

Abstract
Aims: Glutathione (GSH) is the main antioxidant against cell damage. Several pathological states course with reduced nucleophilic tone and perturbation of redox homeostasis due to changes in the 2GSH/GSSG ratio. Here, we investigated the regulation of the rate-limiting GSH biosynthetic heterodimeric enzyme γ-glutamyl-cysteine ligase (GCL) by microRNAs (miRNAs). Results: “In silico” analysis of the 3′- untranslated regions (UTRs) of both catalytic (GCLc) and regulatory (GCLm) subunits of GCL enabled an identification of miR-433 as a strong candidate for the targeting of GCL. Transitory overexpression of miR-433 in human umbilical vein endothelial cells (HUVEC) showed a downregulation of both GCLc and GCLm in a nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-independent manner. Increases in pro-oxidant stimuli such as exposure to hydrogen peroxide or GSH depletion in endothelial and hepatic cells caused an expected increase in GCLc and GCLm protein expression and abrogation of miR-433 levels, thus supporting a cross-regulation of these pathways. Treatment of HUVEC with miR-433 resulted in reduced antioxidant and redox potentials, increased S-glutathionylation, and reduced endothelial nitric oxide synthase activation. In vivo models of renal and hepatic fibrosis were associated with transforming growth factor β1 (TGF-β1)-related reduction of GCLc and GCLm levels that were miR-433 dependent. Innovation and Conclusion: We describe for the first time an miRNA, miR-433, capable of directly targeting GCL and promoting functional consequences in endothelial physiology and fibrotic processes by decreasing GSH levels. Antioxid. Redox Signal. 23, 1092–1105.
Introduction
Glutathione (GSH) is considered the quintessential endogenous antioxidant largely due to its relatively high intracellular concentrations (0.5–10 mM) (40). While ubiquitous, it is preferentially synthesized in the liver as a tripeptide (γ-glutamyl-L-cysteinyl-glycine) and it pairs with the disulfide form (GSSG) in a relatively constant molar ratio (2GSH/GSSG) of 100–300:1 corresponding to redox potentials from −220 to −240 mV (56), although this relationship has been recently challenged (42). GSH contributes to detoxify deleterious metabolites, regulating the cell cycle and, above all, maintaining redox homeostasis by preserving nucleophilic tone, redox potential and facilitating redox signaling [see Ref. (37) for review]. Synthesis of GSH takes place in a two-step reaction. In the first step, glutamate is conjugated with cysteine through the carboxyl group in the γ-position of the glutamate and the amino group of the cysteine, while in the second one the γ-glutamyl-L-cysteine reacts with the glycine to form γ-glutamyl-cysteinyl-glycine. Both reactions require adenosine triphosphate (ATP). The enzymes involved in the synthesis are γ-glutamyl-cysteine ligase (GCL), also called γ-glutamyl-cysteine synthetase and GSH synthetase. GCL catalyzes the first and also limiting step in the synthesis of GSH. This enzyme is composed of two subunits (heavy, Mr 73,000; and light, Mr 31,000) with the heavy subunit (GCLc) accounting for both the catalytic activity of the isolated enzyme (28) and the feedback inhibition by GSH (59). The regulation of GCLc expression takes place at the transcriptional and post-transcriptional levels and has been discussed in detail elsewhere (15, 37). Hormones such as insulin (29), rapid liver growth, or antioxidants (38) are well-known inducers of GCLc transcription. On the other hand, transforming growth factor β1 (TGF-β1) is one of the major repressors of GCL expression and subsequent GSH depletion (27, 33, 34, 55).
Innovation.
The regulation of glutathione biosynthesis is of critical importance for cellular redox homeostasis. γ-Glutamyl-cysteine ligase (GCL) is the enzyme in charge of the rate-limiting step in this process. So far, very little information is available on the post-transcriptional regulation of GCL. Here, we describe miR-433 as the first microRNA that directly targets both subunits of GCL, also resulting in reduced GSH biosynthesis, thus adding a new regulatory mode for GCL expression. Functionally, downregulation of GCL by miR-433 is relevant for endothelial homeostasis and hormetic responses involved in fibrogenesis, as observed in two different models of organ fibrosis.
It has been proposed that these regulations are ruled mainly by the binding of transcription factors to antioxidant response element (ARE) boxes within the GCLc promoter (27, 36, 53, 71). Several transcription factors, including nuclear factor (erythroid-derived 2)-like 2 (Nrf2), the activator protein 1 (AP-1) family, and nuclear factor κB (NFκB), are involved in this interaction (54, 72).
Nrf2, as the essential transcription factor binding to ARE (20), takes central stage in the regulation of GCL expression. The activity of GCLc is also regulated by phosphorylation at the post-translational level (37, 62). The light subunit (GCLm) does not exhibit catalytic activity by itself, but contributes to the function of the heavy subunit, critically affecting interactions of the substrate at the active site (22, 42). Oxidative stress can orchestrate the induction of both GCLc and GCLm and this induction, like that promoted by certain xenobiotics, is linked to the Nrf2-ARE pathway. However, other pro-oxidants such as ethanol and TGF-β1 do not affect GCLm expression (17). Interestingly, hepatic toxicity induced by either litocholic acid (72) or bile duct ligation (BDL) (45, 63) triggers a biphasic response in the expression of both subunits, causing an initial upregulation and further repression of the enzymes (72).
MicroRNAs (miRNAs) are short (20–24 nt) noncoding RNAs involved in post-transcriptional regulation of gene expression by affecting both stability and translation of mRNA. More than half of all mRNAs are estimated to be targets of miRNAs, and each miRNA is predicted to regulate approximately hundreds of targets [for review, see Refs. (5, 26)]. Consistent with this special capacity, miRNAs regulate a broad range of biological processes, including cell proliferation, apoptosis, differentiation, and tissue repair (2, 4, 24, 31, 68, 70). There is now strong evidence that aberrant miRNA expression or function can lead to the development and progression of multiple human pathophysiological processes, including diabetes, cancer, and cardiovascular diseases (39, 58). There are still huge gaps in our understanding of redox homeostasis particularly with regard to its regulation via miRNAs, even though the term “redoximiR” has been recently coined. This set of miRNAs is mostly related to the regulation of the Nrf2 pathway, although increasing examples of specific targeting by miRNAs of cytosolic or mitochondrial genes are appearing [see Ref. (11) for review]. In the particular case of GCL subunits (here termed globally GCLs), there are very few reports about the potential involvement of miRNAs in their post-transcriptional regulation but with limited functional data (1). Hence, we sought to identify miRNAs that could directly target GCLs and interfere with their expression, in the belief that the final regulation of GSH balance could represent a major consequence of their action. We found that miR-433 targets both GCLc and GCLm and significantly affects their expression. Furthermore, we provide data supporting that this effect has a profound impact on redox-related pathophysiological processes such as endothelial dysfunction and organ fibrosis.
Results
Identification and confirmation of miR-433 as a candidate miRNA targeting GCLc and GCLm
“In silico” analysis of the 3′-untranslated region (UTR) of GCLc and GCLm genes with the targetscan.org and mirWalk.org databases provided a list of candidate miRNAs (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars) with complementary sequences in these regions. We selected miR-144 and miR-433 as the most interesting candidates. miR-144 has a predicted seed region in both 3′ UTRs, and it has been described as a redox-sensitive miRNA in sickle cell disease and in neurons by regulating Nrf2 (44, 57). In contrast, miR-433 has not been previously described as a redoximiR and it contains two highly conserved seed sequences in each GCL 3′-UTR. Thus, we focused on the analysis of miR-433.
