

Abstract
Aim: Temporal and spatial variations in shear stress are intimately linked with vascular metabolic effects. Autophagy is tightly regulated in intracellular bulk degradation/recycling system for maintaining cellular homeostasis. We postulated that disturbed flow modulates autophagy with an implication in mitochondrial superoxide (mtO2•−) production. Results: In the disturbed flow or oscillatory shear stress (OSS)-exposed aortic arch, we observed prominent staining of p62, a reverse marker of autophagic flux, whereas in the pulsatile shear stress (PSS)-exposed descending aorta, p62 was attenuated. OSS significantly increased (i) microtubule-associated protein light chain 3 (LC3) II to I ratios in human aortic endothelial cells, (ii) autophagosome formation as quantified by green fluorescent protein (GFP)-LC3 dots per cell, and (iii) p62 protein levels, whereas manganese superoxide dismutase (MnSOD) overexpression by recombinant adenovirus, N-acetyl cysteine treatment, or c-Jun N-terminal kinase (JNK) inhibition reduced OSS-mediated LC3-II/LC3-I ratios and mitochondrial DNA damage. Introducing bafilomycin to Earle's balanced salt solution or to OSS condition incrementally increased both LC3-II/LC3-I ratios and p62 levels, implicating impaired autophagic flux. In the OSS-exposed aortic arch, both anti-phospho-JNK and anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) staining for DNA damage were prominent, whereas in the PSS-exposed descending aorta, the staining was nearly absent. Knockdown of ATG5 with siRNA increased OSS-mediated mtO2•−, whereas starvation or rapamycin-induced autophagy reduced OSS-mediated mtO2•−, mitochondrial respiration, and complex II activity. Innovation: Disturbed flow-mediated oxidative stress and JNK activation induce autophagy. Conclusion: OSS impairs autophagic flux to interfere with mitochondrial homeostasis. Antioxid. Redox Signal. 23, 1207–1219.
Introduction
Autophagy is an evolutionarily conserved process. Cellular components, including soluble and aggregated proteins, as well as organelles, are sequestered in autophagosomes and degraded in lysosomes to adapt to nutrient restriction or to eliminate modified macromolecules and damaged organelles (9, 25, 41, 42, 77). While autophagy is essential for cell survival, differentiation and development, dysfunctional autophagy is associated with a number of pathological conditions, including cardiovascular and neurodegenerative diseases, muscular dystrophies, and cancer (32, 48, 50). Impaired autophagy promotes vascular inflammatory responses and atherogenesis (70), myocardial contractile dysfunction, and heart failure (6, 32, 63). Increasing evidence supports reactive oxygen species (ROS), oxidized lipoproteins, and endoplasmic reticulum stress as autophagy inducers (35, 62, 77); however, the mechanotransduction mechanisms underlying hemodynamic shear stress and autophagy remain elusive.
Innovation.
Spatial and temporal variations in shear stress regulate vascular metabolic effects. We hereby elucidate the hemodynamic mechanisms underlying disturbed flow and impaired autophagic flux. We demonstrate that oscillatory shear stress (OSS) significantly increases microtubule-associated protein light chain 3 (LC3)-II/LC-I ratios and p62 expression, whereas pulsatile shear stress (PSS) minimally increases LC3 ratios and downregulates p62. In corollary, the OSS-exposed aortic arch is preferentially prominent for p62 accumulation in association with markers for JNK activation and mitochondrial DNA damage, as opposed to the PSS-exposed descending aorta. Thus, disturbed flow-associated OSS induces autophagy, but impairs autophagic flux to perturb mitochondrial homeostasis.
Autophagy is a highly regulated process associated with the activation of autophagy-related (ATG) genes (61). The initiation of autophagy is influenced by the UNC-51-like kinase (ULK)-Atg17 complex, which is inhibited by the mechanistic target of rapamycin (mTOR) (24). Inhibition of mTOR by rapamycin results in the activation of autophagy (24). The ATG8/microtubule-associated protein light chain 3 (LC3) family and ATG5 play key roles in autophagosome biogenesis by building protein scaffolds (3, 14, 68, 75) and by mediating expansion of the lipid membrane of the autophagosome (61). Conversion of LC3-I (ATG8) to LC3-II allows the anchorage of ATG8/LC3 to the phagophore and is essential for its expansion to form autophagosomes (61). Autophagy substrates are targeted for degradation by associating with p62/SQSTM1, a multidomain protein that functions as a selective autophagy receptor, which physically link autophagic cargo to ATG8/MAP1-LC3/GABARAP family members located on the forming autophagic membranes (71). Deficiency in autophagy leads to intracellular accumulation of p62, a marker for an incomplete autophagy process known as impaired autophagic flux (45).
Shear stress imparts both metabolic and mechanical effects on vascular endothelial cells (10), with a pathophysiological relevance in the focal and eccentric nature of atherosclerotic lesions (11, 18, 20, 21, 28, 34, 36, 37, 79). A complex flow profile develops at arterial bifurcations; namely, flow separation and migrating stagnation points create low and oscillatory shear stress (OSS). At the lateral walls of bifurcations, disturbed flow, including oscillatory flow (bidirectional and axially misaligned flow), preferentially induces oxidative stress to initiate atherosclerosis, whereas in the medial walls of bifurcations, pulsatile flow (unidirectional and axially aligned flow) downregulates inflammatory responses and oxidative stress (33). Specifically, pulsatile shear stress (PSS) increased endothelial mitochondrial membrane potential (ΔΨm), accompanied by a decrease in mitochondrial superoxide production (mtO2•−) (51), whereas OSS and oxidized low-density lipoprotein (ox-LDL) increased mtO2•− production and apoptosis (73, 74). In this context, spatial (∂τ/∂x) and temporal (∂τ/∂t) variations in shear stress largely determine the focal nature of vascular oxidative stress and proinflammatory state.
Oxidative stress is considered to be a major inducer of autophagy (35, 41). ox-LDL induces autophagy accompanied by mitochondrial DNA (mtDNA) damage (22). Unlike nuclear DNA, the lack of protective histones in mitochondria renders mtDNA vulnerable to oxidative stress (76). A significantly higher incidence of mtDNA with 4977 bp deletion mutation is present in circulating blood cells and atherosclerotic lesions (7). ApoE-null mice deficient in protein kinase ATM (ataxia telangiectasia mutated) also exhibit an increase in frequency of mtDNA damage and a decrease in oxidative phosphorylation, leading to defects in cellular proliferation and initiation of atherosclerosis (60). OSS activates NADPH oxidase to increase cytosolic superoxide (O2•−) production, which in turn activates c-Jun NH2-terminal kinase (JNK), leading to mtO2•− production (19, 73, 74). Increased mtO2•− production impairs the electron transport chain and promotes mtDNA damage (38).
While biomechanical forces have been reported to induce autophagy (42), whether spatial and temporal variations in shear stress modulate autophagy to influence mtO2•− production remains undefined. We demonstrated prominent staining for p62, a reverse marker of autophagic flux, in the disturbed flow-exposed aortic arc, but attenuated p62 in the PSS-exposed descending aorta. OSS significantly increased the LC3-II/LC3-I ratios and p62 levels, whereas PSS minimally increased LC3 ratios and decreased P62 expression. Both anti-phospho-JNK and anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) staining for DNA damage were prominent in the OSS-exposed aortic arch, whereas the staining was nearly absent in the PSS-exposed descending aorta. Our results indicate that OSS-mediated oxidative stress and JNK activation induced autophagy, but impaired autophagic flux to promote mtO2•− production, mtDNA damage, and mitochondrial dysfunction in the disturbed flow-exposed regions.
