

Abstract
Significance: Enhanced levels of reactive oxygen species (ROS) have been associated with different disease states. Most attempts to validate and exploit these associations by chronic antioxidant therapies have provided disappointing results. Hence, the clinical relevance of ROS is still largely unclear. Recent Advances: We are now beginning to understand the reasons for these failures, which reside in the many important physiological roles of ROS in cell signaling. To exploit ROS therapeutically, it would be essential to define and treat the disease-relevant ROS at the right moment and leave physiological ROS formation intact. This breakthrough seems now within reach. Critical Issues: Rather than antioxidants, a new generation of protein targets for classical pharmacological agents includes ROS-forming or toxifying enzymes or proteins that are oxidatively damaged and can be functionally repaired. Future Directions: Linking these target proteins in future to specific disease states and providing in each case proof of principle will be essential for translating the oxidative stress concept into the clinic. Antioxid. Redox Signal. 23, 1171–1185.
Introduction
Reactive oxygen species (ROS) regulate several essential physiological processes (63), including cell proliferation and differentiation, vascular tone, the innate immune response, and inflammation (4, 63). Conversely, aberrant ROS formation may trigger disease either by reaching concentrations that exceed cellular antioxidant defense mechanisms or by more subtle changes, such as ROS production in inappropriate cellular compartments (e.g., a subcellular localization that physiologically does not produce ROS) or a shift in the type of ROS being formed (e.g., superoxide instead of hydrogen peroxide) (32). This may then alone or in combination with other factors contribute to various cardiovascular, (45) neurological, or metabolic pathologies (48, 60, 147) or cancer (32, 137), all diseases with high socioeconomical impact and medical need (77).
Different pharmacological strategies have been pursued to prevent or restore such supposedly systemic redox imbalances and to improve disease outcomes, typically with antioxidant drugs or vitamins. However, intervention trials with small molecules, especially antioxidants, have been mostly ineffective (78) or even harmful (122). Hence, no direct antioxidant approach is currently part of any evidence guideline and the oxidative stress hypothesis still awaits validation in humans.
One crucial reason for these failures may reside in the dichotomy between disease-triggering and beneficial ROS and differences in this between humans and animal models of disease (132). To target redox-dependent diseases safely and effectively, physiological ROS sources that are relevant for signaling need to remain untouched, while disease-triggering ROS should be effectively reduced.
Instead of antioxidants, a more recent and innovative approach uses pharmacological agents that selectively suppress the activity of ROS-forming or toxifying enzymes (2, 3) whose activity or expression is increased under pathological conditions. These include the ROS generators, such as nitric oxide synthase (NOS), monoamine oxidase (MAO), xanthine oxidase (XO), and NADPH oxidase (NOX), or the ROS toxifier, myeloperoxidase (MPO) (Fig. 1). In addition, we will review a third and possibly synergistic strategy to functionally repair proteins that have been damaged by ROS (see related review by Dao et al. in this Forum on the New ROS Pharmacology). It is also possible to reinforce redox homeostasis by targeting the transcription factor Nrf2, a master regulator of the antioxidant control (122). We jointly review these ROS-based interventions of high clinical potential and place them into context. However, all targets included in this review are described at different levels based on their clinical relevance and maturity in drug development.
FIG. 1.

Sources and targets of reactive oxygen species. Largely beneficial enzymes or enzymes that downregulate cyclic GMP (cGMP) signaling include nitric oxide synthase, Fe(II) heme-containing guanylate cyclase; PKG, cGMP-dependent protein kinase; largely detrimental or cGMP downregulating enzymes, include uc-NOS, uncoupled, NOS apo-sGC converted from sGC; PDE, phosphodiesterases; NOX, NADPH oxidase; MPO, myeloperoxidase; Mito, mitochondria; LPO, lipid peroxidase; XO, xanthine oxidase converted from XD, xanthine dehydrogenase; MAO, monoamine oxidase. Antioxidant proteins include SOD, superoxide dismutase; Nrf2, nuclear factor (erythroid-derived 2)-like 2; ARE, antioxidant response element; T/Prx, Thio/Peroxyredoxin. Drugs are depicted next to oblique lines (arrows indicate activation; blocks, inhibition) either in bold (in clinical use: PDEi, PDE inhibitors; sGCs, sGC stimulators; NOd, NO donors; NOSr, NOS recoupling agents; MAOi, monoamine oxidase inhibitors; XODi, XOD inhibitors) or italics (in pre/clinical development: MPOi, MPO inhibitors; sGCa, sGC activators; NOSi, NOS inhibitors; Nrf2a, Nrf2 agonists; NOXi, NOX inhibitors). Proteins in the lower part indicate targets and biomarkers of cGMP and reactive oxygen species signaling: PPase, phosphatases; TRP, transient receptor potential channels; Tyr/Trp-NO2−, tyrosine or tryptophan-nitrated proteins; Tyr-Cl, tyrosine-chlorinated proteins (38a).
Comparison between pharmacological inhibition of enzymes and changes observed in knockout (KO) animals is detailed in the review. In most cases, a KO represents a de novo deficiency possibly leading to adaptive responses. However, most drug interventions are initiated after onset of the disease (see related review by Dao et al. in this Forum on the New ROS Pharmacology). Therefore, although both approaches are not always comparable, they are needed for targeting validation.
Physiology of ROS
While the existence of endogenous antioxidant enzymes (i.e., superoxide dismutase, glutathione peroxidase, catalase) suggests that the capacity to eliminate ROS is of evolutionary benefit (121), evidence that ROS fulfill equally essential physiological functions stems from the existence of the NOX enzyme family that has no other known function than to produce ROS (72). Table 1 lists the major enzymatic sources of ROS, ROS toxifiers, and their biological effects. Importantly, these physiological effects, which can be ascribed to a specific ROS source or toxifying enzyme, need to be kept in mind as potential side effects of enzyme inhibitors during chronic therapy.
Table 1.
Physiological Role of Reactive Oxygen Species Sources and Reactive Oxygen Species Toxifiers
| Enzyme | Function | Potential side effect |
|---|---|---|
| NOX1 | GI epithelial immune defense | GI infections |
| NOX2 | Innate immune response | CGD, immune suppression |
| NOX3 | Otoconia formation | Balance problems |
| NOX4 | Angiogenesis | Preconditioning, increased sensitivity to ischemic damage |
| NOX5 | Sperm motility | Male infertility, immunosuppression |
| DUOX | Thyroid hormone formation | Thyroid suppression |
| MPO | Immune defense | Immune suppression |
| XO | Catabolism of purines | Mild |
| MAO | Breakdown of neurotransmitters | GI diseases and skin reaction |
Potential side effects of drugs targeting these enzymes.
CGD, chronic granulomatous disease; DUOX, dual oxidase; GI, gastrointestinal; MAO, monoamine oxidase; MPO, myeloperoxidase; NOX, nicotinamide adenine dinucleotide phosphate oxidase; XO, xanthine oxidase.
From chemical antioxidants to defined enzyme targets
Antioxidants may have a benefit in acute parenteral treatment, but evidence in chronic therapy is lacking (132). One conceptual problem with the antioxidant approach is the fact that it overlooks that ROS also may have beneficial effects. Thus, scavenging ROS systemically may interfere with physiological as well as with pathological processes. However, three alternative approaches to ROS scavenging have been described, including the targeting of the relevant sources of ROS, ROS toxifiers, or repairing previously oxidized proteins (e.g., oxidized soluble guanylate cyclase [sGC] or endothelial nitric oxide synthase [eNOS]). In fact, targeting disease-relevant enzymatic sources of ROS is one of the most promising options. In this strategy, NOXs are a major target. All other enzymes generate ROS together with other products (e.g., MAO) or start to form ROS as a result of a biochemical accident induced by their proteolytic or oxidative modifications. (125a) The latter include XO (57), uncoupled endothelial NOS (uc-eNOS from eNOS) (95), and several mitochondrial enzymes, particularly respiratory chain complexes. Another important category of enzyme targets includes ROS toxifiers (70). We define them as enzymes that convert relatively nontoxic ROS such as hydrogen peroxide (H2O2) to more reactive species. A typical example is MPO, which converts H2O2 into hypochlorous acid (HOCl) (150). In a particular disease condition, specific inhibition of either a source of ROS or an ROS toxifier may become an effective and safe intervention. Recently, proof of principle has also been shown for a surprising third alternative, that is, the functional repair of oxidatively damaged proteins (see related review by Dao et al. in this Forum on the New ROS Pharmacology).