The human GCLc 3′-UTR is 1414 bp long. The two seed target sites for miR-433 are situated in the extremes of the region (Fig. 1A). The first site is an 8-mer only present in Homo sapiens. The second site is a 7-mer conserved among several mammalian species (Fig. 1A). The human GCLm 3′-UTR is almost twice as long and also bears two targeting sites for miR-433 in its 3′-UTR (Fig. 1B). The first site is a poorly conserved 7-mer-1A seed sequence and the second site, which corresponds to the only functional site, is a 6-mer present in the human 3′-UTR. To verify the functional behavior of this targeting, the 3′-UTR's were cloned in a luciferase reporter vector. For GCLc, one intact and two mutated constructs, point mutation 1 (PM1) (C240G and A241U) and PM2 (including PM1 +C1204G and A1205U) were used. For GCLm, three different constructs named GCLmS1 (180–186), GCLmS2 (2417–2422), and GCLm S2 PM (C2419G and A2420U) were used. miR-433 was capable of decreasing luciferase activity by ∼50% in the case of GCLc 3′-UTR (Fig. 2A) and 35% in the case of GCLm. This downregulation was partially absent in the case of GCLc PM1 and completely abrogated for GCLc PM2. Mutation of the functional site in the GCLm seed sequence (GCLm S2 PM) resulted in only a 20% reduction in luciferase activity (Fig. 2B).
FIG. 1.

Schematic of the 3′-UTRs from GCLc and GCLm mRNAs. (A) The GCLc 3′-UTR is 1414 kb long and contains two seed sites for miR-433 (depicted on top). The first seed site (poorly conserved) is only present in humans, and the second one is an 8-mer seed site conserved across several mammalian species. (B) The GCLm 3′-UTR is 3.7 kb long, and it also contains two target sites for miR-433 at the beginning and end of the sequence (indicated on top). The first one is poorly conserved across diverse species, and the second one is only present in humans. Hsa, Homo Sapiens; Ptr, Pan troglodytes; Mmu, Mus musculus; Rno, Rattus norvegicus; GCLc, glutamyl-cysteine ligase catalytic subunit; GCLm, glutamyl-cysteine ligase modifier subunit; UTR, untranslated region.
FIG. 2.

miR-433 targets the 3′-UTRs of GCLc and GCLm. Luciferase reporter assays in the 3′-UTRs of GCLc and GClm were carried out in COS-7 cells. COS-7 were transfected with their respective psiCHECK2–3′-UTR (125 ng) constructs and miR-433 (40 nM) or its mutated versions for 24 h. (A) The bar graph represents relative luciferase activity of GCLc 3′-UTR referred to as negative control (miR-NC). PM1 is a two-base change in the seed sequence (positions 237–344) for miR-433. PM2 is a mutant that contains both 3′-UTR seed sites for miR-433 mutated in the same bases (positions 237–244 and 1002–1004). (B) The bar graph represents relative luciferase activity of GCLm 3′-UTR referred to as negative control (miR-NC). GCLm site 1 (S1) is a 1 kb sequence that contains the first site for miR-433 (positions 180–186). The 3′-UTR-GCLm site 2 (S2) is a 1 kb construction that contains the second seed sequence (positions 2417–2422). 3′-UTR-GCLm PM2 (S2 PM) contains a two-base change in the S2 construction of GCLm. Data correspond to the mean±SEM of three experiments. *p<0.05; **p<0.01 with regard to control. PM, point mutation.
miR-433 decreases GCLs protein expression, GSH levels, and 2GSH/GSSG ratio while it promotes S-glutathionylation in endothelial cells
To evaluate the effect of miR-433 in endothelial cells, we transfected human umbilical vein endothelial cells (HUVEC) with the synthetic mature miRNA forms corresponding to a negative control (miR-NC), miR-433, or miR-144. miR-433 overexpression induced a significant downregulation of both GCLc and GCLm protein levels, an effect that was not detectable with miR-144 transfection (Fig. 3A). Consistently, expression levels of GCLc and GCLm mRNAs were downregulated in miR-433 transfected cells (Fig. 3B). To test whether the downregulation of GCLs was also detectable in the presence of oxidative stimuli, we treated HUVEC with hydrogen peroxide (H2O2) for different periods of time in the presence and absence of exogenous miR-433. As shown in Figure 3C, exposure to miR-433 was associated with an inhibitory response in the expected upregulation of GCLc and GCLm.
FIG. 3.

miR-433 downregulates the expression of GCL subunits. (A) HUVEC were transfected with 40 nM miRNA of either miR-433 or miR-144 and were lysed after 48 h. GCLc and GCLm protein levels (30 μg) were analyzed by immunoblot. Shown is a representative blot and the quantitative densitometric analysis of n=3 experiments after correction by β-actin. (B) Bar graph represents mean±SEM of GCLc and GCLm mRNA levels determined by qPCR after 48 h of transfection with a negative control (miR-NC, 40 nM), miR-144 (40 nM), or miR-433 (40 nM), n≥3. (C) GCLs downregulation is maintained after exposure to H2O2. HUVEC were kept in starvation without FBS for at least 6 h before treatment with 200 μM H2O2. The bar graph depicts a time course representing mean±SEM of n≥3 densitometric quantifications. *p<0.05; **p<0.01; ***p<0.001 with regard to control; and p<0.05 with regard to its corresponding control time point. FBS, fetal bovine serum; H2O2, hydrogen peroxide; HUVEC, human umbilical vein endothelial cells; GCLs, GCL subunits; microRNA, miRNA; qPCR, quantitative polymerase chain reaction.
To test the repercussion of GCLs downregulation induced by miR-433 on the final product of the biosynthetic pathway, we measured GSH levels in HUVEC, and evaluated the possible consequences of the formation of GSSG. As shown in Figure 4A, miR-433-treated cells showed a 50% reduction in GSH levels, with no significant effect on the oxidized form. In keeping with this result, the 2GSH/GSSG ratio, as determined with the fluorescent probe roGFP, markedly decreased (Fig. 4B). In addition, the redox potential determined according to the Nernst equation (41) was clearly affected (Fig. 4B, left panel). Decreased 2GSH/GSSG ratio is associated with potentially increased S-glutathionylation (48, 56). To verify this, HUVEC were labeled with biotinylated glutathione ethyl ester (BIOGEE) in the presence or absence of S-nitrosoglutathione (GSNO), an inductor of S-thiolation (23). Treatment with miR-433 resulted in a conspicuous increase in the number of S-glutathionylated proteins (Fig. 4B, right panel) and specific endothelial nitric oxide synthase (eNOS)-S-glutathionylation (Supplementary Fig. S2A); see Supplementary Methods. This effect was observed both in basal conditions and after exposure to GSNO. Taken together, these results strongly support that miR-433 has profound consequences for GSH synthesis, altering the balance toward the formation of GSSG and mixed disulfides. We also evaluated whether the functional effects associated with miR-433-related GSH depletion could cause endothelial dysfunction. In HUVEC transfected with miR-433, we observed a significant reduction in eNOS activating phosphorylation in response to short treatments with H2O2 (Fig. 4C), which suggests that miR-433 has a deleterious effect on vascular function caused by GSH depletion (10, 30).
FIG. 4.