Results
Spatial variations in shear stress modulate p62/SQSTM1 immunostaining in rabbit aorta
Atherosclerosis preferentially develops in the branch points and curvature of arterial trees where disturbed flow, including OSS, occurs (11, 18, 28, 36, 79). To examine whether there is a relationship between spatial variations in shear stress and autophagy, we stained cross sections of the rabbit aortic arch and descending aorta with an antibody against the autophagy-related gene p62, an ubiquitin-binding scaffold protein to promote autophagosome formation. Computational fluid dynamics (CFD) was employed to illustrate both spatial (∂τ/∂x) and temporal variations (∂τ/∂t) in wall shear stress at an instantaneous moment in systole and diastole (Fig. 1A). Immunohistochemistry revealed prominent endothelial p62 staining in the OSS-exposed aortic arch (Fig. 1B), but p62 staining was nearly absent in the PSS-exposed descending aorta (Fig. 1C) (65, 72). This observation suggests a link between spatial variations in shear stress and p62 staining and provides a basis to determine whether temporal variations in shear stress (∂τ/∂t), namely OSS versus PSS, differentially induce autophagy.
FIG. 1.

Spatial variations in shear stress differentially increased p62 accumulation in rabbit aorta. (A) Cross section in rabbit aorta highlights instantaneous spatial variations in WSS in the aortic arch versus descending aorta during systole. The instantaneous WSS is low in the lesser curvature of aortic arch, but is circumferentially high in the descending aorta. Immunohistochemistry reveals more prominent p62 staining in the lesser curvature of aortic arch (B) where ECs were exposed to disturbed flow, including OSS, when compared with the descending aorta (C) where ECs were exposed to PSS. ECs, endothelial cells; OSS, oscillatory shear stress; PSS, pulsatile shear stress; WSS, wall shear stress. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Temporal variations (PSS vs. OSS) in shear stress modulate LC3-II to LC3-I ratios and autophagosome formation
Human aortic endothelial cells (HAECs) were exposed to OSS versus PSS for 4 h, and autophagy was assessed by normalizing the LC3-II to LC3-I ratios to the static condition. The LC3-II to LC3-I ratios significantly increased in response to OSS by 120% (p<0.001 vs. static control, n=5) and 77% over PSS (p<0.001, n=5), whereas PSS minimally increased LC3 ratios by 23% (p<0.05, n=5) (Fig. 2A). To further assess the completion of the autophagy process, we examined the p62 levels, a reverse marker for autophagy flux. OSS increased p62 by 37% (p<0.01, n=4), whereas PSS did not significantly change p62 level (Fig. 2A). In parallel, OSS significantly increased autophagosome formation, as illustrated by green fluorescent protein (GFP)-LC3 puncta or dots/cell, but not PSS (Fig. 2B, arrows). Earle's balanced salt solution (EBSS) to induce autophagy by starvation also increased GFP-LC3 puncta (Fig. 2B). Our data indicate that OSS significantly induced autophagy by nearly twofold and thus provided a basis to focus on OSS modulation of autophagy.
FIG. 2.

Temporal variations in shear stress (OSS vs. PSS) differentially induced autophagy in HAECs. (A) HAECs were exposed to OSS versus PSS for 4 h. Autophagy was assessed by normalizing the ratios of LC3-II to LC3-I with the static condition and comparing p62 levels by Western blotting. OSS significantly increased the LC3-II/LC3-I ratios (p<0.001, n=5) when compared with static condition and PSS, whereas PSS minimally increased LC3 ratios (p<0.05, n=5) in comparison with static condition. OSS also increased protein levels of p62 against static condition and PSS (p<0.01, n=4), whereas there was no significant change between static condition and PSS. (B) HAECs were infected with recombinant LC3-GFP adenovirus (1:20) overnight, and then were exposed to OSS versus PSS for 4 h. LC3-GFP puncta or dots/cell (arrows) indicated that OSS significantly increased the number of autophagosomes in comparison with static control (p<0.01, n=3) similarly to EBSS-induced autophagy (p<0.01, n=3). PSS only minimally increased LC3-GFP puncta versus static condition (p<0.05, n=3). (C) HAECs were treated with either OSS or EBSS in the presence or absence of 1 nM of bafilomycin (Baf). While EBSS increased LC3 ratios (p<0.05, n=6) and decreased p62 levels (p<0.05, n=6) over static condition, bafilomycin treatment significantly increased LC3 ratios and p62 to a lesser extent in OSS-exposed cells (LC3: p<0.05, n=6; p62: p<0.01, n=6) than EBSS-treated cells (LC3: p<0.001, n=6; p62: p<0.001, n=6). EBSS, Earle's balanced salt solution; GFP, green fluorescent protein; HAEC, human aortic endothelial cell; LC3, microtubule-associated protein light chain 3. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
OSS impairs autophagic flux
To further determine OSS modulation of autophagic flux, we compared the LC3 ratios and p62 levels in the presence of autolysosome inhibitor, bafilomycin. EBSS-induced autophagy increased C3-II to LC3-I ratios by 28% (p<0.05, n=6), but decreased p62 levels by 10% (p<0.05, n=6), and cotreatment of EBSS and bafilomycin increased LC3-II to LC3-I ratios by 55% (p<0.05 vs. EBSS, n=6) and p62 levels by 120% (p<0.01 vs. EBSS, n=4) (Fig. 2C). Cotreatment of OSS and bafilomycin increased LC3-II to LC3-I ratios by 47% (p<0.05 vs. OSS, n=6) and p62 levels by 41% (p<0.01 vs. OSS, n=6) (Fig. 2C). The increase in p62 levels in response to introducing bafilomycin to EBSS or OSS indicates an incomplete autophagy process, supporting the notion that OSS induces autophagy by an increase in LC3-II to LC3-I ratios, but impairs autophagic flux by a concomitant increase in p62 levels.
OSS-induced oxidative stress and JNK signaling increase LC3-II to LC3-I ratios
Previous studies demonstrated that OSS induces endothelial O2•− production via activation of the NADPH oxidase enzyme system and c-Jun NH2-terminal kinase (JNK-1 and JNK-2) signaling (36, 37, 56, 73, 74). In this study, we transfected HAECs with control (Adv-LacZ) or recombinant manganese superoxide dismutase (MnSOD) adenovirus (Adv-MnSOD) to reduce mitochondrial superoxide production (O2•−) (74). Overexpression of MnSOD significantly attenuated OSS-induced LC3-II to LC3-I ratios (p<0.05 vs. static condition, n=4) (Fig. 3A). Similarly, treatment with antioxidant N-acetyl cysteine (NAC) and JNK inhibitor, SP600125, significantly mitigated OSS-induced LC3-II to LC3-I ratios (NAC: p<0.01, n=4; SP600125: p<0.05, n=4) (Fig. 3B, C). Thus, OSS-mediated O2•− production and JNK phosphorylation induce autophagy.
FIG. 3.