These three approaches hold great therapeutic promise for chronic therapy as long as they are optimally targeted, dosed, and leave physiological ROS formation intact. All are currently in clinical development with the aim to (i) prevent exacerbated ROS production by enzyme inhibition (NOX and XO); (ii) prevent the toxification of ROS such as H2O2 to secondary reactive products (e.g., by MPO); and (iii) promote functional repair of proteins that were damaged by ROS. However, these three approaches require thorough knowledge about the target proteins in addition to possible pharmacological inhibition (see related review by Dao et al. in this Forum on the New ROS Pharmacology). In this review, we focus on these target enzymes and the possible future clinical indications for drugs targeting them.
Disease-Relevant Enzymatic Sources of ROS
NADPH oxidases
NOXs are multiprotein complexes, which contain six or seven transmembrane-spanning domains (72). The NOX enzyme family contains seven members, NOX1-5 and dual oxidase (DUOX)1-2 (also termed NOX6-7). Each isoform has a particular pattern of activity regulation, tissue expression, type of ROS produced, and function (Table 2) (12, 81). The catalytic core of all NOXs contains one multimodular NADPH binding site at the C-terminus and a bimodular flavin adenine dinucleotide (FAD) binding site, as well as four conserved histidine residues involved in the binding of two heme moieties in the membrane. NOXs use NADPH as an electron donor and proximal or extracellular oxygen as an electron acceptor (27). Most NOX family members have similar redox centers (68) as well as the mechanism to generate O2− as the main product. However, NOX4 and DUOX produce H2O2 as their primary product (86, 126). Other membrane, cytosolic, and regulating domains are involved in NOX activity. While NOX1-3 needs docking of cytosolic factors for complete activation, NOX4 seems to produce ROS constitutively. Yet, NOX5 and DUOX are activated by elevated cellular Ca2+ concentrations via N-terminal EF-hand domains (12).
Table 2.
NOXs: Isoforms and Tissue Expression
| Isoforms | Tissue/cell expression | Loss of function | Primary ROS formed |
|---|---|---|---|
| NOX1 | Colon, aorta | Not reported (mouse KO has no obvious phenotype) | O2− |
| NOX2 | Phagocytes, endothelium | Susceptibility to infection (mouse, human), inflammation (mouse, rat, human) | O2− |
| NOX3 | Inner ear | Absence of otoconia (mouse, rat) | O2− |
| NOX4 | Kidney, almost all tissues | Not reported (mouse KO has no obvious phenotype) | H2O2 O2− |
| NOX5 | Spleen, testis, endothelium | Not reported (absent in rodents) | O2− |
| DUOX1/NOX6 | Thyroid, gland, lung, epithelia | Not reported (mouse KO has no phenotype) | H2O2 |
| DUOX2/NOX7 | Thyroid, gland, lung, epithelia | Hypothyroidism H2O2 (mouse, human) |
DUOX, dual oxidase; O2−, superoxide anion radical; H2O2, hydrogen peroxide; KO, knockout; NOX, nicotinamide adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species.
Because NOX is the only known enzyme family with the sole function to produce ROS (unlike XO, uc-eNOS, and mitochondria), it may represent the primary disease mechanism and thus targets for mechanism-based prevention of oxidative damage (12, 110). Moreover, some NOX isoforms are critically regulated by Ser/Thr kinases (e.g., PKC) (15), although PKC itself is upregulated by ROS (74). Of the seven isoforms, three are best studied: NOX1, NOX2, and NOX4. NOX3 appears to have a very limited organ-specific role both in physiology and pathophysiology. It is mostly expressed in the vestibular system of the inner ear where it controls the formation of otoconia, small biomineral particles (103). Mutations affecting NOX3 activity have not been described so far in humans, but its loss of function leads to severe imbalance in the head tilt mouse (69). NOX5 is not expressed in mice and rats and is thus understudied and remains the big unknown when it comes to translating animal data toward human pathology.
An involvement of NOX2 has been suggested in many disease states (76). The complete loss of function of NOX2 results in chronic granulomatous disease (CGD), which is characterized by susceptibility to certain fungal and bacterial infections (120). Foremost, CGD carriers are prone to developing autoimmune diseases, such as polyarthritis and lupus erythematosus (59, 120). Whether it may therefore become a safety risk for pharmacological inhibition of NOX2 and thereby compromise the innate immune response remains to be tested.
With respect to NOX1, it seems to be implicated in systemic hypertension (148). NOX1-deficient mice show a decreased angiotensin II-induced hypertensive response (42, 87). One study also found a significant effect of NOX1 deletion on basal blood pressure (43). Similarly, NOX1 overexpression potentiates angiotensin II-induced hypertension (31). NOX1-deficient mice were also protected from angiotensin II-induced aortic aneurysms (43) and diabetic vasculopathies (Fig. 2), both in the retina (147) and in large vessel atherosclerosis (48). Taken together, NOX1 may be involved in a whole range of vascular diseases and remodeling of the vascular wall.
FIG. 2.

Therapeutic indications for NOX inhibitors and potential unwanted side effects. Highly validated physiological and pathological roles of different NOX isoforms based on gene knockout experiments are based on original data in stroke (73), diabetic atherosclerosis (48), diabetic nephropathy (60), diabetic retinopathy (147), and osteoporosis (46a). For details, see text. Based on this, NOXi seem well suited in treating acute ischemic stroke and diabetic complications.
NOX4 is the most widely distributed isoform and can also contribute to diabetic end-organ damage, especially in the kidney (60). NOX4 is also upregulated under hypoxic conditions (94) and can function as an oxygen sensor (99). Pathologically, stroke is one of the best-validated disease indications for NOX4 inhibition (73, 109) (Fig. 2). Moreover, NOX4 expression and activity are strongly increased following TGF-β stimulation of human fibroblasts. This can lead to the transformation of not only normal fibroblasts into myofibroblasts, a key feature of wound healing, but also of chronic fibrotic diseases of the lungs, kidney, or liver. The fact that NOX4 inhibition mitigates myofibroblast transformation in vitro was first shown in cardiac and lung fibroblasts (28, 53) and later in the bleomycin model of pulmonary fibrosis using NOX4-deficient mice (23, 52). Interestingly, the benefit of NOX4 inhibition appears to be tissue specific as NOX4 deletion does not confer protection in urinary obstruction-induced kidney fibrosis (8), a condition where NOX4 inhibition even seems to be deleterious (100). Although the direct connection of NOX4-derived ROS and the tissue-specific fibroblast phenotypic changes are unclear, NOX4 inhibition in idiopathic lung fibrosis represents another promising indication for a pharmacological intervention targeting NOX4.
Finally, the NOX5 isoform, which is not expressed in mice or rats (11), is unique as it is directly activated by calcium (10) and may thus directly link cellular calcium overload to oxidative stress. In adults, NOX5 is found mostly in the spleen, lymph node, and the reproductive and vascular systems (39); in disease, it may play a role in coronary artery disease (49).
DUOX enzymes are expressed at high levels in the thyroid glands and generate H2O2 at the apical membrane. Mutations in DUOX2 and DUOXA2 lead to defects in thyroidal H2O2 generation, congenital hypothyroidism, and euthyroid goiter (61).
Despite the rich genetic evidence for distinct roles of different NOX isoforms (3), the development of isoform-specific inhibitors is lagging behind (see related review by Dao et al. in this Forum on the New ROS Pharmacology). The first generation of inhibitors was highly unspecific, that is, not even specific for NOX (56, 149), while, more recently, the achieved differences in IC50 of the second-generation NOX-specific compounds are hardly relevant in vivo (4). However, with the increasing interest in this target both in pharmaceutical and biotech industries, a third generation of inhibitors, specific and isoform selective, is on the horizon (see related review by Dao et al. in this Forum on the New ROS Pharmacology).
NOS
In this review, we consider nitric oxide (NO) a member of the ROS family. NO is generated from l-arginine, contains an unpaired electron (making it a free radical), and reacts with O2−• in a diffusion-limited manner to form the highly reactive peroxynitrite. Three NOS isoforms exist: NOS1 is predominantly present in the central and peripheral nervous system (thus it is also named nNOS); NOS2, in macrophages; and NOS3 in eNOS (123). Importantly, NOS3 has also to be considered as a target of ROS, leading to reversible uncoupling (see chapter below on proteins reversibly damaged by ROS).