miR-433 shifts the redox balance of vascular endothelial cells toward a pro-oxidative state. (A) HUVEC were transfected with miR-433 (40 nM, 48 h) or control miRNA (miR-NC), and GSH (left) and GSSG (right) levels were determined as indicated in methods. Bar graph represents mean±SEM of at least n=3 experiments (B) The 2GSH/GSSG ratio and redox potential (Ero GFP) were determined in HUVEC after transfection with miR-433 (40 nM, 48 h) or miR-NC as indicated in methods. Bar graphs represent mean±SEM of n≥3. Global S-glutathionylation (right panel) was studied by labeling cells with BIOGEE. Cells were transfected with miR-433 or miR-NC and treated with GSNO (50 μM, 1 h) before labeling. Pull-downs of samples were analyzed with antistreptavidin. The blot is representative of n=3. (C) HUVEC were transfected with miR-433 (40 nM, 48 h), were kept without FBS for at least 6 h before treatment with H202 (200 μM) for the indicated time points, and analyzed by Western blot against p-eNOS (S1177) and total eNOS. The bar graph (right panel) represents the quantification of the ratio p-eNOS/eNOS in n=4 blots. *p<0.05; **p<0.01 with regard to control. †p<0.05 with regard to the corresponding control time point. BIOGEE, biotinylated glutathione ethyl ester; eNOS, endothelial nitric oxide synthase; GSSG, oxidized form of glutathione; GSH, glutathione.
miR-433 induces nitroxidative stress
To analyze whether miR-433 also generated an increase in reactive oxygen species (ROS) production and therefore an imbalance in redox state, we measured ROS production with a probe for general ROS detection, 2′,7′-dichlorofluorescein; see Supplementary Methods. As shown in Figure 5A, miR-433 increased cellular oxidative stress, in both the presence and absence of H2O2.
FIG. 5.

miR-433 overexpression increases nitroxidative stress. (A) HUVEC were transfected with miR-433 (40 nM, 48 h) or control miRNA (miR-NC); then, cells were treated with H2O2 (500 μM) for 5 min; and HUVEC were incubated for 30 min at 37°C with the DCF probe (2.5 μM). Cells were analyzed by fluorescence-activated cell sorting. Bar graph represents fluorescence levels in arbitrary units (mean±SEM, n=4) (B) Representative Western blot for 3-Nitro-Tyr staining. HUVEC were transfected with miR-NC or miR-433 (40 nM) for 48 h and were kept without FBS at least 6 h before treatment with SIN-1 (500 μM) for 1 h. The bar graph (right panel) represents the densitometric quantification, mean±SEM of n=4. *p<0.05 with regard to control in the absence of any treatment; ‡p<0.05 with regard to miR-433 treatment without SIN-1. DCF, 2′,7′-Dichlorofluorescein.
To further confirm the pro-oxidative action of miR-433, we studied its potential effect on tyrosine nitration in human endothelial cells. In basal conditions, overexpression of miR-433 was associated with a visible increase in global protein nitration, which was enhanced with SIN-1 treatment (Fig. 5B). Taken together, these data support that miR-433 is able to promote an increase in nitroxidative stress by itself.
Opposite regulation of GCLs and miR-433 expression by pro-oxidative stimuli
To evaluate the effect of oxidative stress on the expression of GCLs and miR-433, we tested the effect of GSH depletion in HUVEC by using the chemical inhibitor of GSH synthesis, L-buthionine-sulfoximine (L-BSO), that was effective in significantly depleting reduced GSH levels by 80% (data not shown) and, as expected, increased protein levels of GCLm significantly (Fig. 6A). The mRNA levels of GCLc experimented similar changes (Fig. 6B). This has been attributed to the regulatory feedback between the expression of GCLs and the final product of the reaction, GSH (15, 37). Of interest, L-BSO drastically reduced miR-433 levels (Fig. 6C), suggesting that depletion of GSH levels may also trigger responses directed toward the suppression of molecules inhibiting GCLs expression. Liver injury has also been related to redox damage and GSH depletion (53, 73). To explore the role of miR-433 in this context, we exposed HepG2, a hepatic cell line, to oxidative stress promoted by increasing concentrations of H2O2. As shown in Supplementary Figure S3, H2O2 significantly induced the expression of GCLc and GCLm to 8 h with progressive decrease after this time point (Supplementary Fig. S3A). In concordance with these observations, H2O2 also significantly decreased miR-433 expression in a time-dependent fashion to 8 h (Supplementary Fig. S3B), consistently supporting an inverse regulation of GCLs and miR-433 by oxidative stimuli.
FIG. 6.

GSH depletion upregulates GCLs while it represses miR-433 levels. HUVEC were treated with 500 μM L-BSO for the indicated time points or for 8 h where not shown. (A) Representative Western blot of n≥3 depicting GCLc and GCLm protein levels. Bar graph (right) represents densitometric analyses (mean±SEM). (B) Bar graph represents mRNA levels of GCLs from at least 3 different experiments (mean±SEM). (C) Bar graph represents mean±SEM of miR-433 levels quantified by qPCR after exposure of HUVEC to L-BSO, *p<0.05 with regard to control. L-BSO, L- buthionine-sulfoximine.
miR-433 decreases the expression of both GCLs by an Nrf2-independent mechanism
Nrf2 is a transcription factor that plays a fundamental role in antioxidant responses (20). Several miRNAs have been shown to interfere with the Nrf2-Keap1-Bach-ARE pathways (7, 11, 20). Among them, miR-144 has been described to downregulate GCLs expression by an Nrf2-dependent mechanism in neuronal (44) and erythroid cells (57). Thus, we asked whether Nrf2 may also mediate the inhibitory effect of miR-433 on GCLs expression in endothelial cells. To confirm or refute whether the effect of miR-433 on GCLs levels was independent from Nrf2, we determined mRNA and protein levels of this transcription factor after overexpression with miR-433 in HUVEC and compared its potential effects with those of miR-144. As shown in Figure 7A, miR-433 had no effect on the protein or mRNA levels of Nrf2. miR-144 has been described to modify Nrf2 levels (44, 57). In our hands, we detected a tendency in this direction at both the mRNA and protein levels that did not reach statistical significance. These data suggest that the downregulation of GCLc and GCLm induced by miR-433 is independent from Nrf2. However, it is important to emphasize that Nrf2 remains a key element controlling the expression of GCLs, as its knockdown was associated with a clear decrement in the basal levels of both GCLs, more evident in the case of GCLm (Fig. 7B upper panels). In addition, Nrf2 appears to play a role in governing the expression of miR-433, as its absence results in reduced miRNA levels (Fig. 7B lower panel). This is also consistent with the observation that L-BSO treatment was able to abrogate the expression of miR-433 (Fig. 6C) as an absence of antioxidant pathways such as Nrf2 should also promote the reduction of inhibitory molecules resulting in reduced GSH levels, such as miR-433.
FIG. 7.