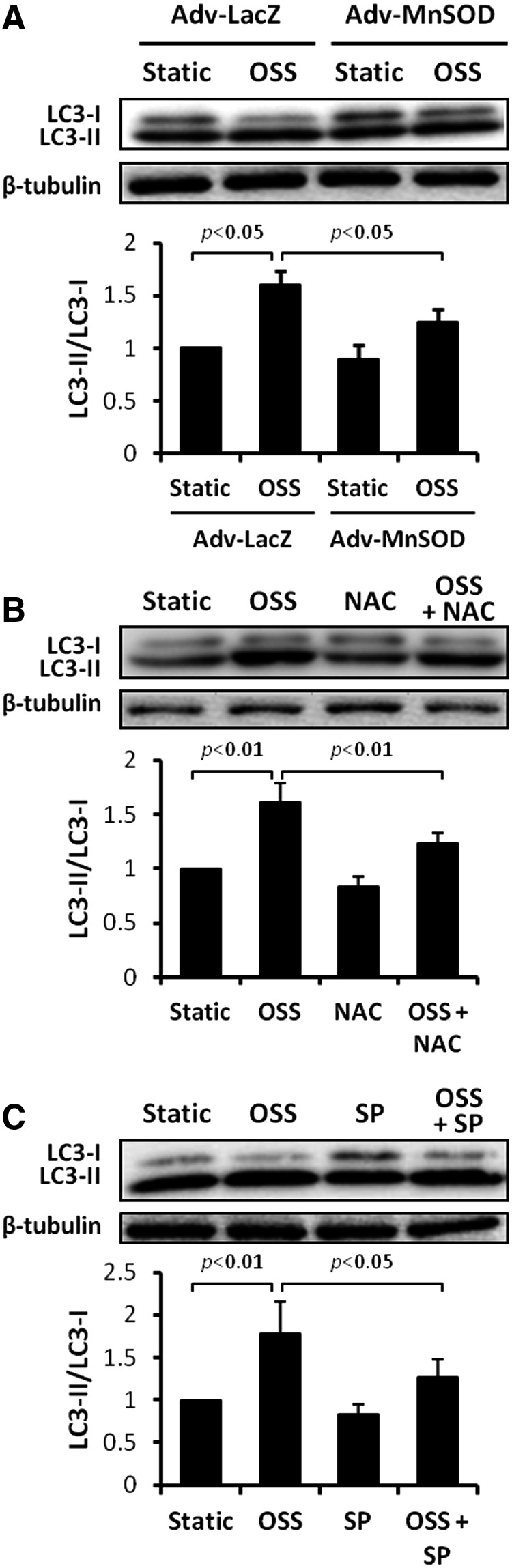
OSS induced oxidative stress and JNK activation to activate autophagy. (A) HAECs were infected with control or recombinant MnSOD adenovirus (MOI 1:100) overnight, and then were exposed to static condition or OSS for 4 h. Overexpression of MnSOD significantly attenuated OSS-induced LC3-II/LC3-I ratios (OSS: p<0.05, n=4; MnSOD: p<0.05, n=4). (B) HAECs were exposed to static condition or OSS for 4 h in the presence or absence of NAC at 5 mM or (C) a JNK inhibitor, SP600125 (SP), at 2 μM. Both antioxidant NAC and JNK inhibitor significantly attenuated OSS-induced LC3-II/LC3-I ratios (NAC: p<0.01; SP: p<0.05, n=4). JNK, c-Jun N2-terminal kinase; MnSOD, manganese superoxide dismutase; MOI, multiplicity of infection; NAC, N-acetyl cysteine.
EBSS or rapamycin-induced autophagy mitigates OSS-induced mitochondrial superoxide (mtO2•−) production
OSS significantly increased mtO2•− production in HAECs, as measured via MitoSOX Red fluorescent intensity quantification by flow cytometry (p<0.05, n=4) (Fig. 4A). EBSS attenuated OSS-induced inhibition of autophagy flux (p<0.05, n=6) (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars) and mtO2•− production (p<0.05, n=4) (Fig. 4A). Treatment of HAECs with rapamycin resulted in a similar finding (Supplementary Fig. S2), whereas ATG5 knockdown with siRNA (siATG5) accentuated MitoSOX Red intensities (p<0.05, n=4) (Fig. 4B). Thus, induction of autophagy attenuated OSS-mediated mtO2•− production.
FIG. 4.

EBSS-activated autophagy mitigated OSS-induced mitochondrial O2•− production. (A) HAECs were exposed to OSS in DMEM/1% FBS (control) or in EBSS for 4 h. MtO2•− production was measured by the MitoSox Red fluorescent intensity. EBSS attenuated OSS-induced MitoSox Red intensity (p<0.05, n=4). (B) HAECs were transfected with scrambled siRNA or ATG5 siRNA for 48 h, and then were exposed to static condition or OSS. Knockdown of ATG5 augmented OSS-induced MitoSox Red intensity (p=0.059, n=4). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
EBSS or rapamycin-induced autophagy restores OSS-mediated reduction in mitochondrial respiration
OSS significantly reduced mitochondrial respiration by 46% (p<0.01, n=4). EBSS-induced autophagy completely restored OSS-mediated reduction in mitochondrial respiration (p<0.01, n=4) (Fig. 5A). Mitochondrial complex II generates ROS at high rates during oxidative phosphorylation (69). OSS reduced complex II activity in HAECs by 43% (p<0.05, n=3), which was restored by EBSS (p<0.05, n=3) (Fig. 5B). Rapamycin-induced autophagy also completely restored an OSS-mediated decrease in complex II activity (p<0.05, n=4) (Supplementary Fig. S3). Collectively, induction of autophagy restores mitochondrial respiration.
FIG. 5.
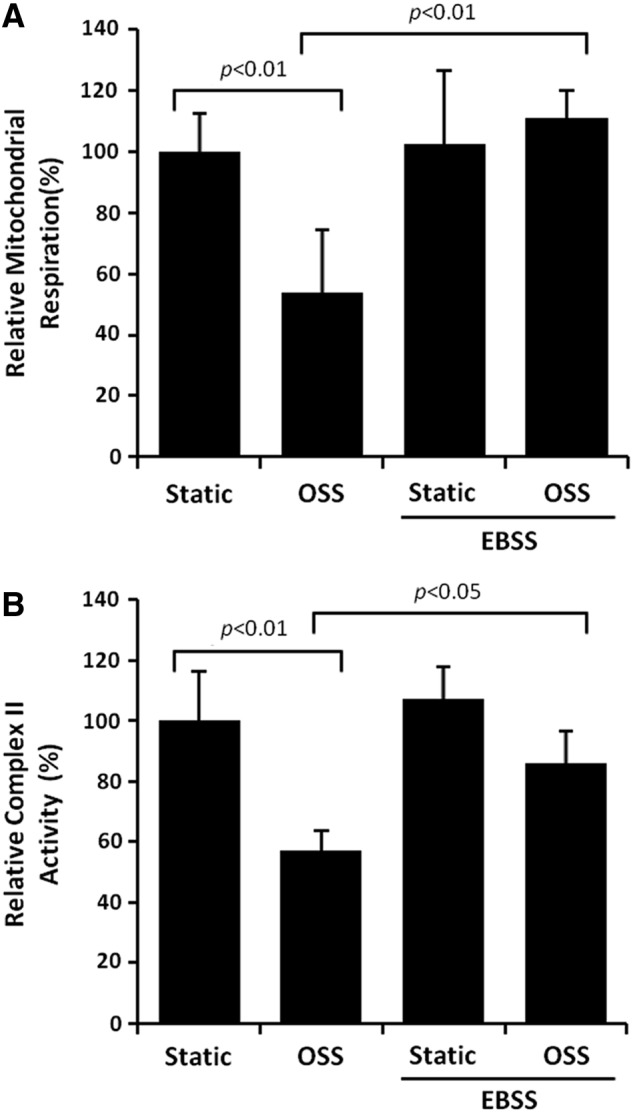
EBSS-activated autophagy restored OSS-mediated reduction in mitochondrial respiration. HAECs were exposed to OSS in DMEM/1% FBS (control) or in EBSS for 4 h. (A) Mitochondrial respiration was assessed by Seahorse Flux Analyzer. EBSS restored OSS-mediated reduction in mitochondrial respiration (OSS: p<0.01, n=4; EBSS: p<0.01, n=4). (B) EBSS restored OSS-mediated reduction in mitochondrial complex II activity (p<0.05, n=4).