Mice with genetic deficiencies in one of the NOS isoforms are viable. Nos1−/− mice show impaired cognitive performance (145), dramatic enlargement of the stomach, and significantly reduced brain damage after cerebral ischemia (84). Nos2−/− mice suffer from impaired host defense against pathogens and are prone to severe infections. However, they are protected from life-threatening hypotension in septic shock (84). Nos3−/− mice display impaired vasodilation, elevated blood pressure, diminished cardiac contractility (84), and, under stress conditions, impaired adaptation, for example, increased atherogenesis under high-fat diet and accelerated development of diabetic complications (18). Double knockout mice that lack both NOS1 and NOS3 display abnormalities in hippocampal long-term potentiation, a model for learning and memory (7). Triple knockout mice are severely insulin resistant (nephrogenic diabetes insipidus), display a number of cardiovascular risk factors, including hypertension and hypertriglyceridemia, and develop spontaneous myocardial infarction, supporting a critical role of NO in maintaining cardiovascular homeostasis (7, 138). Clinically, many reports suggested therapeutic benefit from inhibiting NOS1 or NOS2, for example, asthma (51), migraine (139), or cardiovascular diseases (CVDs) (2). Currently, the clinically most advanced therapeutic approach for NOS inhibition is in traumatic brain injury (133) (Fig. 3).
FIG. 3.

VAS203 treatment against brain traumatic injury. (A) NADPH donates electrons to the reductase domain of NOS. They are transferred via FAD and FMN to the oxygenase domain where they reduce heme-bound oxygen and an intermediate role of BH4. Activated oxygen oxidizes a guanidine nitrogen of l-arginine to produce NO and l-citrulline (1a). VAS203 is an analog of the physiological NOS cofactor tetrahydrobiopterin, which enables blockade of NOS activity. VAS203 exhibits more suitable properties than other classical arginine NOS inhibitors (i.e., L-N6-Nitroarginine methyl ester [L-NAME]). (B) VAS203-treated patients significantly reduced the therapeutic intensity level after a 6-day observation period. (C) The median level in the extended Glasgow Outcome Score after 6 months was 1.5 score points higher under VAS203 treatment compared with placebo (133). NOS, nitric oxide synthase. *Statistically significant difference between VAS203 (30 mg/kg) treatment and TL-placebo
XO
XO is defined as an enzyme activity; it utilizes oxygen as the electron acceptor to form reduced ROS (89) according to the following:
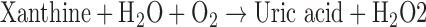 |
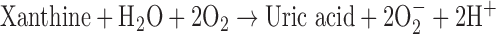 |
XO is derived from xanthine dehydrogenase (XDH, encoded by Xdh) by reversible sulfhydryl oxidation or by irreversible proteolytic modification (57, 98). As the terminal enzyme in the catabolism of purines, XDH activity utilizes NAD+ as the electron acceptor to convert hypoxanthine to xanthine and the latter to uric acid, according to the following:
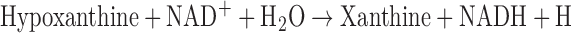 |
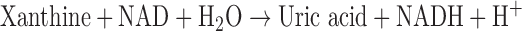 |
In some mammals, such as mice, uric acid is metabolized further by uricase to form allantoin. As XO may arise from XDH by sulfhydryl oxidation, XO activity can be a direct consequence of increased oxidative stress that further contributes to the pathogenesis of various diseases as a feed-forward mechanism of ROS-induced ROS.
Homozygous Xdh−/− mice show early neonatal lethality and display renal dysplasia (106), while heterozygous Xdh+/− mice have disrupted formation of the milk fat globule, underlining the importance of XDH to lactation (38). Reduced expression of Xdh in mice augments lipid accumulation in adipocytes, accompanied by an increase in oxidative stress, and induces obesity with insulin resistance in older age groups (96).
Inhibition of XO has been clinically applied for decades for the treatment of hyperuricemia and gout (35). In addition, xanthine oxidase inhibitors (XOi) has recently been explored for cardiovascular therapy (Fig. 4) based on animal (124, 135) and clinical studies in patients with type 2 diabetes and idiopathic dilated cardiomyopathy (21, 22). While several studies show clinical efficacy of XOi in CVD, others do not, such as in the case of heart failure patients with hyperuricemia (46). A possible explanation for these contradicting results is that XO inhibition might be a double-edged sword. Thus, while XO generates O2−/H2O2 as by-products, its final metabolite, uric acid, is also an antioxidant. Depending on the disease condition, one of these opposite effects of XO (ROS or antioxidant production) may prevail. This notion is consistent with the observation that plasma uric acid concentrations associate inversely with some diseases (144), whereas they are independently and significantly associated with other diseases (37).
FIG. 4.

Role of XO in ischemia–reperfusion-induced oxidative stress. During ischemia, ATP causes accumulation of its catabolite, hypoxanthine. Ischemia also induces conversion of XD into XO. During reperfusion, with oxygen available again, hypoxanthine is oxidized to uric acid, while molecular oxygen is concomitantly reduced to O2−. Scheme derived from (89).
MAO
MAOs are flavoenzymes (located at the outer mitochondrial membrane) that catalyze the oxidative deamination of both endogenous and exogenous amines, including neurotransmitters and several drugs. They exist as two isoforms, A and B, differing with respect to their tissue distribution, substrate preference, and inhibitor specificity (111). MAO-A reacts preferentially with tyramine, serotonin, and norepinephrine, while dopamine and phenylethylamine are preferential substrates for MAO-B. The imine products are coupled to the reduction of a covalently bound FAD, which in turn is reoxidized by oxygen leading to H2O2. In mitochondria, MAOs thus generate a significant percentage of total H2O2 in addition to that formed by the electron transport chain (5, 64). On the other hand, the imine product can also spontaneously hydrolyze, generating the corresponding aldehyde and ammonia (33, 111). Of note, all the three products of MAO catalysis are potentially toxic, especially at the level of mitochondria (64). In this regard, H2O2 and aldehydes can particularly synergize (62) leading to mitochondrial dysfunction. Moreover, they are directly related to endothelial dysfunction, heart function, and muscular dystrophy (Fig. 5). In addition, ammonia can stimulate further ROS formation by dihydrolipoyl dehydrogenase, the E3 component of pyruvate and oxoglutarate dehydrogenase (67).
FIG. 5.

Schematic representation of the mechanisms underlying the contribution of MAO activity to pathological conditions. Although the scheme focuses on cardiovascular and muscular diseases, especially heart failure, endothelial dysfunction, or muscular dystrophy, similar pathological mechanisms based upon mitochondrial dysfunction are likely to act also in other organs.
Patients and mice lacking MAO-A activity are characterized by borderline mental retardation and aggressive behavior (17, 19, 20), whereas polymorphisms in the MAO-A gene have been associated with bipolar disorder (40, 83, 114). On the other hand, variations in MAO-B activity in patients have been associated with psychotic disorders, depression, impulsivity, behavioral disinhibition, and attention-deficit/hyperactivity disorder (1, 82, 85, 112). MAO inhibitors have been used for the treatment of affective disorders and their mood-enhancing effect of MAO inhibition is likely related to an increased availability of serotonin, norepinephrine, and dopamine since their decrease is associated with depression (154). Deficit in both MAO-A and -B activity causes severe developmental and intellectual deficits, autistic-like behavior, and stereotypical movements (25, 97, 127, 128, 146).
The emphasis on MAO substrates (i.e., neurotransmitters) has curtailed the attention on the relevance of MAO products. Increased MAO-B activity has been correlated with Parkinson's disease (13, 66, 117). MAO expression increases in aging (88, 118) and an increased expression is associated with endothelial dysfunction (134), postoperative atrial fibrillation, muscular dystrophy (91), and prostate cancer (152). A common denominator among all these pathologies is altered ROS. In agreement with this, beneficial effects of MAO inhibition have been demonstrated in these conditions as well as in myocardial ischemia/reperfusion injury (14, 30), heart failure (62, 65, 140), and neurodegenerative disorders (16).
ROS Toxifiers
ROS toxifiers include different peroxidases such us eosinophil peroxidase, lactoperoxidase, and thyroid peroxidase. These enzymes share several similarities with their ortholog, MPO. However, here we mainly focus on MPO due to its clinical relevance and promising preclinical data.
MPO
MPO, a heme peroxidase present in circulating neutrophils, monocytes, and some tissue macrophages, plays an important role in killing invading microbes (71). MPO generates a number of reactive chlorinating and brominating oxidants, including nonradical species (two-electron oxidants) and radical species (50). In fact, MPO acts as a toxifier since in the presence of halides (Cl−, Br−), it transforms the relatively weak two-electron oxidant H2O2 into the more reactive hypohalous acids, (hypochlorous acid, [HOCl]; and hypobromous acid, [HOBr]) (150), as well as chloramines (104). MPO is also a major contributor to protein nitration since inflamed tissues of MPO-deficient mice contain significantly less 3-nitrotyrosine than those in wild-type mice (41). In addition to their role in the innate immune response, MPO-derived oxidants have the potential to cause host tissue injury by promoting post-translational protein modification (107, 150) and lipid oxidation (119). MPO was demonstrated to promote CVD and pharmacological inhibition or genetic deletion partially prevented these adverse effects (79, 141). However, it should be noted that MPO also fulfills an important role in host defense against pathogens and genetic deletion increased the severity of infections in animal models, although no clear increase in susceptibility to infections was observed in humans with MPO polymorphisms (34). Therefore, although MPO is detrimental in the context of CVDs, it also plays a major role in defense against pathogens, thus partial inhibition may be better than a complete blockage of the enzyme (see related review by Dao et al. in this Forum on the New ROS Pharmacology).