miR-433 does not regulate Nrf2, but Nrf2 is necessary for the full expression of GCLs and miR-433. (A) Shown on top is a Western blot depicting Nrf2 protein levels in HUVEC transfected with a negative control (miR-NC), miR-144, or miR-433 (all 40 nM, 48 h). Bar graphs represent mean±SEM of n≥3 experiments for protein (left) and mRNA levels (right). (B) Effect of Nrf2 RNA silencing on GCLc and GCLm expression (protein and mRNA levels) in HUVEC. Representative Western blot (top left) of Nrf2 and GCLs protein levels after treatment with siRNA control (SC) or siRNA for Nrf2 (40 nM, 48 h). Bar graphs represent GCLs mRNA (top right panel) and miR-433 (lower panel) levels after Nrf2 silencing, mean±SEM of at least three experiments, *p<0.05 with regard to siRNA control. Nrf2, nuclear factor (erythroid-derived 2)-like 2; siRNA, small-interfering RNA.
miR-433 regulates redox responses associated to injury and fibrogenesis
In order to investigate the potential role of miR-433 in pathophysiological contexts, we addressed its function in two different models of injury. The first one consisted of inducing liver injury by BDL, a procedure which triggers cholestasis, liver dysfunction, and, eventually, fibrosis that courses with GSH depletion (53, 72). The drop in the levels of GCLc and GCLm was visible after 1 day and reflected in lesser quantified protein abundance and mRNA levels (GCLm) at 21 and 7–14 days after the surgical procedure, respectively (Fig. 8A, B). The levels of miR-433 were significantly increased at 14 and 21 days after BDL (Fig. 8C), thus supporting a relationship between the decrement of GCLs and upregulation of the miRNA. To evaluate the cellular responses that could underlie the effects described earlier, we employed the hepatic cell line Huh7 and exposed it to TGF-β1, the quintessential profibrotic cytokine. Treatment of Huh7 hepatoma cells with TGF-β1 induced a reduction in the expression of GCLs and also diminished GSH levels at 48 h (Supplementary Fig. S4A, B). In addition, TGF-β1 treatment at 48 h dramatically increased miR-433 levels (Supplementary Fig. S4C). Pretreatment with the inhibitory antagomir (antimiR-433) for miR-433 rescued TGF-β1-induced inhibition of GCLs expression (Fig. 9A). As could be expected, TGF-β1 was capable of inducing the expression of the fibrotic marker αSMA and interestingly, transfection with the antagomir prevented α-SMA TGF-β1-dependent increase (Fig. 9B).
FIG. 8.

Bile duct ligation induces GCLs downregulation and increases miR-433 levels. (A) Western blot showing GCLc and GCLm protein levels of whole livers at 1, 7, 14, and 21 days postoperation. Bar graph represents densitometric analyses from at least three mice (mean±SEM, *p<0.05 with regard to sham-operated mice). (B) GCLc and GCLm mRNA expression in operated livers analyzed by qPCR. (C) miR-433 expression in operated livers analyzed by qPCR. Bar graphs represent RNA levels, mean±SEM n≥3 *p<0.05; **p<0.01 with regard to 1 day time point.
FIG. 9.

Antagonism of miR-433 prevents TGF-β-induced GCLs downregulation and fibrogenic phenotype in hepatic cells. (A) GCLc and GCLm protein analysis in Huh7 cells transfected with antimiR control (antimiR-NC) or antimiR-433 (both 40 nM, 48 h) and treated or not (0 h) with 5 ng/ml TGF-β1 for 8 and 24 h. Bar graphs represent densitometric quantifications of protein levels (n≥3, mean±SEM). (B) The blot depicts α-SMA protein levels in Huh7 cells transfected with 40 nM antagomiRs (48 h) and treated with 5 ng/ml TGF-β1 for 8 and 24 h. Bar graphs represent densitometric analyses of protein levels, mean±SEM n≥3; *p<0.05 with regard to control untreated antimiR-NC; ‡p<0.05 with regard to control untreated antimiR-433. †p<0.05; ††p<0.01; †††p<0.001 with regard to corresponding control time points. αSMA, α-smooth muscle actin; TGF-β1, transforming growth factor β1.
The second model, unilateral ureteral obstruction (UUO), results in obstructive uropathy of one kidney and abolition of its function due to renal fibrosis (65, 66). This fibrotic process is characterized by the activation of the TGF-β pathway. We found that the expression of GCLs was significantly decreased in the obstructed kidney after 5, 10, and 15 days of UUO (Fig. 10A, B and Supplementary Fig. S5B). As expected, TGF-β and fibronectin levels were significantly higher after 5 days (Fig. 10B right panel). To evaluate further renal damage, two novel described biomarkers, kidney injury molecule 1 (Kim-1), related to the transition from acute to chronic renal damage (25, 47) and neutrophil gelatinase-associated lipocalin (Ngal), an early marker of kidney disease (12), were also evaluated. Ngal and Kim-1 gene expression became increased at 5 and 10 days after UUO surgery compared with control mice (Supplementary Fig. S5A). Importantly, miR-433 was also significantly upregulated in the obstructed kidney at 5 days post-UUO (Fig. 10C), supporting the notion that miR-433 is involved in the reduction of GCLs levels promoted by UUO. Collectively, these results suggest not only that both UUO and BDL are good models for exploring TGF-β-mediated inhibition of GCLs but also, and more importantly, that miR-433 reveals itself as a potential mediator of injury and fibrogenesis through its inhibitory action on GCLs expression.
FIG. 10.

Renal fibrosis caused by UUO is associated to GCLc and GCLm dowregulation and elevated levels of miR-433. (A) Representative Western blot of GCLc and GCLm renal expression levels in UUO model at 5 days postoperation compared with nonobstructed contralateral (CON) kidney. Bar graphs correspond to densitometric analyses from at least four mice from each group (mean±SEM). (B) Renal gene expression of GCLc, GCLm, TGF-β, and FN with UUO during 5 days was analyzed by qPCR. (C) Renal miR-433 expression was determined by qPCR in contralateral (CON) or obstructed (UUO) kidneys at 5 days postsurgery. Bar graphs represent quantifications of RNA levels, mean±SEM, n≥3; *p<0.05; **p<0.01; and ***p<0.001 with regard to contralateral kidney. FN, fibronectin; UUO, unilateral ureteral obstruction.
Discussion
Since their discovery, miRNAs have progressively moved to central stage in our understanding of the post-transcriptional regulation of gene expression (1, 4, 5, 18); it is now difficult to conceive a cellular pathway in which these small molecules do not play an important regulatory role. However, only very recently has the contribution of miRNAs been formally addressed in the field of redox biology (11, 46). It is now clear that redox stress modulates miRNAs biogenesis (43, 52, 67) and, indeed, miRNAs control redox homeostasis at multiple levels, including antioxidant responses. Our results add new insights into the balance of the major endogenous redox pair, 2GSH/GSSG, by identifying miR-433 as a new regulatory player of GSH synthesis through the specific targeting of GCL.
As the rate limiting enzyme involved in GSH synthesis, the regulation of GCL has been the object of thorough study. Both transcriptional and post-transcriptional mechanisms are important, and their role has been reviewed in depth elsewhere (15, 36, 37). Information on the involvement of miRNAs in these enzymes' final level of expression is limited. miR-144 has been studied in detail and demonstrated to interfere with GCLs' abundance in several reports (44, 57), which concur in the notion that the Nrf2 pathway plays an important role as an intermediate target and, hence, the mechanism of action of miR-144 appears to be indirect. Our data are consistent with an effect by which miR-433 downregulates GCLs expression in an Nrf2-independent manner. This is sustained by the fact that overexpression of miR-433 in HUVEC does not modify Nrf2 mRNA or protein levels (Fig. 7A). A regulatory role of Nrf2 in miR-433 cannot be excluded, as its knockdown appears to reduce the levels of the miRNA. However, an extensive analysis of 10 kb of the miR-433 promoter region (human chromosome 14) did not reveal evident ARE binding sites dosing blast algorithm (data not shown), thus suggesting an indirect mechanism for this downregulation. It is possible that the potential depletion of the antioxidant systems (GSH in particular) promoted by miR-433 or Nrf2 depletion and the unbalance in the nucleophilic tone (14) toward a more oxidative state could tend to reduce the expression of pro-oxidant molecules such as miR-433 as a part of the cellular homeostatic response. This is in line with our observations that increased levels of H2O2, well above its signaling range, promote the repression of miR-433 (Supplementary Fig. S3). Clearly and importantly, as shown by others and confirmed here, Nrf2 is essential for the transcriptional regulation of GCLs (9, 20, 37) (Fig. 7B).