OSS-mediated JNK signaling and mtO2•− production induce mtDNA damage
Unlike nuclear DNA, mtDNA is not bound directly to the histone family in the mitochondrial matrix (13, 40) and is susceptible to oxidative DNA damage (76). OSS-induced mtDNA damage was assessed by accumulation of 8-OHdG (40) and was predominantly colocalized with mitochondria (anti-cytochrome C, red fluorescence) (Fig. 6A). OSS induced a 4.5-fold increase in mtDNA damage (p<0.001 vs. control, n=4) (Fig. 6B). OSS-induced nuclear DNA damage was statistically insignificant, as measured by the Comet assay (p=0.9, n=3) (Fig. 6C). Furthermore, OSS-induced mtDNA damage was significantly mitigated by overexpression of MnSOD with recombinant adenoviruses (p<0.05, n=4) (Fig. 7A), treatment with antioxidant NAC (p<0.05, n=4) (Fig. 7B), and the JNK inhibitor, SP600125 (p<0.01, n=4) (Fig. 7C). These observations demonstrate that OSS-induced mtO2•− production and JNK signaling promote mtDNA damage.
FIG. 6.
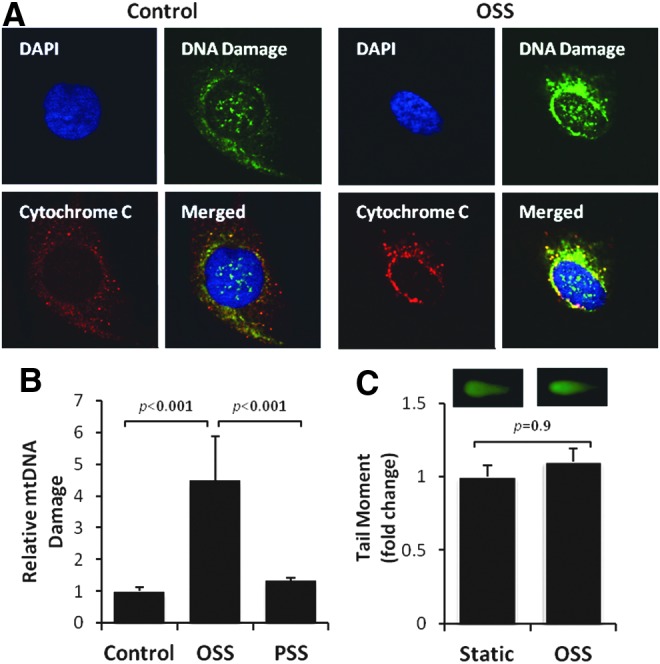
OSS induced mtDNA damage. (A) Immunofluorescent staining revealed mtDNA damage. HAECs exposed to OSS (4 h) developed a prominent intensity for anti 8-OHdG (green) as an indicator for DNA damage, which colocalized with anti-cytochrome C (red) for mitochondria. DAPI (blue) stained for nuclei. (B) HAECs were exposed to OSS versus PSS for 4 h, and mtDNA damage was measured by quantitative PCR. OSS induced a 4.5-fold increase in mtDNA damage (p<0.001, n=4), and PSS induced an insignificant increase (p>0.05, n=3). (C) OSS induces negligible nuclear DNA damage. Tail moment analysis of Comet assay (CometScore software) revealed an insignificant difference in nuclear DNA damage between static and OSS conditions (p=0.9, n=3). 8-OHdG, 8-hydroxy-2′-deoxyguanosine; mtDNA, mitochondrial DNA. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 7.
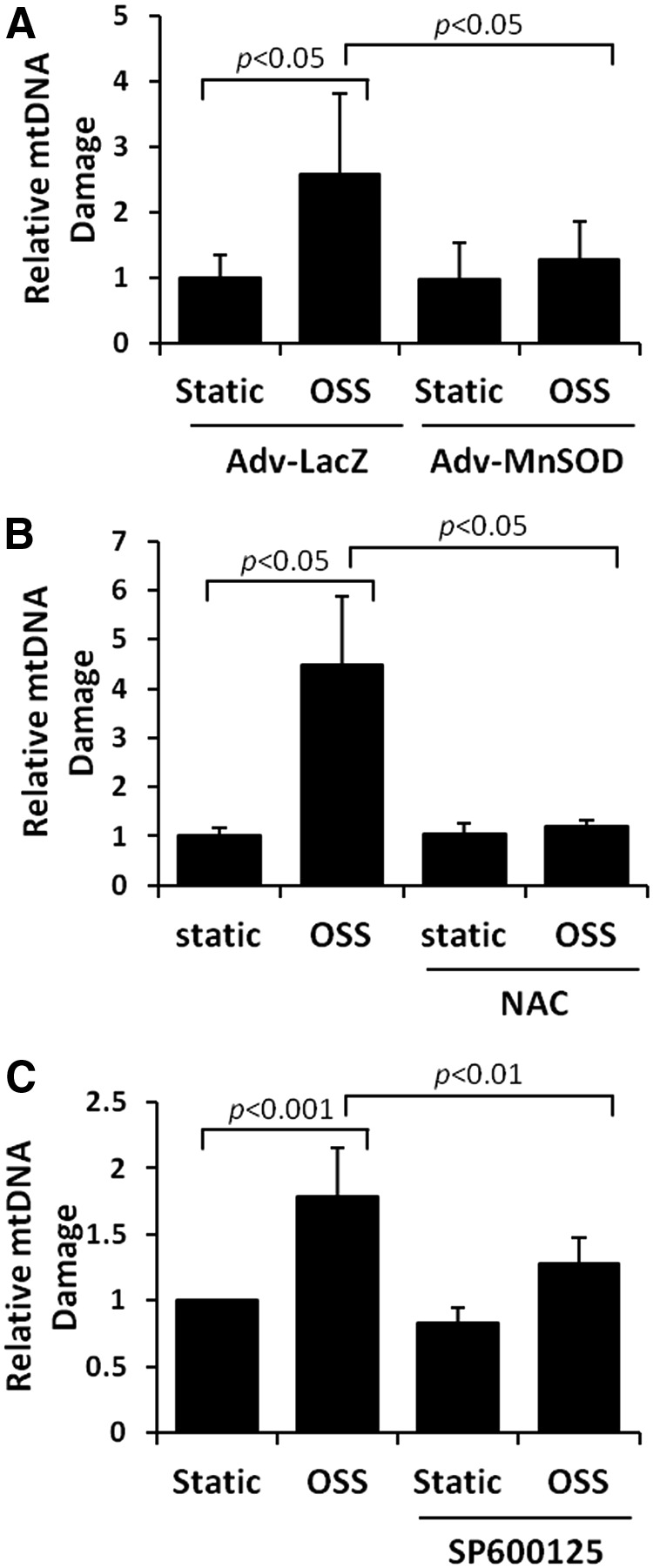
Antioxidants and JNK inhibitor reversed OSS-induced mtDNA damage. (A) HAECs were infected with control or recombinant MnSOD adenovirus (MOI 1:100) overnight, and then were exposed to OSS for 4 h. MnSOD overexpression significantly reversed OSS-induced mtDNA damage (p<0.05, n=4). (B) HAECs were exposed to static condition or OSS for 4 h in the presence or absence of 5 mM of antioxidant (NAC) or (C) 10 μM of JNK inhibitor, SP600125 (SP). Both NAC and SP significantly reversed OSS-induced mtDNA damage (NAC: p<0.05; SP: p<0.01, n=4).