MPO is predominately located in inflamed tissue where it is found within or nearby infiltrated neutrophils and certain macrophages. Upon activation, phagocytes release MPO. In the case of circulating neutrophils, released MPO can bind to the endothelium, translocate, and be deposited in the subendothelial space (101). As a consequence of its localization and production of highly reactive oxidants, MPO is thought to contribute to a wide range of chronic inflammatory diseases as well as cardiovascular and neuroinflammatory diseases.
In addition to inhibiting MPO activity directly, an alternative therapeutic strategy is to displace MPO from the vascular endothelium and subendothelial space, that is, the sites where MPO released from circulating phagocytes is thought to bind to and reside. MPO binds to endothelial cells via heparin sulfate glycosaminoglycan, and heparin prevents and reverses such binding (9). The removal of MPO by heparin may help explain its anti-inflammatory actions. In fact, infusion of heparin increases the plasma concentration of MPO and increases flow-mediated dilatation (115), a major indicator of endothelial nitric oxide bioavailability.
Targeting MPO is in the early stages of clinical development, for example, as treatment for neurodegenerative, cardiovascular, and pulmonary diseases. Therefore, the next few years will be crucial in providing a definitive answer on whether inhibition of this toxifier enzyme is a valid strategy for preventing or alleviating various inflammatory diseases.
Proteins Damaged by ROS
In addition to preventing ROS-induced damage, its functional repair is much more than an option and has already entered clinical practice. A key example of this is impaired NO-cyclic guanosine monophosphate (cGMP) signaling.
uc-eNOS, NO scavenging, and apo-sGC
NO is an important cellular signaling molecule, which is involved in many physiological processes (58). NO production and ROS activate PKC, which contributes to cellular proliferation, neoplasia, and cancer (75, 113). Besides, NO interacts with different receptors (i.e., NMDAR and G-protein-coupled receptors) promoting the release of zinc ions from metallothioneins mediated by the nNOS/NO pathway (116). Accumulation of zinc is related to mood disorders, schizophrenia, and both neurological and neurodegenerative diseases (108).
However, almost all physiological effects of NO are mediated through its receptor enzyme, sGC, a heterodimeric heme protein comprising of a larger α subunit and a smaller heme-binding β subunit. Upon binding of NO to sGC heme, the conversion of guanosine-5′-triphosphate to the intracellular signaling molecule, cGMP, is activated. cGMP in turn regulates cGMP-dependent protein kinases and ion channels and is degraded by phosphodiesterases (29). The resulting effects include (acutely) inhibition of blood vessel contraction, improved perfusion, antithrombosis, neurotransmission, and memory formation, as well as (chronically) antiproliferation, antiremodeling, and anti-inflammation effects (123).
Oxidative stress can lead to the deregulation of NO-cGMP signaling (90) either by oxidizing and uncoupling NOS, by chemical scavenging of NO, or by oxidation and loss of heme in sGC. NOS3/eNOS, NOS1/nNOS (93), and to a lesser extent NOS2/iNOS (153) can be oxidatively damaged. This involves a highly redox-sensitive cofactor, tetrahydrobiopterin (H4B). In uc-NOS, oxygen activation is uncoupled from arginine-to-NO metabolism and NOSs become themselves ROS-forming enzymes; another example of ROS-induced ROS formation.
Impaired NO-sGC-cGMP signaling can thus be caused by reduced NO bioavailability and/or decreased responsiveness to NO and has been implicated in the pathogenesis of many cardiovascular, pulmonary, endothelial, renal, and neurological diseases (90, 148). Clinical evidence for a role of oxidative H4B depletion is based on recoupling and improvement of endothelial function in chronic smokers by BH4, but not by tetrahydroneopterin (H4N), which shares the antioxidant properties of H4B, but is not a cofactor for NOS3/eNOS (54, 55). Likewise, supplementation with the BH4 analog, folic acid, improves endothelial function in human subjects (6, 47). In fact, in experimental hypertension as well as atherosclerosis treatment with the H4B precursor, sepiapterin restores endothelial function (80, 125). In addition, many studies have reported a positive effect of l-arginine supplementation in endothelial dysfunction (136). This is surprising as arginine plasma levels by far exceed the Km of eNOS for l-arginine. A possible explanation for the protective effects of high-dose l-arginine administration may reside in the competition of l-arginine with an endogenous competitive inhibitor at the l-arginine binding site, asymmetric dimethyl l-arginine (ADMA), or the normalization of intracellular ADMA levels (24). Because of lower bioavailability of l-arginine in humans versus rodents, l-citrulline may be a better alternative and is subject to ongoing trials (Australian New Zealand Clinical Trials Registry ACTRN12609000882224).
In addition to NOS, ROS can also affect the bioavailability of NO by direct chemical scavenging and the redox state of sGC resulting in the oxidation of its heme iron to Fe3+ and/or ultimately in loss of the sGC heme (36, 131). The resulting apo-sGC is completely unresponsive to NO and rapidly degrades (92). Both pathomechanisms can be functionally reversed. So-called sGC stimulators sensitize sGC for lower NO concentrations to yield the same cGMP stimulatory effects as physiological NO levels would cause; sGC activators bind to the oxidized/heme-free form of sGC and reactivate the enzyme to the same Vmax levels as NO-stimulated heme-containing sGC.
sGC stimulators have entered the clinic. The PATENT-1 and PATENT-2 clinical trials in pulmonary arterial hypertension patients showed an increased walking distance (45). Moreover, the CHEST-1 trial in chronic thromboembolic pulmonary hypertension, for which otherwise pulmonary endarterectomy has been the only other curative option, showed improved exercise capacity, mean pulmonary artery pressure, cardiac output, and decreased clinically relevant pulmonary vascular resistance. A second sGC stimulator, vericiguat, is now in clinical development for different forms of heart failure (105). In addition, preclinical data suggest that sGC stimulators may be of benefit in chronic kidney disease (130) and hypertension (26, 129, 142, 143).
The development of sGC activators lags behind that of sGC stimulators due to initial pharmacokinetic setbacks (44), but preclinical data suggest benefit in cardiac hypertrophy (26) and type 2 diabetic nephropathy (102). Importantly, these effects seem to occur at doses that do not affect mean arterial pressure and heart rate and may thus involve preferential microvascular dilation.
Conclusions
For several decades redox imbalances have been suggested to have relevance in neurodegenerative, cardiovascular, metabolic, and neoplastic diseases. Therapeutically, most attempts to translate ROS scavenging by antioxidants into the clinic have yielded mostly disappointing results. However, pharmacological modulation of protein targets to either decrease ROS overproduction or toxification, as well as functional reversal of ROS-induced damage, has lead to several therapeutic breakthroughs (Table 3).
Table 3.
Pathological Role of Enzymatic Reactive Oxygen Species Sources and Their Current Clinical Status
| Target | Pathology | Current status of clinical translation |
|---|---|---|
| NOX | Type 2 diabetes mellitus associated with diabetic nephropathy (68) | Reduction in both liver enzyme and inflammatory marker levels, primary efficacy endpoints, albuminuria, not achieved (NCT 02010242) |
| NOS | Septic shock (91) | Failure in treatment against septic shock (91) |
| Asthma (58) | No improvement of respiratory functions (58) | |
| Acute migraine (66, 145) | Ineffective in the treatment of acute migraine (66, 145) | |
| Cardiogenic shock complicating acute myocardial infarction (2) | Did not reduce mortality in patients with refractory cardiogenic shock (2) | |
| Traumatic brain injury (139) | Phase II clinical trial complete | |
| XO | CVD (129, 141), type 2 diabetes (24, 25) | Improves endothelial function in patients with CVD (64) |
| Gout (12) | More effective than allopurinol in gout patients (12, 13, 27) | |
| MPO | Multiple sclerosis and COPD (28) | Phase I clinical trial for COPD and multiple sclerosis |
| Parkinson's disease (115) | Phase IIA clinical trial in patients with Parkinson's disease | |
| CVD (89) | Positive results in preclinical animal models (89) | |
| NOS | CVD | Reverses pulmonary hypertension (43) |
| sGC | PAH (18) | Entered in the clinic (18, 52) |
| Heart failure (112) | Phase II clinical trial. Still open—recruiting | |
| Acute heart failure (51) | Still in clinical development. Phase IIb clinical trial complete (40, 51) | |
| Acute heart failure (51) | Still in clinical development | |
| MAO | Parkinson's disease, dementia, and depression (5) | In the clinic |
| Parkinson's disease, dementia, and depression (5) | In the clinic |
CVD, cardiovascular disease; COPD, chronic obstructive pulmonary disease; MAO, monoamine oxidase; MPO, myeloperoxidase; NOS, nitric oxide synthase; NOX, nicotinamide adenine dinucleotide phosphate oxidase; PAH, pulmonary arterial hypertension; sGC, soluble guanylate cyclase; XO, xanthine oxidase.