The importance of redox unbalance in vascular and endothelial dysfunction has been well known for at least two decades and reviewed elsewhere (6, 60). In the particular case of GSH, several reports have shown that reduction in the levels of this antioxidant results in endothelial dysfunction (8, 10, 30). Recent and enlightening evidence was provided by the genetic deficiency of GCLm in a mouse model (69). The authors of this study provided conclusive evidence that haploinsufficiency of GCLm is associated to alteration of aortic vascular reactivity in classical organ bath experiments where aortic strips precontracted with phenylephrine showed reduced vasodilatation in response to acetylcholine. Significantly, complete deficiency of GCLm was associated to enhanced vasorelaxation but greater sensitivity to vasoconstriction, perhaps indicating that only precise levels of GSH allow appropriate homeostatic responses. Our results are consistent with the idea that reduced levels of GSH mediated by miR-433 result in functional biochemical changes associated to endothelial dysfunction such as increased global and specific S-glutathionylation (Fig. 4B and Supplementary Fig. S2) and diminished eNOS activation (Fig. 4C). These data suggest that GCL is a key enzyme whose intact functionality is indispensable for a correct endothelial and vascular response.
Beyond the direct effects of miR-433 on endothelial function derived from the abrogation of GSH synthesis and reduced nucleophilic tone, we have explored other potential pathophysiological consequences. Here, we focused on the process of fibrogenesis for several reasons: (1) There is an established link between oxidative stress and fibrosis mainly due to the reciprocal relationship between TGF-β, a major profibrotic cytokine, and redox homeostasis (21, 50); (ii) The ability of TGF-β to downregulate the expression of GCL has been known for over a decade. In this regard, TGF-β has been shown to decrease GSH in various cell types. The mechanism appears to relate to the inhibition of GCL gene expression due to the hindrance of Nrf2 binding to the ARE sequence promoted by the competition of Smad3 or small Maf proteins (3, 16, 27, 53, 55). (iii) A recent important report has shown that miR-433 is upregulated by TGF-β1 and is relevant to the development of renal fibrosis (32). By using two different experimental models of organ fibrosis in the kidney and liver, UUO and BDL respectively, we confirmed the repression of GCL expression under the induction of fibrogenesis (Figs. 8 and 10A, B). These observations were recapitulated by the exposure of liver-derived cells to TGF-β1. An important novelty is that antagonism of miR-433 prevented the downregulation of GCLs in this cell type (Fig. 9A). We believe that this finding adds new mechanistic insights to the reports mentioned earlier and allows us to propose a model (Fig. 11) in which fibrotic stimuli, such as TGF-β1, downregulate GCLs and GSH levels by specifically inducing miR-433. We are tempted to propose a scenario in which a biphasic response is mounted in the context of inflammation, leading to fibrosis. In the early stage of damage, the expression of GCLs is quickly upregulated, mediated by Nrf2 activation. When the exposure to injury persists, as is the case in profibrotic TGF-β-dependent processes, miR-433 expression increases progressively, reducing GCLs expression and GSH levels. This, in turn, results in an unbalance in the nucleophilic cellular tone in the cell and could contribute toward limiting miR-433 expression. However, persistent damage overcomes this situation, leading to deposition of extracellular matrix (ECM) proteins and fibrosis.
FIG. 11.
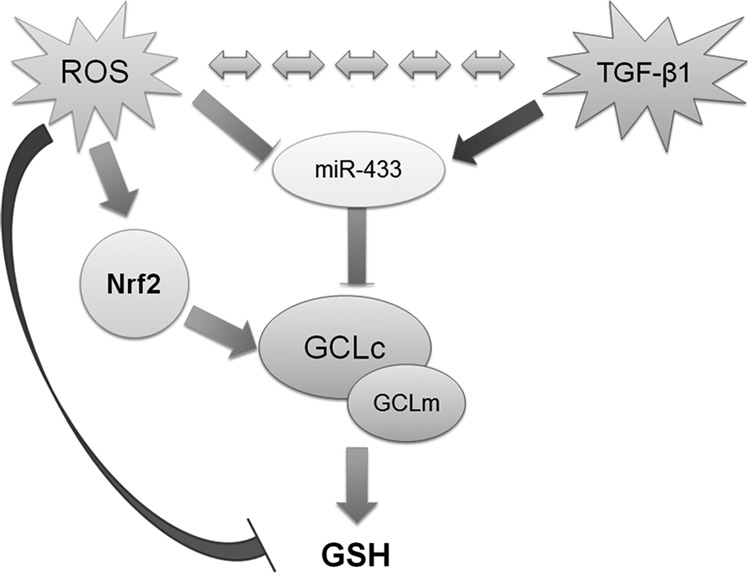
Proposed model of interaction of miR-433 with regulators and targets. miR-433 is susceptible to regulation by ROS and TGF-β. ROS induces activation of the Nrf2 pathway, an important mediator in the expression of GCLs. On the other hand, ROS and TGF-β are reciprocally regulated. An increase in the activation in TGF-β results in elevated miR-433 levels targeting GCLc and GCLm, contributing to both reduced GSH levels and nucleophilic tone. ROS, reactive oxygen species.
The term “redoximiRs” has been recently proposed to describe a set of miRNAs that participate in redox responses both as direct regulatory molecules of the post-transcriptional expression of several pathways and as indirect modulators of the redox homeostatic response (11). In addition, the term “fibromiRs” has been coined to suggest the orchestrated influence of a panoply of miRNAs participating in the regulation of fibrogenesis through several mechanisms (51). As expected, and even though this has not yet been formally established, these two functional families of miRNAs overlap. We have now identified a new member, miR-433, which we suggest belongs to both subgroups of miRNAs due to its capacity to regulate the levels of a critical target, GCL, that is essential for homeostatic and hormetic redox responses, including those involved in fibrogenesis (27). We believe that changes observed in the levels of miR-433 in response to variations in the redox state are specific. As shown in Supplementary Figure S6, both the redoximir miR-34a (61, 64) and the fibromir miR-21 (49, 51) behave similarly to miR-433 when exposed to BSO. However, when Nrf2 was silenced, miR-34a and miR-21 exhibited a tendency to increase. In the UUO model, both miRNAs were not significantly upregulated, in contrast to miR-433 (74). Corroboration of this belief should arrive from the study of further pathophysiological scenarios where redox responses are already known to be key players in the process of organ fibrosis.
Materials and Methods
Reagents
H2O2, L-BSO, N-ethylmaleimide (NEM), and GSNO were purchased from Sigma. BIOGEE was purchased from Life's Technologies. TGF-β1 was purchased from R & D systems.