DNA damage and JNK activation develop in the disturbed flow-exposed aortic arch
To recapitulate spatial variations in OSS-induced JNK activation and mtDNA damage in vivo, we performed immunohistochemistry staining in both the aortic arch and descending aorta of New Zealand White rabbits fed a normal chow diet. Both anti-8-OHdG antibody staining for oxidative DNA damage and anti-phospho-JNK staining were prominent in the OSS-exposed aortic arch, but were nearly absent in the PSS-exposed descending aorta (Fig. 8). Taken together, spatial (lesser vs. greater curvature vs. descending aorta) and temporal variations (OSS vs. PSS) in shear stress differentially induce autophagy and autophagic flux (Fig. 1) to modulate mitochondrial homeostasis (Fig. 6).
FIG. 8.

Spatial variations in shear stress differentially promoted DNA damage and JNK activation in New Zealand White rabbits. Cross sections of the aortic arch versus descending aorta were stained with anti-8-OHdG for DNA damage and anti-phospho-JNK for JNK activation. Prominent anti-8-OHdG staining was observed in the aortic arch, but nearly absent in the descending aorta. Phospho-JNK staining was prominent in the aortic arch, but nearly absent in the descending aorta. The red squares denote enlarged areas (100× magnification). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Discussion
Spatial and temporal variations in hemodynamic shear stress modulate endothelial metabolic states to support the focal and eccentric nature of atherosclerotic lesions. While the mechanotransduction mechanisms underlying fluid shear stress and oxidative stress have been well elucidated (15–18, 29), we hereby provide new metabolic insights into the disturbed flow-mediated (i) LC3-II to LC3-I ratios to promote autophagosome biogenesis and (ii) p62 accumulation to impair autophagic flux. We further elucidate OSS-mediated oxidative stress and JNK signaling underlying activation of autophagy to modulate mitochondrial homeostasis (Fig. 9).
FIG. 9.
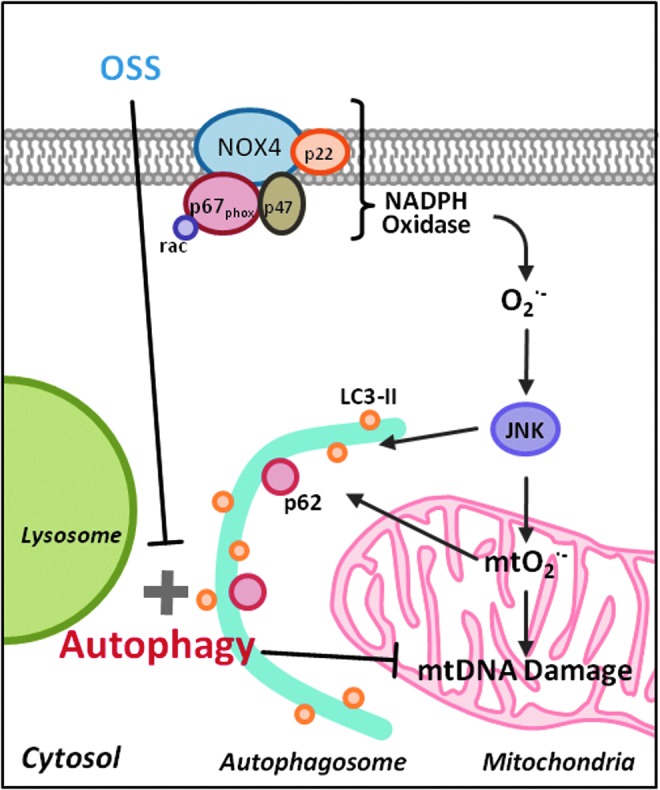
Scheme of OSS modulation of autophagy. OSS-induced oxidative stress activates JNK, which plays a dual role in inducing mitochondrial dysfunction and initiating autophagy. However, sustained OSS impairs autophagic flux, leading to accumulation of p62, increased mtO2•− production, and mtDNA damage. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Organisms experience frequent changes in their biophysical and biochemical environment. Induction of basal autophagy preserves cellular function and adapts to stress responses via delivery of proteins, macromolecules, organelles, and microbes to the lysosome for digestion (12, 31, 46, 54, 57, 59, 66, 78). Macrophage-specific ATG5-null mice develop an increase in atherosclerotic lesions accompanied by elevated inflammatory markers, supporting the impairment of autophagy as an underlying mechanism in the initiation of atherosclerosis (70). In our dynamic flow system, ATG5 knockdown accentuated OSS-induced mtO2•− production, whereas EBSS or rapamycin activated autophagy to attenuate OSS-induced mtO2•− production (Fig. 4A and Supplementary Fig. S2).
In the disturbed flow-exposed regions of the adult porcine aorta, both proinflammatory and antioxidative endothelial transcription profiles coexist (64). While disturbed flow promotes inflammatory responses, the antioxidative transcript profiles, including MnSOD expression, prevent initiation of atherosclerosis (64). Shear stress-responsive MnSOD is located in the mitochondrial matrix to dismutate the conversion of superoxide anion (O2•−) to hydrogen peroxide (H2O2) (2, 43). MnSOD+/− mice exhibit impaired endothelium-dependent vasodilation (26). ApoE−/− MnSOD+/− mice display earlier mtDNA damage and accelerated atherosclerosis compared with ApoE−/− MnSOD+/+ mice (4). In our dynamic HAEC model, induction of autophagy mitigates OSS-induced mtO2•− production in the absence of hyperlipidemia. EBSS or rapamycin-induced autophagy mitigates OSS-mediated JNK activation and mtO2•− production to reduce mtDNA damage, which initiates atherogenesis (22, 58, 60). Overexpression of MnSOD and treatment with NAC or JNK inhibitor significantly attenuate OSS-induced mtDNA damage (Fig. 7). In corollary, overexpression of MnSOD attenuates JNK-mediated MnSOD protein degradation and ubiquitination, leading to an increase in cleaved caspase-3 (74). Preferential accumulation of p62 levels in the OSS-exposed aortic arch rather than PSS-exposed descending aorta further supports an incomplete autophagy process known as impaired autophagic flux (45) (Fig. 1) (44, 47). Thus, the combination of impaired autophagic flux and mtDNA damage in the disturbed flow-exposed regions is conducive to initiate endothelial dysfunction (Fig. 8).
It is well recognized that ROS activate antibacterial autophagy and autophagic cell death through distinct mechanisms, depending on the specific cell types in the microenvironment (35, 41). Induction of autophagy by pharmacological intervention, overexpression of ATG genes, and exercise-mediated shear stress augmentation confer cellular protection against the aggregate-prone proteins associated with neurodegeneration (1, 67). In this study, we demonstrate that spatial (∂τ/∂x) and temporal variations (∂τ/∂t) in shear stress induce cytosolic O2•− production, JNK activation, and mitochondrial metabolic changes to increase LC3-II to LC3-I ratios.