Outlook
With the introduction of sGC stimulators for pulmonary hypertension, repurposing of XOi and monoamine oxidase inhibitors for ROS-related cardiovascular indications and the successful development of several new principles such as NOXi, MPOi, and NOSi into phase III translational ROS research are at the verge of major breakthroughs. Several of these candidate compounds are currently in clinical development and are likely to dramatically reshape the perception of the field of ROS and oxidative stress.
Abbreviations Used
- ADMA
asymmetric dimethyl-l-arginine
- apo-sGC
heme-free soluble guanylate cyclase
- H4B
tetrahydrobiopetrin
- cGMP
nicotinamide adenine dinucleotide phosphate
- CVD
cardiovascular disease
- CGD
chronic granulomatous disease
- DUOX
dual oxidase
- eNOS
endothelial nitric oxide synthase
- FAD
flavin adenine dinucleotide
- H2O2
hydrogen peroxide
- IC50
half-maximal inhibitory concentration
- iNOS
inducible nitric oxide synthase
- Km
Michaelis constant
- KO
knockout
- LPO
lipid peroxidase
- MAO
monoamine oxidases
- MPO
myeloperoxidase
- NAD
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- H4N
tetrahydroneopterin
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOS1
nitric oxide synthase 1
- NOS2
nitric oxide synthase 2
- NOX
nicotinamide adenine dinucleotide phosphate oxidases
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- O2−
superoxide anion
- PAH
pulmonary arterial hypertension
- ROS
reactive oxygen species
- sGC
soluble guanylate cyclase
- sGCa
soluble guanylate cyclase activators
- sGCs
soluble guanylate cyclase stimulators
- uc-eNOS
uncoupled endothelial nitric oxide synthase
- XO
xanthine oxidase
- XOi
xanthine oxidase inhibitors
- XDH
xanthine dehydrogenase
Acknowledgments
A.I.C., A.D., F.D.L., V.J., T.S., K.H.K., M.G.L., A.C., P.G., and H.H.H.W.S. were supported by the European Cooperation in Science and Technology (COST Action BM1203/EU-ROS). N.K. is supported by an EFSD/Sanofi Award. H.H.H.W.S. is the recipient of an ERC Advanced Grant and a Marie Curie IRG. R.S. is supported by a Senior Principal Research Fellowship from the National Health and Medical Research Council of Australia.
References
- 1.Adolfsson R, Gottfries CG, Oreland L, Wiberg A, and Winblad B. Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci 27: 1029–1034, 1980 [DOI] [PubMed] [Google Scholar]
- 1a.Alderton WK, Cooper CE, and Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 357: 593–615, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander JH, Reynolds HR, Stebbins AL, Dzavik V, Harrington RA, Van de Werf F, and Hochman JS. Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock: the TRIUMPH randomized controlled trial. JAMA 297: 1657–1666, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, and Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci 69: 2327–2343, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altenhofer S, Radermacher KA, Kleikers PW, Wingler K, and Schmidt HH. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal 23: 406–427, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson EJ, Efird JT, Davies SW, O'Neal WT, Darden TM, Thayne KA, Katunga LA, Kindell LC, Ferguson TB, Anderson CA, Chitwood WR, Koutlas TC, Williams JM, Rodriguez E, and Kypson AP. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc 3: e000713, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, and Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 114: 1193–1201, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Atochin DN. and Huang PL. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch 460: 965–974, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schroder K, and Brandes RP. Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 53: 842–853, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Baldus S, Eiserich JP, Mani A, Castro L, Figueroa M, Chumley P, Ma W, Tousson A, White CR, Bullard DC, Brennan M-L, Lusis AJ, Moore KP, and Freeman BA. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J Clin Invest 108: 1759–1770, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, and Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 276: 37594–37601, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Bedard K, Jaquet V, and Krause KH. NOX5: from basic biology to signaling and disease. Free Radic Biol Med 52: 725–734, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Bedard K. and Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Bialecka M, Klodowska-Duda G, Honczarenko K, Gawronska-Szklarz B, Opala G, Safranow K, and Drozdzik M. Polymorphisms of catechol-0-methyltransferase (COMT), monoamine oxidase B (MAOB), N-acetyltransferase 2 (NAT2) and cytochrome P450 2D6 (CYP2D6) gene in patients with early onset of Parkinson's disease. Parkinsonism Relat Disord 13: 224–229, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, Seguelas MH, Nistri S, Colucci W, Leducq N, and Parini A. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112: 3297–3305, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Bokoch GM, Diebold B, Kim JS, and Gianni D. Emerging evidence for the importance of phosphorylation in the regulation of NADPH oxidases. Antioxid Redox Signal 11: 2429–2441, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bortolato M, Chen K, and Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev 60: 1527–1533, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bortolato M. and Shih JC. Behavioral outcomes of monoamine oxidase deficiency: preclinical and clinical evidence. Int Rev Neurobiol 100: 13–42, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bressler J, Pankow JS, Coresh J, and Boerwinkle E. Interaction between the NOS3 gene and obesity as a determinant of risk of type 2 diabetes: the atherosclerosis risk in communities study. PLoS One 8: e79466, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunner HG, Nelen M, Breakefield XO, Ropers HH, and van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 262: 578–580, 1993 [DOI] [PubMed] [Google Scholar]
- 20.Brunner HG, Nelen MR, van Zandvoort P, Abeling NG, van Gennip AH, Wolters EC, Kuiper MA, Ropers HH, and van Oost BA. X-linked borderline mental retardation with prominent behavioral disturbance: phenotype, genetic localization, and evidence for disturbed monoamine metabolism. Am J Hum Genet 52: 1032–1039, 1993 [PMC free article] [PubMed] [Google Scholar]
- 21.Butler R, Morris AD, Belch JJ, Hill A, and Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension 35: 746–751, 2000 [DOI] [PubMed] [Google Scholar]
- 22.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marban E, and Hare JM. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation 104: 2407–2411, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, and Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal 15: 607–619, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Closs EI, Ostad MA, Simon A, Warnholtz A, Jabs A, Habermeier A, Daiber A, Forstermann U, and Munzel T. Impairment of the extrusion transporter for asymmetric dimethyl-l-arginine: a novel mechanism underlying vasospastic angina. Biochem Biophys Res Commun 423: 208–223, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Collins FA, Murphy DL, Reiss AL, Sims KB, Lewis JG, Freund L, Karoum F, Zhu D, Maumenee IH, and Antonarakis SE. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am J Med Genet 42: 127–134, 1992 [DOI] [PubMed] [Google Scholar]
- 26.Costell MH, Ancellin N, Bernard RE, Zhao S, Upson JJ, Morgan LA, Maniscalco K, Olzinski AR, Ballard VL, Herry K, Grondin P, Dodic N, Mirguet O, Bouillot A, Gellibert F, Coatney RW, Lepore JJ, Jucker BM, Jolivette LJ, Willette RN, Schnackenberg CG, and Behm DJ. Comparison of soluble guanylate cyclase stimulators and activators in models of cardiovascular disease associated with oxidative stress. Front Pharmacol 3: 128, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cross AR. and Segal AW. The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim Biophys Acta 1657: 1–22, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, and Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 97: 900–907, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Derbyshire ER. and Marletta MA. Structure and regulation of soluble guanylate cyclase. Annu Rev Biochem 81: 533–559, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Di Lisa F, Kaludercic N, Carpi A, Menabo R, and Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol 104: 131–139, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, and Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112: 2668–2676, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Edmondson DE, Mattevi A, Binda C, Li M, and Hubalek F. Structure and mechanism of monoamine oxidase. Curr Med Chem 11: 1983–1993, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, Castro L, Lusis AJ, Nauseef WM, White CR, and Freeman BA. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 296: 2391–2394, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Elion GB. The purine path to chemotherapy. Science 244: 41–47, 1989 [DOI] [PubMed] [Google Scholar]