Cell culture
HUVEC were obtained from umbilical cords of normal deliveries (after approval by the ethics committee of the Hospital “Ruber Internacional”). For isolation, umbilical cords were digested with 0.1% collagenase at 37°C for 20 min. Endothelial cells were collected and grown on 0.2% gelatin with Medium EGM2 (Lonza) at 37°C in 5% CO2. Human tumor hepatic cells, HepG2 and Huh7, were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2% penicillin-streptomycin, and L-glutamine and nonessential amino acids at 37°C in 5% CO2. COS-7 cells were grown in DMEM supplemented with 10% FBS, 2% penicillin-streptomycin at 37°C in 5% CO2.
Transfection of miRNA mimics and miRNA antimiRNAs in human cells. HUVEC at 70% confluence were transfected with 40 nM mirVana mimic (miR-433) or with 40 nM antimiR miRNA inhibitor (antimiR-433) (Life Technologies) by utilizing Lipofectamine 2000 (Invitrogen) and studied 48 h later. In all experiments, an equal concentration of a nontargeting control mimic sequence (miR-NC) or an antimiR negative control (antimiR-NC) sequence was used as a control for nonsequence-specific effects in miRNA experiments.
3′-UTR luciferase reporter assays
cDNA fragments corresponding to the 3′-UTRs of human GCLc and GCLm were amplified by reverse transcription-polymerase chain reaction (PCR) from genomic DNA extracted from HUVEC with XhoI and NotI linkers. The PCR products were directionally cloned downstream of the Renilla luciferase open reading frame in the psiCHECK2 vector (Promega), which also contains a constitutively expressed firefly luciferase gene, which is used to normalize transfections. Site-directed mutations in the seed region of predicted miR-433 sites within the 3′-UTRs were generated by using the Multisite-QuikChange directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. All constructs were sequenced before use to confirm their proper structure. COS-7 cells were plated into 12-well plates and cotransfected with 125 μg of the indicated 3′-UTR luciferase reporter vectors and the miR-433 mimic or negative control by using Lipofectamine 2000. Luciferase activity was measured by using the Dual-Glo luciferase assay system (Promega). Renilla luciferase activity was normalized to the corresponding firefly luciferase activity and plotted as a percentage of the control. Experiments were performed in triplicate samples at least thrice.
Western blot and densitometric analysis
After treatment, cells were washed in phosphate-buffered saline (PBS) and lysed in RIPA lysis buffer (25 mM Tris/HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate solution, 1 mM PMSF, 0.2 mM Na3VO4, and 0.2 mM NaF). Cells were harvested by scraping, and proteins were analyzed by Western blot as previously described. Blots were probed with anti-GCLc (generous gift of Dr. Kavanagh's laboratory), anti-GCLm and anti-Nrf2 (Santa Cruz Biotechnology), and anti-β-actin antibody (Sigma). eNOS and p-eNOS (Ser1177) antibodies were purchased from Cell Signaling. Secondary antibodies against rabbit and mouse were from LI-COR. Densitometry of n≥3 Western blots was done using β-actin or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as housekeeping controls for the total levels of protein loaded. p-eNOS analysis was corrected by the levels of eNOS, after normalization with β-actin. These analyses were represented in bar graphs showing the mean±SEM of protein with regard to controls.
Real-time PCR
Total RNA and miRNA were isolated with miRNeasy Mini Kit (Qiagen). Reverse transcription was performed with 1 μg/μl mRNA sample using iScript cDNA synthesis kit (Bio-Rad) or in the case of miRNAs, Universal cDNA Synthesis and Master Mix (Exiqon). cDNA expression was analyzed by determining SYBR green-based real-time quantitative PCR with Bio-Rad master mix, using specific primers (Sigma). miRNA expression was determined using specific miRNA Locked Nucleic Acid (LNA™) PCR primers (Exiqon) or for tissue samples, specific Taqman probes (Life Technologies). The relative quantification of gene expression was determined using the 2−ΔΔCt method (35). Using this method, we obtained the fold changes in gene expression normalized to an internal control gene, GAPDH or U6 snRNA, respectively.
Small-interfering RNA design and transfection
HUVEC were transfected with specific small-interfering RNA (siRNA)-targeting constructs as previously described and analyzed for the presence of Nrf2 (40 nM, 48 h), GCLc, and GCLm (40 nM, 48 h). Duplex siRNA-targeting constructs were purchased from Ambion (Life Technologies).
GSH assay
Reduced and oxidized forms of GSH were measured in HUVEC utilizing GSH-Glo assay and 2GSH/GSSG assay (Promega) according to their protocol.
roGFP2 probes
Redox status was measured using ro-GFP adenovirus probe (19, 41). HUVEC were transfected with the appropriate miRNA as described, left for 24 h, infected for 2 h, and washed with PBS and the medium was replaced. 2GSH/GSSG ratio was analyzed after 24 h by fluorescence-activated cell sorting after blocking with NEM 100 μM for 5 min. They were then fixed with paraformaldehyde 4% at 37°C for 5 min. The samples were excited by FL1 (485 nm) and FL8 (405 nm) lasers, and emission was recorded at 552 nm. The ratio was calculated by analyzing the values of fluorescence obtained with the reduced form (485 nm) divided by those of the oxidized form (405 nm).
Protein S-glutathionylation assay
HUVEC were transiently transfected for 48 h, and were deprived of FBS 16 h before treatments. They were treated with GSNO 50 μM for 1 h, and then incubated with BIOGEE for 30 min. Cells were lysed in RIPA lysis buffer. S-Glutathionylated proteins were precipitated with A-Sepharose beads (Thermo Scientific) and analyzed by Western blot in nonreducing conditions with antistreptavidin antibody (LI-COR).
Protein 3-nitro-tyrosine assay
HUVEC were transiently transfected for 48 h, and were deprived of FBS at least 6 h before treatments. They were treated with SIN-1 500 μM for 1 h. Cells were lysed in RIPA lysis buffer and analyzed by Western blot in nonreducing conditions with anti-3-Nitro-Tyr antibody (Millipore).
Bile duct ligation
Studies were conducted according to the NIH Guide for the Care and Use of Laboratory Animals. Male C57BL/6 mice (3 months old) were anesthesized and bile duct was ligated, as described (73). In addition, another group of mice was subjected to surgery without ligation (Sham). Animals were sacrificed at 1, 7, 14, and 21 days after surgery. Liver was snap frozen in liquid nitrogen for RNA and protein studies.
Unilateral ureteral obstruction
Studies were conducted according to the NIH Guide for the Care and Use of Laboratory Animals. Male C57BL/6 mice (12–14-week-old) were anesthesized, the left ureter was ligated, and the kidney was obstructed (n=4–8/group), using the contralateral kidney as control (nonobstructed), as previously described (13). Animals were sacrificed at 5, 10, and 15 days after surgery. Kidneys were perfused in situ with cold saline before removal. Half a kidney was snap frozen in liquid nitrogen for RNA and protein studies.
Statistical analysis
Statistical analysis was performed with GraphPadPrism (GraphPad Software). Data are expressed as means±SEM. Differences among groups with one experimental condition were assessed with Kruskal–Wallis test with Dunn's correction. Two-way ANOVA with Bonferroni correction was used to analyze differences among groups exposed to more than one condition. Differences between only two groups were analyzed with Mann–Whitney two-tailed test.