NADPH oxidase cytosolic O2•−-JNK phosphorylation signaling was implicated in OSS-mediated mtO2•− production (73). Immunostaining of aorta from New Zealand White (NZW) rabbits fed a normal diet supports the notion that DNA damage and JNK activation coexist in the OSS-exposed region or lesser curvature of the aortic arch (Fig. 8). In our dynamic HAEC model, OSS increased LC3-II/LC3-I ratios and GFP-LC3 dots/cell to a greater extent than did PSS, and OSS-mediated cytosolic O2•−-JNK phosphorylation signaling induces autophagy (Fig. 3). Conversely, EBSS and rapamycin-induced autophagy mitigate OSS-induced mtO2•− production, whereas ATG5 siRNA-inhibited autophagy increased OSS-induced mtO2•− production. We further observed that autophagy restored OSS-mediated mitochondrial respiration (Fig. 5A).
Due to the lack of protective histones, mtDNA is vulnerable to oxidative damage (76). Oxidative phosphorylation is coupled with respiratory chain complexes I to II, III, and IV (23, 30). While the transfer of electrons from ubisemiquinone through the mitochondrial respiratory chain is more than 98% efficient, 1.5%–2% of electrons leak out to form O2•− (27). For these reasons, autophagy modulates OSS-mediated changes in mitochondrial respiration and complex activity to mitigate mtO2•− production and mtDNA damage (Fig. 4A and Supplementary Figs S1 and S2).
mtDNA damage is frequently observed in blood cells and the vascular wall. Despite its low abundance, a specific 4977 bp common deletion (mtDNA [4977]) is associated with mitochondrial dysfunction (7). mtDNA damage primes the initiation of atherosclerosis (60). Our laboratory and others have demonstrated that OSS activates NADPH oxidase, which, in turn, induces an increase in cytosolic O2•− production to activate JNK and to increase mtO2•− production (19, 73, 74). Thus, OSS induces the JNK signaling pathway to initiate mtO2•− production and mtDNA damage.
Overall, disturbed flow modulates the cross talk between autophagy and mitochondrial function (5, 22, 35, 49, 55). In the athero-prone or OSS-exposed arterial regions, basal endothelial homeostasis may be dependent on the balance between OSS-induced mtDNA damage and autophagy. In the presence of cardiovascular risk factors, disturbed flow impairs autophagic flux to initiate inflammatory responses and atherosclerosis. We hereby provide a dynamic model with translational implications to modulate mechanosensitive tissues to maintain cellular homeostasis.
Materials and Methods
Immunohistochemistry staining
p62, also known as sequestosome (SQSTM1), is an ubiquitin-binding scaffold protein. p62 accumulates when autophagy is inhibited and decreases when autophagy is induced (44, 47). Cross sections of the aortic arch and descending aorta from NZW rabbits were stained with anti-p62 antibody (Boster Biological Technologies) to assess changes in autophagy in vivo. Staining of the aortic arch and descending aorta was performed with anti-8-OHdG (Cayman Chemicals) and anti-phospho-JNK (Cell Signaling) to assess DNA damage by oxidation.
Three-dimensional CFD simulation
Aortic arch geometry was extracted from an angiogram video of the aorta. Diameters were measured every 5 mm along the length of the aorta and cross sections were assumed to be circular at every measured site. The aorta was reconstructed in SolidWorks (Concord). The pulsatile arterial profiles were captured from the MRI images and the video frames were extracted by ImageJ (National Institute of Health). Arterial velocity profiles were used as inlet boundary condition and the outlet was defined at 0 Pa, static pressure. CFD simulation was performed with SolidWorks Flow Simulation (Concord) with 3176 fluid mesh cells and 6082 partial mesh cells. The simulation was run until the defining 2 s physical time with a time step size of 0.01. SolidWorks Flow Simulation automatically sets convergence conditions with changing iteration numbers. The governing equations were solved by laminar incompressible blood with nonslip unsteady flow condition.
Endothelial cell culture
HAECs were cultured in endothelial cell growth medium (Cell Applications) and used between passages 5 and 9. For the autophagy study in response to shear stress, cells were seeded on gelatin-coated glass slides (25×75 mm) and grown to confluent monolayers for 48 h in 5% CO2 at 37°C before flow was applied. For inhibitor and stimulator studies, the cells were pretreated with c-Jun NH2-terminal Kinase (JNK) inhibitor, SP600125 (2–10 μM), antioxidant NAC (5 mM), or rapamycin (1 μM) before shear stress exposure.
Dynamic flow system to simulate PSS versus OSS profiles
A dynamic flow channel was used to recapitulate hemodynamics in human carotid arterial bifurcations (2, 39). The flow system was designed to generate well-defined flow profiles across the width of the parallel flow chamber at various temporal gradients (∂τ/∂t), frequencies, and amplitudes. HAECs were exposed to three conditions in DMEM/1% FBS unless otherwise stated: (i) control at static conditions, (ii) PSS at τavg=23 dyn·cm−2 and ∂τ/∂t at±8 dyn·cm−2·s−1 at 1 Hz, and (iii) OSS at τavg=0.02 dyn·cm−2 and ∂τ/∂t at±3 dyn·cm−2·s−1 at 1 Hz.
Western blot analyses
After flow exposure, HAECs were rinsed with phosphate-buffered saline and lysed using RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentration was measured using the Bio-Rad DC assay and 50 μg of protein was loaded for Western blotting essentially as previously described (53). Antibodies against autophagy-associated genes were purchased from Abcam for LC3 and from Boster Biological Technologies for p62. Parallel blots were performed with anti-β-tubulin (Millipore, Inc.) for loading normalization. Densitometry was performed to quantify blot bands as previously described (53).
Autophagosome visualization with GFP-LC3
HAECs seeded onto glass slides were infected with recombinant GFP-LC3 adenoviruses, kindly provided by Dr. Junichi Sadoshima from Rutgers New Jersey Medical School, at the multiple of infection (MOI) of 1:20. The cells were then applied to EBSS, static condition, or shear stress for 4 h. Fluorescent images were acquired using an inverted microscope (Olympus) and a DSLR camera (Canon). The puncture autophagosome structures were assessed by quantifying the dots inside cells. Dots were averaged from 50 cells for each condition as an indication of autophagosome formation.
Mitochondrial respiration assay
HAECs treated with dynamic shear stress were collected by trypsinization and seeded on XF24 V7 microplates at 25,000 cells/well. The cells were allowed to adhere to the plate for 2 h, then analyzed for mitochondrial respiration using the Seahorse XF24 analyzer as previously described (69). Mitochondrial respiration was measured and normalized to protein levels using the Bio-Rad protein assay.