- 36.Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, and Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov 5: 755–768, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang J. and Alderman MH. Serum uric acid and cardiovascular mortality the NHANES I epidemiologic follow-up study, 1971–1992. National Health and Nutrition Examination Survey. JAMA 283: 2404–2410, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Fini MA, Monks J, Farabaugh SM, and Wright RM. Contribution of xanthine oxidoreductase to mammary epithelial and breast cancer cell differentiation in part modulates inhibitor of differentiation-1. Mol Cancer Res 9: 1242–1254, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38a.Frijhoff J WPG, Stocker R, Cheng D, Davies SS, Knight A, Taylor E, Ruskovska T, Gasparovic AC, Oettrich J, Zarkovic N, Weber D, Poulsen HE, Cuadrado A, Grune T, Schmidt HH, and Ghezzi P. Clinical relevance of biomarkers of oxidative stress. Antioxid Redox Signal 23: 1144–1170, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal 11: 2443–2452, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furlong RA, Ho L, Rubinsztein JS, Walsh C, Paykel ES, and Rubinsztein DC. Analysis of the monoamine oxidase A (MAOA) gene in bipolar affective disorder by association studies, meta-analyses, and sequencing of the promoter. Am J Med Genet 88: 398–406, 1999 [PubMed] [Google Scholar]
- 41.Gaut JP, Byun J, Tran HD, Lauber WM, Carroll JA, Hotchkiss RS, Belaaouaj A, and Heinecke JW. Myeloperoxidase produces nitrating oxidants in vivo. J Clin Invest 109: 1311–1319, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, and Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580: 497–504, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Gavazzi G, Deffert C, Trocme C, Schappi M, Herrmann FR, and Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension 50: 189–196, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Gheorghiade M, Greene SJ, Filippatos G, Erdmann E, Ferrari R, Levy PD, Maggioni A, Nowack C, and Mebazaa A. Cinaciguat, a soluble guanylate cyclase activator: results from the randomized, controlled, phase IIb COMPOSE programme in acute heart failure syndromes. Eur J Heart Fail 14: 1056–1066, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, and Rubin LJ. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 369: 330–340, 2013 [DOI] [PubMed] [Google Scholar]
- 46.Givertz MM, Anstrom KJ, Redfield MM, Deswal A, Haddad H, Butler J, Tang WH, Dunlap ME, LeWinter MM, Mann DL, Felker GM, O'Connor CM, Goldsmith SR, Ofili EO, Saltzberg MT, Margulies KB, Cappola TP, Konstam MA, Semigran MJ, McNulty SE, Lee KL, Shah MR, and Hernandez AF. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation 131: 1763–1771, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46a.Goettsch C, Babelova A, Trummer O, Erben RG, Rauner M, Rammelt S, Weissmann N, Weinberger V, Benkhoff S, Kampschulte M, Obermayer-Pietsch B, Hofbauer LC, Brandes RP, and Schroder K. NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest 123: 4731–4738, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gori T, Burstein JM, Ahmed S, Miner SE, Al-Hesayen A, Kelly S, and Parker JD. Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: a human in vivo study. Circulation 104: 1119–1123., 2001 [DOI] [PubMed] [Google Scholar]
- 48.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, and Jandeleit-Dahm KA. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127: 1888–1902, 2013 [DOI] [PubMed] [Google Scholar]
- 49.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, and Harrison DG. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol 52: 1803–1809, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hampton MB, Kettle AJ, and Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92: 3007–3017, 1998 [PubMed] [Google Scholar]
- 51.Hansel TT, Kharitonov SA, Donnelly LE, Erin EM, Currie MG, Moore WM, Manning PT, Recker DP, and Barnes PJ. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics. FASEB J 17: 1298–1300, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, and Thannickal VJ. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 6: 231ra47, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, and Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, and Munzel T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res 86: E36–E41, 2000 [DOI] [PubMed] [Google Scholar]
- 55.Heitzer T, Krohn K, Albers S, and Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 43: 1435–1438, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, and Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51: 211–217, 2008 [DOI] [PubMed] [Google Scholar]
- 57.Hille R. and Nishino T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J 9: 995–1003, 1995 [PubMed] [Google Scholar]
- 58.Hou YC, Janczuk A, and Wang PG. Current trends in the development of nitric oxide donors. Curr Pharm Des 5: 417–441, 1999 [PubMed] [Google Scholar]
- 59.Hultqvist M, Olsson LM, Gelderman KA, and Holmdahl R. The protective role of ROS in autoimmune disease. Trends Immunol 30: 201–208, 2009 [DOI] [PubMed] [Google Scholar]
- 60.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, and Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25: 1237–1254, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson KR, Marden CC, Ward-Bailey P, Gagnon LH, Bronson RT, and Donahue LR. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol Endocrinol 21: 1593–1602, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Kaludercic N, Carpi A, Nagayama T, Sivakumaran V, Zhu G, Lai EW, Bedja D, De Mario A, Chen K, Gabrielson KL, Lindsey ML, Pacak K, Takimoto E, Shih JC, Kass DA, Di Lisa F, and Paolocci N. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid Redox Signal 20: 267–280, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaludercic N, Deshwal S, and Di Lisa F. Reactive oxygen species and redox compartmentalization. Front Physiol 5: 285, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, and Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol 73: 34–42, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, Pacak K, Kass DA, Di Lisa F, and Paolocci N. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 106: 193–202, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang SJ, Scott WK, Li YJ, Hauser MA, van der Walt JM, Fujiwara K, Mayhew GM, West SG, Vance JM, and Martin ER. Family-based case-control study of MAOA and MAOB polymorphisms in Parkinson disease. Mov Disord 21: 2175–2180, 2006 [DOI] [PubMed] [Google Scholar]
- 67.Kareyeva AV, Grivennikova VG, Cecchini G, and Vinogradov AD. Molecular identification of the enzyme responsible for the mitochondrial NADH-supported ammonium-dependent hydrogen peroxide production. FEBS Lett 585: 385–389, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawahara T, Quinn MT, and Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol 7: 109, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kiss PJ, Knisz J, Zhang Y, Baltrusaitis J, Sigmund CD, Thalmann R, Smith RJ, Verpy E, and Banfi B. Inactivation of NADPH oxidase organizer 1 results in severe imbalance. Curr Biol 16: 208–213, 2006 [DOI] [PubMed] [Google Scholar]
- 70.Klebanoff SJ. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J Bacteriol 95: 2131–2138, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klebanoff SJ. Oxygen metabolism and the toxic properties of phagocytes. Ann Intern Med 93: 480–489, 1980 [DOI] [PubMed] [Google Scholar]
- 72.Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S, Radermacher KA, Janssen B, Gorlach A, and Schmidt HH. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl) 90: 1391–1406, 2012 [DOI] [PubMed] [Google Scholar]
- 73.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, and Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8: pii: , 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Knapp LT. and Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem 275: 24136–24145, 2000 [DOI] [PubMed] [Google Scholar]
- 75.Korichneva I, Hoyos B, Chua R, Levi E, and Hammerling U. Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J Biol Chem 277: 44327–44331, 2002 [DOI] [PubMed] [Google Scholar]