Supplementary Material
Abbreviations Used
- αSMA
α-smooth muscle actin
- AP-1
activator protein 1
- ARE
antioxidant response element
- ATP
adenosine triphosphate
- BDL
bile duct ligation
- BIOGEE
biotinylated glutathione ethyl ester
- CON
contralateral
- DCF
2′,7′-Dichlorofluorescein
- DMEM
Dulbecco's modified Eagle's medium
- ECM
extracellular matrix
- eNOS
endothelial nitric oxide synthase
- FBS
fetal bovine serum
- FN
fibronectin
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GCL
γ-glutamyl-cysteine ligase
- GCLc
glutamyl-cysteine ligase catalytic subunit
- GCLm
glutamyl-cysteine ligase modifier subunit
- GS
glutathione synthase
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- GSSG
oxidized form of glutathione
- H2O2
hydrogen peroxide
- Hsa
Homo sapiens
- HUVEC
human umbilical vein endothelial cells
- Kim-1
kidney injury molecule 1
- L-BSO
L-buthionine-sulfoximine
- microRNA
miRNA
- Mmu
Mus musculus
- NEM
N-ethylmaleimide
- NFκB
nuclear factor κB
- Ngal
neutrophil gelatinase-associated lipocalin
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PM
point mutation
- Ptr
Pan troglodytes
- Rno
Rattus norvegicus
- ROS
reactive oxygen species
- SIN-1
3-morpholinosydnonimine
- siRNA
small-interfering RNA
- TGF-β1
transforming growth factor β1
- UTR
untranslated region
- UUO
unilateral ureteral obstruction
Acknowledgments
Funding agencies: Ministerio de Economía y Competitividad (MINECO): SAF 2012-31338 (S.L.), CSD 2007-00020 (S.L.), Instituto de Salud Carlos III RD12/0021/0009 (S.L. and M.R.-O.), PI11/01854 (M.R.-O.), Comunidad de Madrid “Fibroteam” S2010/BMD-2321 (S.L. and M.R.-O.), and Fundación Renal “Iñigo Alvarez de Toledo,” all of which are in Spain. This study was supported by COST actions BM-1203 (EU-ROS) and BM-1005 (ENOGAS). C.E.-D is a fellow of the FPI program from MINECO. The authors are indebted to all the members of their lab (S.L.) for helpful discussions.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Ambros V. microRNAs: tiny regulators with great potential. Cell 107: 823–826, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Baker AH. and van Rooij E. miRNA overexpression induces cardiomyocyte proliferation in vivo. Mol Ther 21: 497–498, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakin AV, Stourman NV, Sekhar KR, Rinehart C, Yan X, Meredith MJ, Arteaga CL, and Freeman ML. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding Phase II detoxifying proteins. Free Radic Biol Med 38: 375–387, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breton-Romero R. and Lamas S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol 2: 529–534, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryan HK, Olayanju A, Goldring CE, and Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 85: 705–717, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Crabtree MJ, Brixey R, Batchelor H, Hale AB, and Channon KM. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J Biol Chem 288: 561–569, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapple SJ, Siow RC, and Mann GE. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol 44: 1315–1320, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, and Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng X, Ku CH, and Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic Biol Med 64: 4–11, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Devarajan P. Neutrophil gelatinase-associated lipocalin (NGAL): a new marker of kidney disease. Scand J Clin Lab Invest Suppl 241: 89–94, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esteban V, Lorenzo O, Ruperez M, Suzuki Y, Mezzano S, Blanco J, Kretzler M, Sugaya T, Egido J, and Ruiz-Ortega M. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol 15: 1514–1529, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Forman HJ, Davies KJ, and Ursini F. How do nutritional antioxidants really work: nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic Biol Med 66: 24–35, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, and Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med 30: 86–98, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franklin CC, Rosenfeld-Franklin ME, White C, Kavanagh TJ, and Fausto N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. FASEB J 17: 1535–1537, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Fu Y, Zheng S, Lu SC, and Chen A. Epigallocatechin-3-gallate inhibits growth of activated hepatic stellate cells by enhancing the capacity of glutathione synthesis. Mol Pharmacol 73: 1465–1473, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo H, Ingolia NT, Weissman JS, and Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, and Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5: 553–559, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Hayes JD. and Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39: 199–218, 2014 [DOI] [PubMed] [Google Scholar]
- 21.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, and Thannickal VJ. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 6: 231ra47, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang CS, Chang LS, Anderson ME, and Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem 268: 19675–19680, 1993 [PubMed] [Google Scholar]
- 23.Huang KP. and Huang FL. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem Pharmacol 64: 1049–1056, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, and Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem 67: 129–139, 2011 [DOI] [PubMed] [Google Scholar]
- 25.Humphreys BD, Xu F, Sabbisetti V, Grgic I, Naini SM, Wang N, Chen G, Xiao S, Patel D, Henderson JM, Ichimura T, Mou S, Soeung S, McMahon AP, Kuchroo VK, and Bonventre JV. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 123: 4023–4035, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huntzinger E. and Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12: 99–110, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Jardine H, MacNee W, Donaldson K, and Rahman I. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. J Biol Chem 277: 21158–21166, 2002 [DOI] [PubMed] [Google Scholar]
- 28.Krejsa CM, Franklin CC, White CC, Ledbetter JA, Schieven GL, and Kavanagh TJ. Rapid activation of glutamate cysteine ligase following oxidative stress. J Biol Chem 285: 16116–16124, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langston W, Circu ML, and Aw TY. Insulin stimulation of gamma-glutamylcysteine ligase catalytic subunit expression increases endothelial GSH during oxidative stress: influence of low glucose. Free Radic Biol Med 45: 1591–1599, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laursen JB, Boesgaard S, Trautner S, Rubin I, Poulsen HE, and Aldershvile J. Endothelium-dependent vasorelaxation in inhibited by in vivo depletion of vascular thiol levels: role of endothelial nitric oxide synthase. Free Radic Res 35: 387–394, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Leonardo TR, Schultheisz HL, Loring JF, and Laurent LC. The functions of microRNAs in pluripotency and reprogramming. Nat Cell Biol 14: 1114–1121, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R, Chung AC, Dong Y, Yang W, Zhong X, and Lan HY. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-beta/Smad3-Azin1 pathway. Kidney Int 84: 1129–1144, 2013 [DOI] [PubMed] [Google Scholar]
- 33.Liu RM, Liu Y, Forman HJ, Olman M, and Tarpey MM. Glutathione regulates transforming growth factor-beta-stimulated collagen production in fibroblasts. Am J Physiol Lung Cell Mol Physiol 286: L121–L128, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Liu RM, Vayalil PK, Ballinger C, Dickinson DA, Huang WT, Wang S, Kavanagh TJ, Matthews QL, and Postlethwait EM. Transforming growth factor beta suppresses glutamate-cysteine ligase gene expression and induces oxidative stress in a lung fibrosis model. Free Radic Biol Med 53: 554–563, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ. and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 36.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med 30: 42–59, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu SC. Glutathione synthesis. Biochim Biophys Acta 1830: 3143–3153, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu SC. and Ge JL. Loss of suppression of GSH synthesis at low cell density in primary cultures of rat hepatocytes. Am J Physiol 263: C1181–C1189, 1992 [DOI] [PubMed] [Google Scholar]