Complex II activity assay
Mitochondrial complex II activity was measured with an assay kit from Abcam. Briefly, HAECs were collected after flow and resuspended at 5.5 mg/mL. The cells were lysed with the lysis buffer provided in the kit. After 30 min of incubation, the lysates were centrifuged at 25,000 g for 20 min at 4°C. Supernatants were collected for the assay following the manufacturer's instructions. Relative complex II activities were normalized to protein levels.
mtDNA damage assay
mtDNA damage was assessed by quantifying a large base pair deletion along the major arch of the mitochondrial genome by quantitative PCR. A common 4977 bp deletion in mtDNA was quantified by quantitative PCR (8) using primers flanking the deletion and normalized to the mitochondrial COX1 gene. The pair of primers for detection of the 4977 bp deletion was forward: CCTTACACTATTCCTCATCACC; reverse: TGTGGTCTTTGGAGTAGAAACC; and the pair of primers for mitochondrial COX1 was forward: TTCGCCGACCGTTGACTATTCTCT; reverse: AAGATTATTACAAATGCATGGGC.
siRNA transfection
Validated siRNA for ATG5 was purchased from Qiagen, Inc. Negative control siRNA or ATG5 siRNA (30 nM) was transfected to HAECs with Lipofectamine RNAiMAX (Life Technologies) following the manufacturer's instructions. Real-time PCR was used for confirmation of gene knockdown at 48 h after transfection.
Overexpression of MnSOD
Overexpression of the mitochondrial antioxidant enzyme, MnSOD, was done by transfecting HAECs with MnSOD adenovirus at an MOI of 1:100 for 1 h. HAECs were allowed to recover for 24 h before flow exposure, as previously described (73).
Immunofluorescence staining
Following flow exposure, HAECs were fixed with 4% PFA and stained with antibody against 8-OHdG for DNA oxidation and anti-cytochrome C for mitochondria. Fluorescent images were acquired using an inverted microscope (Olympus) and a DSLR camera (Canon).
Flow cytometry analysis to quantify mtO2•− production
Mitochondrial superoxide (mtO2•−) production was assessed by staining with MitoSOX Red, as previously described (73, 74), and was quantified using flow cytometry as previously described (52).
Nuclear DNA damage assay (Comet assay)
Nuclear DNA damage was assessed by quantifying nuclear DNA fragmentation using the Comet assay kit (Trevigen) according to the manufacturer's suggested procedures. Briefly, HAECs were trypsinized and immobilized on a glass slide with low-melting point agarose. The cells were lysed, treated with alkaline buffer, and stained with an SYBR green nucleic acid stain. The sample was then run at 28 V for 30 m, comet tails were visualized by fluorescence microscopy, and the tail moments were quantified using CaspLab Comet Assay Software.
Statistical analysis
Data were expressed as mean±standard deviation and compared among separate experiments. Comparisons of multiple values were made by one-way analysis of variance (ANOVA), and statistical significance for pair-wise comparisons was determined post hoc using Tukey's method. p-Values of <0.05 were considered statistically significant.
Supplementary Material
Abbreviations Used
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- CFD
computational fluid dynamic
- EBSS
Earle's balanced salt solution
- GFP
green fluorescent protein
- HAEC
human aortic endothelial cell
- JNK
c-Jun N2-terminal Kinase
- LC3
microtubule-associated protein light chain 3
- MnSOD
manganese superoxide dismutase
- MOI
multiplicity of infection
- mtDNA
mitochondrial DNA
- mtO2•−
mitochondrial superoxide
- mTOR
mechanistic target of rapamycin
- NAC
N-acetyl cysteine
- NZW
New Zealand White
- OSS
oscillatory shear stress
- ox-LDL
oxidized low-density lipoprotein
- PSS
pulsatile shear stress
- ROS
reactive oxygen species
Acknowledgments
The authors are grateful to Dr. Junichi Sadoshima from Rutgers New Jersey Medical School for providing recombinant GFP-LC3 adenoviruses. The authors thank Dr. Peixiang Zhang from UCLA for the Seahorse assay. This project was supported by National Institutes of Health, HL083015 (T.K.H.), HL118650 (T.K.H.), HL111437 (T.K.H.), DE010742 (D.K.A.), and DE014183 (D.K.A.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Adlard PA, Perreau VM, Pop V, and Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci 25: 4217–4221, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ai L, Rouhanizadeh M, Wu JC, Takabe W, Yu H, Alavi M, Li R, Chu Y, Miller J, and Heistad DD. Shear stress influences spatial variations in vascular Mn-SOD expression: implication for LDL nitration. Am J Physiol Cell Physiol 294: C1576–C1585, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Overvatn A, Kirkin V, and Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 287: 39275–39290, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, and Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation 106: 544–549, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Bess AS, Ryde IT, Hinton DE, and Meyer JN. UVC-induced mitochondrial degradation via autophagy correlates with mtDNA damage removal in primary human fibroblasts. J Biochem Mol Toxicol 27: 28–41, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, and Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123: 5284–5297, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botto N, Rizza A, Colombo MG, Mazzone AM, Manfredi S, Masetti S, Clerico A, Biagini A, and Andreassi MG. Evidence for DNA damage in patients with coronary artery disease. Mutat Res 493: 23–30, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Chen T, He J, Shen L, Fang H, Nie H, Jin T, Wei X, Xin Y, Jiang Y, Li H, Chen G, Lu J, and Bai Y. The mitochondrial DNA 4,977-bp deletion and its implication in copy number alteration in colorectal cancer. BMC Med Genet 12: 8, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, McMillan-Ward E, Kong J, Israels SJ, and Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 15: 171–182, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Cheng C, Tempel D, van Haperen R, de Boer HC, Segers D, Huisman M, van Zonneveld AJ, Leenen PJ, van der Steen A, Serruys PW, de Crom R, and Krams R. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest 117: 616–626, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu JJ, Wang DL, Chien S, Skalak R, and Usami S. Effects of disturbed flow on endothelial cells. J Biomech Eng 120: 2–8, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Choi AM, Ryter SW, and Levine B. Autophagy in human health and disease. N Engl J Med 368: 651–662, 2013 [DOI] [PubMed] [Google Scholar]
- 13.Choi YS, Hoon Jeong J, Min HK, Jung HJ, Hwang D, Lee SW, and Kim Pak Y. Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: identification of mitochondrial histones. Mol Biosyst 7: 1523–1536, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Colecchia D, Strambi A, Sanzone S, Iavarone C, Rossi M, Dall'Armi C, Piccioni F, Verrotti di Pianella A, and Chiariello M. MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy 8: 1724–1740, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham KS. and Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest 85: 9–23, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev 75: 519–560, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med 6: 16–26, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies PF, Dewey CF, Jr., Bussolari SR, Gordon EJ, and Gimbrone MA., Jr. Influence of hemodynamic forces on vascular endothelial function. In vitro studies of shear stress and pinocytosis in bovine aortic cells. J Clin Invest 73: 1121–1129, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Keulenaer GW, Chappell DC, Ishizaka N, Nerem RM, Alexander RW, and Griendling KK. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ Res 82: 1094–1101, 1998 [DOI] [PubMed] [Google Scholar]
- 20.DePaola N, Gimbrone MA, Jr., Davies PF, and Dewey CF., Jr. Vascular endothelium responds to fluid shear stress gradients. Arterioscler Thromb 12: 1254–1257, 1992 [DOI] [PubMed] [Google Scholar]
- 21.Dewey CF, Jr., Bussolari SR, Gimbrone MA, Jr., and Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng 103: 177–185, 1981 [DOI] [PubMed] [Google Scholar]
- 22.Ding Z, Liu S, Wang X, Khaidakov M, Dai Y, and Mehta JL. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci Rep 3: 1077, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duchen MR, Surin A, and Jacobson J. Imaging mitochondrial function in intact cells. Biophotonics, Pt B 361: 353–389, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Dunlop EA. and Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36C: 121–129, 2014 [DOI] [PubMed] [Google Scholar]
- 25.Dutta D, Xu J, Kim JS, Dunn WA, Jr., and Leeuwenburgh C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy 9: 328–344, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forgione MA, Cap A, Liao R, Moldovan NI, Eberhardt RT, Lim CC, Jones J, Goldschmidt-Clermont PJ, and Loscalzo J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation 106: 1154–1158, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Forman HJ. and Azzi A. Production of reactive oxygen species in mitochondria and age-associated pathophysiology: a reality check. In: Understanding the Process of Aging: The roles of mitochondria, Free radicals and Antioxidants, edited by Cadenas E. and Packer L. New York, Basel: Marcel Dekker Inc, 1999, pp. 73–94 [Google Scholar]