- 76.Krause KH, Lambeth D, and Kronke M. NOX enzymes as drug targets. Cell Mol Life Sci 69: 2279–2282, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kreatsoulas C. and Anand SS. The impact of social determinants on cardiovascular disease. Can J Cardiol 26 Suppl C: 8c–13c, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lapchak PA. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert Opin Pharmacother 11: 1753–1763, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brummer J, Rudolph V, Munzel T, Heitzer T, Meinertz T, and Baldus S. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A 102: 431–436, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, and Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103: 1282–1288, 2001 [DOI] [PubMed] [Google Scholar]
- 81.Leto TL, Morand S, Hurt D, and Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal 11: 2607–2619, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li J, Wang Y, Hu S, Zhou R, Yu X, Wang B, Guan L, Yang L, Zhang F, and Faraone SV. The monoamine oxidase B gene exhibits significant association to ADHD. Am J Med Genet B Neuropsychiatr Genet 147: 370–374, 2008 [DOI] [PubMed] [Google Scholar]
- 83.Lim LC, Powell J, Sham P, Castle D, Hunt N, Murray R, and Gill M. Evidence for a genetic association between alleles of monoamine oxidase A gene and bipolar affective disorder. Am J Med Genet 60: 325–331, 1995 [DOI] [PubMed] [Google Scholar]
- 84.Liu VW. and Huang PL. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res 77: 19–29, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mann J. and Chiu E. Platelet monoamine oxidase activity in Huntington's chorea. J Neurol Neurosurg Psychiatry 41: 809–812, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, and Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18: 69–82, 2006 [DOI] [PubMed] [Google Scholar]
- 87.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, and Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation 112: 2677–2685, 2005 [DOI] [PubMed] [Google Scholar]
- 88.Maurel A, Hernandez C, Kunduzova O, Bompart G, Cambon C, Parini A, and Frances B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol Heart Circ Physiol 284: H1460–H1467, 2003 [DOI] [PubMed] [Google Scholar]
- 89.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312: 159–163, 1985 [DOI] [PubMed] [Google Scholar]
- 90.Melichar VO, Behr-Roussel D, Zabel U, Uttenthal LO, Rodrigo J, Rupin A, Verbeuren TJ, Kumar HSA, and Schmidt HH. Reduced cGMP signaling associated with neointimal proliferation and vascular dysfunction in late-stage atherosclerosis. Proc Natl Acad Sci U S A 101: 16671–16676, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, Reggiani C, Di Lisa F, Bonaldo P, and Canton M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum Mol Genet 19: 4207–4215, 2010 [DOI] [PubMed] [Google Scholar]
- 92.Meurer S, Pioch S, Pabst T, Opitz N, Schmidt PM, Beckhaus T, Wagner K, Matt S, Gegenbauer K, Geschka S, Karas M, Stasch JP, Schmidt HH, and Muller-Esterl W. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res 105: 33–41, 2009 [DOI] [PubMed] [Google Scholar]
- 93.Miller RT, Martasek P, Roman LJ, Nishimura JS, and Masters BS. Involvement of the reductase domain of neuronal nitric oxide synthase in superoxide anion production. Biochemistry 36: 15277–15284, 1997 [DOI] [PubMed] [Google Scholar]
- 94.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, and Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 101: 258–267, 2007 [DOI] [PubMed] [Google Scholar]
- 95.Montezano AC. and Touyz RM. Reactive oxygen species and endothelial function—role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin Pharmacol Toxicol 110: 87–94, 2012 [DOI] [PubMed] [Google Scholar]
- 96.Murakami N, Ohtsubo T, Kansui Y, Goto K, Noguchi H, Haga Y, Nakabeppu Y, Matsumura K, and Kitazono T. Mice heterozygous for the xanthine oxidoreductase gene facilitate lipid accumulation in adipocytes. Arterioscler Thromb Vasc Biol 34: 44–51, 2014 [DOI] [PubMed] [Google Scholar]
- 97.Murphy DL, Sims KB, Karoum F, de la Chapelle A, Norio R, Sankila EM, and Breakefield XO. Marked amine and amine metabolite changes in Norrie disease patients with an X-chromosomal deletion affecting monoamine oxidase. J Neurochem 54: 242–247, 1990 [DOI] [PubMed] [Google Scholar]
- 98.Nishino T. The conversion from the dehydrogenase type to the oxidase type of rat liver xanthine dehydrogenase by modification of cysteine residues with fluorodinitrobenzene. J Biol Chem 272: 29859–29864, 1997 [DOI] [PubMed] [Google Scholar]
- 99.Nisimoto Y, Diebold BA, Constentino-Gomes D, and Lambeth JD. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry 53: 5111–5120, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nlandu Khodo S, Dizin E, Sossauer G, Szanto I, Martin PY, Feraille E, Krause KH, and de Seigneux S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol 23: 1967–1976, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Odobasic D, Kitching AR, Yang Y, O'Sullivan KM, Muljadi RC, Edgtton KL, Tan DS, Summers SA, Morand EF, and Holdsworth SR. Neutrophil myeloperoxidase regulates T-cell-driven tissue inflammation in mice by inhibiting dendritic cell function. Blood 121: 4195–4204, 2013 [DOI] [PubMed] [Google Scholar]
- 102.Pullen SS, Lincoln KA, Harrison PC, Chen H, Wang H, Clifford H, Qian H, Wong D, Sarko C. Brenneman J, Fryer R, Richman J, Reinhart GA, and Boustany C. A soluble guanylate cyclase activator protects from diabetic nephropathy beyond standard of care in the ZSF1 rat. BMC Pharmacology and Toxicology 2015. 16(Suppl): A4 [Google Scholar]
- 103.Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, Russ A, Stumm G, Schimenti JC, and Bergstrom DE. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev 18: 486–491, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peskin AV, Midwinter RG, Harwood DT, and Winterbourn CC. Chlorine transfer between glycine, taurine, and histamine: reaction rates and impact on cellular reactivity. Free Radic Biol Med 37: 1622–1630, 2004 [DOI] [PubMed] [Google Scholar]
- 105.Pieske B, Butler J, Filippatos G, Lam C, Maggioni AP, Ponikowski P, Shah S, Solomon S, Kraigher-Krainer E, Samano ET, Scalise AV, Muller K, Roessig L, and Gheorghiade M. Rationale and design of the SOluble guanylate cyclase stimulatoR in heArT failurE Studies (SOCRATES). Eur J Heart Fail 16: 1026–1038, 2014 [DOI] [PubMed] [Google Scholar]
- 106.Piret SE, Esapa CT, Gorvin CM, Head R, Loh NY, Devuyst O, Thomas G, Brown SD, Brown M, Croucher P, Cox R, and Thakker RV. A mouse model of early-onset renal failure due to a xanthine dehydrogenase nonsense mutation. PLoS One 7: e45217, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Podrez EA, Abu-Soud HM, and Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic Biol Med 28: 1717–1725, 2000 [DOI] [PubMed] [Google Scholar]
- 108.Prakash A, Bharti K, and Majeed AB. Zinc: indications in brain disorders. Fundam Clin Pharmacol 29: 131–149, 2015 [DOI] [PubMed] [Google Scholar]
- 109.Radermacher KA, Wingler K, Kleikers P, Altenhofer S, Jr., Hermans J, Kleinschnitz C, and Hhw Schmidt H. The 1027th target candidate in stroke: will NADPH oxidase hold up? Exp Transl Stroke Med 4: 11, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Radermacher KA, Wingler K, Langhauser F, Altenhofer S, Kleikers P, Hermans JJ, Hrabe de Angelis M, Kleinschnitz C, and Schmidt HH. Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress. Antioxid Redox Signal 18: 1418–1427, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramsay RR. Monoamine oxidases: the biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr Top Med Chem 12: 2189–2209, 2012 [DOI] [PubMed] [Google Scholar]
- 112.Ribases M, Ramos-Quiroga JA, Hervas A, Bosch R, Bielsa A, Gastaminza X, Artigas J, Rodriguez-Ben S, Estivill X, Casas M, Cormand B, and Bayes M. Exploration of 19 serotoninergic candidate genes in adults and children with attention-deficit/hyperactivity disorder identifies association for 5HT2A, DDC and MAOB. Mol Psychiatry 14: 71–85, 2009 [DOI] [PubMed] [Google Scholar]
- 113.Rodriguez-Munoz M, de la Torre-Madrid E, Sanchez-Blazquez P, and Garzon J. NO-released zinc supports the simultaneous binding of Raf-1 and PKCgamma cysteine-rich domains to HINT1 protein at the mu-opioid receptor. Antioxid Redox Signal 14: 2413–2425, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rubinsztein DC, Leggo J, Goodburn S, Walsh C, Jain S, and Paykel ES. Genetic association between monoamine oxidase A microsatellite and RFLP alleles and bipolar affective disorder: analysis and meta-analysis. Hum Mol Genet 5: 779–782, 1996 [DOI] [PubMed] [Google Scholar]
- 115.Rudolph TK, Rudolph V, Witte A, Klinke A, Szoecs K, Lau D, Heitzer T, Meinertz T, and Baldus S. Liberation of vessel adherent myeloperoxidase by enoxaparin improves endothelial function. Int J Cardiol 140: 42–47, 2008 [DOI] [PubMed] [Google Scholar]
- 116.Sanchez-Blazquez P, Rodriguez-Munoz M, Bailon C, and Garzon J. GPCRs promote the release of zinc ions mediated by nNOS/NO and the redox transducer RGSZ2 protein. Antioxid Redox Signal 17: 1163–1177, 2012 [DOI] [PubMed] [Google Scholar]
