Scholar]
- Zuellig MP, Kenney AM, Sweigart AL. Evolutionary genetics of plant adaptation: insights from new model systems. Current Opinion in Plant Biology. 2014;18:44–50. doi: 10.1016/j.pbi.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Location information for Mimulus guttatus populations used for the analysis of phenotypic trade-offs.
Table S2 Summary statistics for bulk segregant sequencing and analysis.
Table S3 QTL regions defined by comb-p following correction of P-values for spatial autocorrelation and multiple testing at a false discovery rate of 0.05.
Table S4 Candidate genes in QTL intervals.
Data Availability Statement
Data uploaded to Dryad: doi:10.5061/dryad.76f45.
Trait measurements for wild populations grown in a greenhouse: traits_pops.xls.
Flowering time and stolon number for F2 mapping population: DUN_IM_F2.xls.
Genotype for single-marker analysis for 384 F2 individuals: F2_markers.xls.
Illumina sequence data from the bulk segregant analyses have been deposited in the NCBI Sequence Read Archive (Accession nos. SRX658835, SRX658836, SRX658838, SRX658839).