- 28.Frangos JA, Huang TY, and Clark CB. Steady shear and step changes in shear stimulate endothelium via independent mechanisms—superposition of transient and sustained nitric oxide production. Biochem Biophys Res Commun 224: 660–665, 1996 [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Cardena G. and Gimbrone MA., Jr. Biomechanical modulation of endothelial phenotype: implications for health and disease. Handb Exp Pharmacol 176: 79–95, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Genova ML. and Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta 1887:427–443, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Green DR, Galluzzi L, and Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333: 1109–1112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hariharan N, Ikeda Y, Hong C, Alcendor RR, Usui S, Gao S, Maejima Y, and Sadoshima J. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One 8: e51632, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison D, Griendling KK, Landmesser U, Hornig B, and Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 91: 7A-11A, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Hsiai TK, Hwang J, Barr ML, Correa A, Hamilton R, Alavi M, Rouhanizadeh M, Cadenas E, and Hazen SL. Hemodynamics influences vascular peroxynitrite formation: implication for low-density lipoprotein apo-B-100 nitration. Free Radic Biol Med 42: 519–529, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang J, Lam GY, and Brumell JH. Autophagy signaling through reactive oxygen species. Antioxid Redox Signal 14: 2215–2231, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, Sevanian A, and Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res 93: 1225–1232, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, and Jo H. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem 278: 47291–47298, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, and Majima HJ. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 7: 106–118, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Jen N, Yu F, Lee J, Wasmund S, Dai X, Chen C, Chawareeyawong P, Yang Y, Li R, Hamdan MH, and Hsiai TK. Atrial fibrillation pacing decreases intravascular shear stress in a New Zealand white rabbit model: implications in endothelial function. Biomech Model Mechanobiol 12: 735–745, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kakimoto M, Inoguchi T, Sonta T, Yu HY, Imamura M, Etoh T, Hashimoto T, and Nawata H. Accumulation of 8-hydroxy-2′-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes 51: 1588–1595, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Kiffin R, Bandyopadhyay U, and Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal 8: 152–162, 2006 [DOI] [PubMed] [Google Scholar]
- 42.King JS, Veltman DM, and Insall RH. The induction of autophagy by mechanical stress. Autophagy 7: 1490–1499, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kluge MA, Fetterman JL, and Vita JA. Mitochondria and endothelial function. Circ Res 112: 1171–1188, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komatsu M. and Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 584: 1374–1378, 2010 [DOI] [PubMed] [Google Scholar]
- 45.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, and Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149–1163, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Kubli DA. and Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 111: 1208–1221, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamark T. and Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol 22: 192–198, 2010 [DOI] [PubMed] [Google Scholar]
- 48.Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, and Hill JA. Cardiovascular autophagy: concepts, controversies and perspectives. Autophagy 9, 2013 [DOI] [PubMed] [Google Scholar]
- 49.Lee J, Giordano S, and Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441: 523–540, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levine B. and Kroemer G. Autophagy in the pathogenesis of disease. Cell 132: 27–42, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li R, Beebe T, Cui J, Rouhanizadeh M, Ai L, Wang P, Gundersen M, Takabe W, and Hsiai TK. Pulsatile shear stress increased mitochondrial membrane potential: implication of Mn-SOD. Biochem Biophys Res Commun 388: 406–412, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li R, Jen N, Yu F, and Hsiai TK. Assessing mitochondrial redox status by flow cytometric methods: vascular response to fluid shear stress. Curr Protoc Cytom Chapter 9: Unit9.37, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li R, Ning Z, Cui J, Khalsa B, Ai L, Takabe W, Beebe T, Majumdar R, Sioutas C, and Hsiai T. Ultrafine particles from diesel engines induce vascular oxidative stress via JNK activation. Free Radic Biol Med 46: 775–782, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, and Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15: 545–553, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luo C, Li Y, Wang H, Feng Z, Long J, and Liu J. Mitochondrial accumulation under oxidative stress is due to defects in autophagy. J Cell Biochem 114: 212–219, 2013 [DOI] [PubMed] [Google Scholar]
- 56.Madamanchi NR, Vendrov A, and Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 25: 29–38, 2005 [DOI] [PubMed] [Google Scholar]
- 57.Maiuri MC, Grassia G, Platt AM, Carnuccio R, Ialenti A, and Maffia P. Macrophage autophagy in atherosclerosis. Mediators Inflamm 2013: 584715, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maiuri MC, Zalckvar E, Kimchi A, and Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8: 741–752, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Martinet W. and De Meyer GR. Autophagy in atherosclerosis. Curr Atheroscler Rep 10: 216–223, 2008 [DOI] [PubMed] [Google Scholar]
- 60.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, Vidal-Puig A, Logan A, Murphy MP, and Bennett M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res 107: 1021–1031, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mizushima N, Yoshimori T, and Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132, 2011 [DOI] [PubMed] [Google Scholar]
- 62.Muller C, Salvayre R, Negre-Salvayre A, and Vindis C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ 18: 817–828, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, and Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr., and Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A 101: 2482–2487, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pei ZH, Xi BS, and Hwang NH. Wall shear stress distribution in a model human aortic arch: assessment by an electrochemical technique. J Biomech 18: 645–656, 1985 [DOI] [PubMed] [Google Scholar]
- 66.Perrotta I. The use of electron microscopy for the detection of autophagy in human atherosclerosis. Micron 50: 7–13, 2013 [DOI] [PubMed] [Google Scholar]
- 67.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, and Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118: 2190–2199, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, and Dikic I. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 32: 1733–1744, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, and Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem 287: 27255–27264, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, and Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15: 534–544, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rogov V, Dotsch V, Johansen T, and Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178, 2014 [DOI] [PubMed] [Google Scholar]
- 72.Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, and Giddens DP. Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol 27: 346–351, 2007 [DOI] [PubMed] [Google Scholar]
- 73.Takabe W, Jen N, Ai L, Hamilton R, Wang S, Holmes K, Dharbandi F, Khalsa B, Bressler S, Barr ML, Li R, and Hsiai TK. Oscillatory shear stress induces mitochondrial superoxide production: implication of NADPH oxidase and c-Jun NH2-terminal kinase signaling. Antioxid Redox Signal 15: 1379–1388, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takabe W, Li R, Ai L, Yu F, Berliner JA, and Hsiai TK. Oxidized low-density lipoprotein-activated c-Jun NH2-terminal kinase regulates manganese superoxide dismutase ubiquitination: implication for mitochondrial redox status and apoptosis. Arterioscler Thromb Vasc Biol 30: 436–441, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, and Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 20: 444–454, 2011 [DOI] [PubMed] [Google Scholar]
- 76.Yakes FM. and Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 94: 514–519, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yorimitsu T, Nair U, Yang Z, and Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281: 30299–30304, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Youle RJ. and Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, and Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res 53: 502–514, 1983 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.