- 117.Sandler M, Glover V, Clow A, and Jarman J. Monoamine oxidase-B, monoamine oxidase-B inhibitors, and Parkinson's disease. A role for superoxide dismutase? Adv Neurol 60: 238–241, 1993 [PubMed] [Google Scholar]
- 118.Saura J, Andres N, Andrade C, Ojuel J, Eriksson K, and Mahy N. Biphasic and region-specific MAO-B response to aging in normal human brain. Neurobiol Aging 18: 497–507, 1997 [DOI] [PubMed] [Google Scholar]
- 119.Savenkova ML, Mueller DM, and Heinecke JW. Tyrosyl radical generated by myeloperoxidase is a physiological catalyst for the initiation of lipid peroxidation in low density lipoprotein. J Biol Chem 269: 20394–20400, 1994 [PubMed] [Google Scholar]
- 120.Schappi MG, Jaquet V, Belli DC, and Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol 30: 255–271, 2008 [DOI] [PubMed] [Google Scholar]
- 121.Schieber M. and Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–R462, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmidt HH, Stocker R, Vollbracht C, Paulsen G, Riley DP, Daiber A, and Cuadrado A. Antioxidants in Translational Medicine. Antioxid Redox Signal 23: 1130–1143, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122a.This reference has been deleted. [Google Scholar]
- 123.Schmidt HH. and Walter U. NO at work. Cell 78: 919–925, 1994 [DOI] [PubMed] [Google Scholar]
- 124.Schroder K, Vecchione C, Jung O, Schreiber JG, Shiri-Sverdlov R, van Gorp PJ, Busse R, and Brandes RP. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic Biol Med 41: 1353–1360, 2006 [DOI] [PubMed] [Google Scholar]
- 125.Schuhmacher S, Wenzel P, Schulz E, Oelze M, Mang C, Kamuf J, Gori T, Jansen T, Knorr M, Karbach S, Hortmann M, Mathner F, Bhatnagar A, Forstermann U, Li H, Munzel T, and Daiber A. Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension 55: 897–904, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125a.Schulz E, Wenzel P, Munzel T, and Daiber A. Mitochondrial redox signaling: interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid Redox Signal 20: 308–324, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, and Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sims KB, de la Chapelle A, Norio R, Sankila EM, Hsu YP, Rinehart WB, Corey TJ, Ozelius L, Powell JF, Bruns G, et al. . Monoamine oxidase deficiency in males with an X chromosome deletion. Neuron 2: 1069–1076, 1989 [DOI] [PubMed] [Google Scholar]
- 128.Singh C, Bortolato M, Bali N, Godar SC, Scott AL, Chen K, Thompson RF, and Shih JC. Cognitive abnormalities and hippocampal alterations in monoamine oxidase A and B knockout mice. Proc Natl Acad Sci U S A 110: 12816–12821, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stasch JP, Dembowsky K, Perzborn E, Stahl E, and Schramm M. Cardiovascular actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41–8543: in vivo studies. Br J Pharmacol 135: 344–355, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stasch JP, Schlossmann J, and Hocher B. Renal effects of soluble guanylate cyclase stimulators and activators: a review of the preclinical evidence. Curr Opin Pharmacol 21c: 95–104, 2015 [DOI] [PubMed] [Google Scholar]
- 131.Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY. H S AK, Meurer S, Deile M, Taye A, Knorr A, Lapp H, Muller H, Turgay Y, Rothkegel C, Tersteegen A, Kemp-Harper B, Muller-Esterl W, and Schmidt HH. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest 116: 2552–2561, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol 101: 14d–19d, 2008 [DOI] [PubMed] [Google Scholar]
- 133.Stover JF, Belli A, Boret H, Bulters D, Sahuquillo J, Schmutzhard E, Zavala E, Ungerstedt U, Schinzel R, and Tegtmeier F. Nitric oxide synthase inhibition with the antipterin VAS203 improves outcome in moderate and severe traumatic brain injury: a placebo-controlled randomized Phase IIa trial (NOSTRA). J Neurotrauma 31: 1599–1606, 2014 [DOI] [PubMed] [Google Scholar]
- 134.Sturza A, Leisegang MS, Babelova A, Schroder K, Benkhoff S, Loot AE, Fleming I, Schulz R, Muntean DM, and Brandes RP. Monoamine oxidases are mediators of endothelial dysfunction in the mouse aorta. Hypertension 62: 140–146, 2013 [DOI] [PubMed] [Google Scholar]
- 135.Suzuki H, DeLano FA, Parks DA, Jamshidi N, Granger DN, Ishii H, Suematsu M, Zweifach BW, and Schmid-Schonbein GW. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc Natl Acad Sci U S A 95: 4754–4759, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sydow K. and Munzel T. ADMA and oxidative stress. Atheroscler Suppl 4: 41–51, 2003 [DOI] [PubMed] [Google Scholar]
- 137.Thanan R, Oikawa S, Hiraku Y, Ohnishi S, Ma N, Pinlaor S, Yongvanit P, Kawanishi S, and Murata M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int J Mol Sci 16: 193–217, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tsutsui M, Shimokawa H, Morishita T, Nakashima Y, and Yanagihara N. Development of genetically engineered mice lacking all three nitric oxide synthases. J Pharmacol Sci 102: 147–154, 2006 [DOI] [PubMed] [Google Scholar]
- 139.Van der Schueren BJ, Lunnon MW, Laurijssens BE, Guillard F, Palmer J, Van Hecken A, Depre M, Vanmolkot FH, and de Hoon JN. Does the unfavorable pharmacokinetic and pharmacodynamic profile of the iNOS inhibitor GW273629 lead to inefficacy in acute migraine? J Clin Pharmacol 49: 281–290, 2009 [DOI] [PubMed] [Google Scholar]
- 140.Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, De Paulis D, Couderc B, Spreux-Varoquaux O, Tortosa F, Garnier A, Knauf C, Valet P, Borchi E, Nediani C, Gharib A, Ovize M, Delisle MB, Parini A, and Mialet-Perez J. p53-PGC-1alpha pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal 18: 5–18, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.von Leitner EC, Klinke A, Atzler D, Slocum JL, Lund N, Kielstein JT, Maas R, Schmidt-Haupt R, Pekarova M, Hellwinkel O, Tsikas D, D'Alecy LG, Lau D, Willems S, Kubala L, Ehmke H, Meinertz T, Blankenberg S, Schwedhelm E, Gadegbeku CA, Boger RH, Baldus S, and Sydow K. Pathogenic cycle between the endogenous nitric oxide synthase inhibitor asymmetrical dimethylarginine and the leukocyte-derived hemoprotein myeloperoxidase. Circulation 124: 2735–2745, 2011 [DOI] [PubMed] [Google Scholar]
- 142.Wang-Rosenke Y, Mika A, Khadzhynov D, Loof T, Neumayer HH, and Peters H. Stimulation of soluble guanylate cyclase improves renal recovery after relief of unilateral ureteral obstruction. J Urol 186: 1142–1149, 2011 [DOI] [PubMed] [Google Scholar]
- 143.Wang-Rosenke Y, Mika A, Khadzhynov D, Loof T, Neumayer HH, and Peters H. Impact of biological gender and soluble guanylate cyclase stimulation on renal recovery after relief of unilateral ureteral obstruction. J Urol 188: 316–323, 2012 [DOI] [PubMed] [Google Scholar]
- 144.Weisskopf MG, O'Reilly E, Chen H, Schwarzschild MA, and Ascherio A. Plasma urate and risk of Parkinson's disease. Am J Epidemiol 166: 561–567, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Weitzdoerfer R, Hoeger H, Engidawork E, Engelmann M, Singewald N, Lubec G, and Lubec B. Neuronal nitric oxide synthase knock-out mice show impaired cognitive performance. Nitric Oxide 10: 130–140, 2004 [DOI] [PubMed] [Google Scholar]
- 146.Whibley A, Urquhart J, Dore J, Willatt L, Parkin G, Gaunt L, Black G, Donnai D, and Raymond FL. Deletion of MAOA and MAOB in a male patient causes severe developmental delay, intermittent hypotonia and stereotypical hand movements. Eur J Hum Genet 18: 1095–1099, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wilkinson-Berka JL, Deliyanti D, Rana I, Miller AG, Agrotis A, Armani R, Szyndralewiez C, Wingler K, Touyz RM, Cooper ME, Jandeleit-Dahm KA, and Schmidt HH. NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxid Redox Signal 20: 2726–2740, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, Neff C, Shah AM, Wingler K, and Schmidt HH. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension 56: 490–497, 2010 [DOI] [PubMed] [Google Scholar]
- 149.Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, and Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol 161: 885–898, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Winterbourn CC. and Kettle AJ. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radic Biol Med 29: 403–409, 2000 [DOI] [PubMed] [Google Scholar]
- 151.This reference has been deleted. [Google Scholar]
- 152.Wu JB, Shao C, Li X, Li Q, Hu P, Shi C, Li Y, Chen YT, Yin F, Liao CP, Stiles BL, Zhau HE, Shih JC, and Chung LW. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J Clin Invest 124: 2891–2908, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xia Y. and Zweier JL. Direct measurement of nitric oxide generation from nitric oxide synthase. Proc Natl Acad Sci U S A 94: 12705–12710, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Youdim MB, Edmondson D, and Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7: 295–309, 2006 [DOI] [PubMed] [Google Scholar]