- 39.Magenta A, Greco S, Gaetano C, and Martelli F. Oxidative stress and microRNAs in vascular diseases. Int J Mol Sci 14: 17319–17346, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meister A. Glutathione metabolism and its selective modification. J Biol Chem 263: 17205–17208, 1988 [PubMed] [Google Scholar]
- 41.Meyer AJ. and Dick TP. Fluorescent protein-based redox probes. Antioxid Redox Signal 13: 621–650, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Morgan B, Ezerina D, Amoako TN, Riemer J, Seedorf M, and Dick TP. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat Chem Biol 9: 119–125, 2013 [DOI] [PubMed] [Google Scholar]
- 43.Mori MA, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y, Russell SJ, Kirkland JL, Blackwell TK, and Kahn CR. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab 16: 336–347, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, and Mahimainathan L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS One 7: e51111, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neuschwander-Tetri BA, Nicholson C, Wells LD, and Tracy TF., Jr Cholestatic liver injury down-regulates hepatic glutathione synthesis. J Surg Res 63: 447–451, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Ouyang Y-B, Creed M, Stary CM, White RE, and Giffard RG. The use of microRNAs to modulate redox and immune response to stroke. Antioxid Redox Signal 22: 187–202, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parikh CR. and Devarajan P. New biomarkers of acute kidney injury. Crit Care Med 36: S159–S165, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Pastore A. and Piemonte F. S-Glutathionylation signaling in cell biology: progress and prospects. Eur J Pharm Sci 46: 279–292, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Patel V. and Noureddine L. MicroRNAs and fibrosis. Curr Opin Nephrol Hypertens 21: 410–416, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peshavariya HM, Chan EC, Liu GS, Jiang F, and Dusting GJ. Transforming growth factor-beta1 requires NADPH oxidase 4 for angiogenesis in vitro and in vivo. J Cell Mol Med 18: 1172–1183, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pottier N, Cauffiez C, Perrais M, Barbry P, and Mari B. FibromiRs: translating molecular discoveries into new anti-fibrotic drugs. Trends Pharmacol Sci 35: 119–126, 2014 [DOI] [PubMed] [Google Scholar]
- 52.Poulsen HE, Specht E, Broedbaek K, Henriksen T, Ellervik C, Mandrup-Poulsen T, Tonnesen M, Nielsen PE, Andersen HU, and Weimann A. RNA modifications by oxidation: a novel disease mechanism? Free Radic Biol Med 52: 1353–1361, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Ramani K, Tomasi ML, Yang H, Ko K, and Lu SC. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J Biol Chem 287: 36341–36355, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravuri C, Svineng G, and Huseby NE. Differential regulation of gamma-glutamyltransferase and glutamate cysteine ligase expression after mitochondrial uncoupling: gamma-glutamyltransferase is regulated in an Nrf2- and NFkappaB-independent manner. Free Radic Res 47: 394–403, 2013 [DOI] [PubMed] [Google Scholar]
- 55.Ryoo IG, Shin DH, Kang KS, and Kwak MK. Involvement of Nrf2-GSH signaling in TGFbeta1-stimulated epithelial-to-mesenchymal transition changes in rat renal tubular cells. Arch Pharm Res 2014. [Epub ahead of print]; DOI: 10.1007/s122272-014-0380-y [DOI] [PubMed] [Google Scholar]
- 56.Sanchez-Gomez FJ, Espinosa-Diez C, Dubey M, Dikshit M, and Lamas S. S-glutathionylation: relevance in diabetes and potential role as a biomarker. Biol Chem 394: 1263–1280, 2013 [DOI] [PubMed] [Google Scholar]
- 57.Sangokoya C, Telen MJ, and Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 116: 4338–4348, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santoro MM. and Nicoli S. miRNAs in endothelial cell signaling: the endomiRNAs. Exp Cell Res 319: 1324–1330, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seelig GF. and Meister A. Gamma-glutamylcysteine synthetase. Interactions of an essential sulfhydryl group. J Biol Chem 259: 3534–3538, 1984 [PubMed] [Google Scholar]
- 60.Sena CM, Pereira AM, and Seica R. Endothelial dysfunction—a major mediator of diabetic vascular disease. Biochim Biophys Acta 1832: 2216–2231, 2013 [DOI] [PubMed] [Google Scholar]
- 61.Staszel T, Zapala B, Polus A, Sadakierska-Chudy A, Kiec-Wilk B, Stepien E, Wybranska I, Chojnacka M, and Dembinska-Kiec A. Role of microRNAs in endothelial cell pathophysiology. Pol Arch Med Wewn 121: 361–366, 2011 [PubMed] [Google Scholar]
- 62.Sun WM, Huang ZZ, and Lu SC. Regulation of gamma-glutamylcysteine synthetase by protein phosphorylation. Biochem J 320 (Pt 1): 321–328, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan KP, Yang M, and Ito S. Activation of nuclear factor (erythroid-2 like) factor 2 by toxic bile acids provokes adaptive defense responses to enhance cell survival at the emergence of oxidative stress. Mol Pharmacol 72: 1380–1390, 2007 [DOI] [PubMed] [Google Scholar]
- 64.Truong Do M, Gyun Kim H, Ho Choi J, and Gwang Jeong H. Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1alpha/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic Biol Med 74: 21–34, 2014 [DOI] [PubMed] [Google Scholar]
- 65.Ucero AC, Benito-Martin A, Izquierdo MC, Sanchez-Nino MD, Sanz AB, Ramos AM, Berzal S, Ruiz-Ortega M, Egido J, and Ortiz A. Unilateral ureteral obstruction: beyond obstruction. Int Urol Nephrol 46: 765–776, 2014 [DOI] [PubMed] [Google Scholar]
- 66.Ucero AC, Goncalves S, Benito-Martin A, Santamaria B, Ramos AM, Berzal S, Ruiz-Ortega M, Egido J, and Ortiz A. Obstructive renal injury: from fluid mechanics to molecular cell biology. Open Access J Urol 2: 41–55, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, Csiszar A, Losonczy G, Valcarcel-Ares MN, and Sonntag WE. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci 68: 877–891, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Rooij E, Marshall WS, and Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ Res 103: 919–928, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weldy CS, Luttrell IP, White CC, Morgan-Stevenson V, Bammler TK, Beyer RP, Afsharinejad Z, Kim F, Chitaley K, and Kavanagh TJ. Glutathione (GSH) and the GSH synthesis gene Gclm modulate vascular reactivity in mice. Free Radic Biol Med 53: 1264–1278, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White K, Dempsie Y, Caruso P, Wallace E, McDonald RA, Stevens H, Hatley ME, Van Rooij E, Morrell NW, Maclean MR, and Baker AH. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension 64: 185–194, 2014 [DOI] [PubMed] [Google Scholar]
- 71.Wild AC, Moinova HR, and Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem 274: 33627–33636, 1999 [DOI] [PubMed] [Google Scholar]
- 72.Yang H, Ko K, Xia M, Li TW, Oh P, Li J, and Lu SC. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of glutathione synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology 51: 1291–1301, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang H, Ramani K, Xia M, Ko KS, Li TW, Oh P, Li J, and Lu SC. Dysregulation of glutathione synthesis during cholestasis in mice: molecular mechanisms and therapeutic implications. Hepatology 49: 1982–1991, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou Y, Xiong M, Niu J, Sun Q, Su W, Zen K, Dai C, and Yang J. Secreted fibroblast miR-34a induces tubular cell apoptosis in fibrotic kidney. J Cell Sci, 127(Pt 20): 4494–4506, 2014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.