

Abstract
We investigated the rotational dynamics of single microparticles during their internalization by macrophage cells. The microparticles used were triblock patchy particles that display two fluorescent patches on their two poles. The optical anisotropy made it possible to directly visualize and quantify the orientation and rotation of the particles. We show that particles exhibit a mixture of fast and slow rotation as they are uptaken by macrophages and transiently undergo directional rotation during their entry into the cell. The size of the particles and the surface presentation of ligands exerted a negligible influence on this heterogeneity of particle rotation.
1. Introduction
Understanding the uptake of particles by cells is critical for deciphering the fundamental mechanisms of many biological functions, such as the clearance of pathogens by immune cells.1–3 This understanding is equally necessary for engineering synthetic particles for biomedical applications, from drug and gene delivery to in vivo imaging.4–7 Cellular uptake of particles is a complex process that includes many interactions between the particles, cell membranes, and intracellular proteins. Imaging and tracking the movements of individual particles has been proven a powerful technique for dissecting these complex interactions in this process. This single-particle tracking method allows the translational motions of single particles, viruses, or intracellular organelles to be quantified, and thus makes it possible to probe their dynamics with high spatiotemporal resolutions and reveal information that is otherwise inaccessible with ensemble-average methods.8–17 However, previous studies focused on the translational motion of particles. Little is known about how particles rotate during cellular uptake.
Tracking both the orientation and rotation of single particles is more challenging than conventional methods that track only their center-of-mass. Only a few studies have explored the rotational dynamics of particles in biological systems, and all these studies involved imaging probe particles that were optically anisotropic. Fang and coworkers used gold nanorods as rotational probes because the nanorods exhibit localized surface plasmonic resonance bands that are distinctly different longitudinally and transversely.18 They investigated the rotation of these nanorods upon binding to cell membranes and during intracellular cargo transport in neural cells.19,20 This method offers temporal resolution down to a few milliseconds, but is not suitable for systems with highly scattering background, such as live cells. Quantum dots of various geometries have been utilized in separate studies to measure the orientation of membrane receptors on the surface of living cells.21,22 Tracking of the rotation of fluorescent nanodiamonds inside live cells has also been demonstrated.23 These methods take advantage of the intrinsic optical properties of non-conventional particle probes. A slightly different strategy for visualizing and tracking the rotational dynamics of particles is to create optical anisotropy on otherwise isotropic particles. For instance, to track the orientation and rotation of single viruses, Kukura et al. attached quantum dots onto the outer surface of viruses and measured their orientation by locating the position of both the virus and the quantum dot probe.24 A similar strategy was also used to track the longitudinal rotation of bacteria.25 Kopelman and coworkers investigated the rotation of fluorescent microparticles half-coated with metal, called modulated optical nanoprobes (MOONs), in various non-biological environments.26,27 The Granick group later reported a single-particle tracking method to measure two rotational angles of the MOON particles. They employed the method to study particle rotation in a colloidal glass.28,29 Most of these studies focused on technical demonstrations. The particle rotational dynamics involved in many biological processes, such as in the cellular internalization step, have yet to be explored.
In this paper, we report a quantitative study of the rotational dynamics of single particles during their uptake by macrophages, a process known as phagocytosis. By creating triblock microparticles, which display patches of distinctive fluorescence on their two poles, we directly visualize the rotation of particles as they enter the cell. Our single-particle rotational tracking analysis reveals that particles undergo a mixture of fast and slow rotational movements during macrophage internalization. The effect of surface presentation of the ligand immunoglobulin G (IgG) was explored by coating just one hemisphere of the particle or by completely coating the entire particle with ligand. Our results demonstrate that the surface presentation of ligands has negligible effect on the heterogeneous rotational dynamics.
2. Experimental
2.1 Reagents and cells
Monodisperse silica particles (1.57 μm and 3.14 μm in diameter, 5% w/v) were purchased from Spherotech Inc (Lake Forest, IL). Sylgard 184 silicone elastomer kit was obtained from Dow Corning (Midland, MI). Bovine serum albumin (BSA), biotin N-hydroxysuccinimide ester (biotin-NHS), Immunoglobulin G (IgG) from rabbit serum and penicillin–streptomycin were acquired from Sigma-Aldrich (St. Louis, MO). Alexa Fluor succinimidyl esters were purchased from Life Technologies (Grand Island, NY). VivoTrack 680 for labeling macrophage cell membranes was purchased from Perkin Elmer (Waltham, MA). IgG and BSA were biotinylated using the reaction of biotin-NHS ester with primary amines. BSA or BSA-biotin were fluorescently labeled with Alexa Fluor succinimidyl esters of different colors as indicated. Mus musculus macrophages (RAW 264.7) were purchased from ATCC (Manassas, VA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 100 units mL−1 penicillin from, 100 μg mL−1 streptomycin, 10% fetal bovine serum (FBS) and 0.11 mg mL−1 (1 mM) sodium pyruvate. Imaging buffer (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2·6H2O, 2 mM Na2H2PO4·H2O, 10 mM HEPES and 10 mM glucose) was adjusted to pH 7.2–7.4 and used for all live cell imaging.
2.2 Fabrication of triblock patchy particles
A “sandwich” microcontact printing procedure was used to prepare the triblock particles. Prior to printing proteins onto particles, polydimethylsiloxane (PDMS) stamps and monolayers of silica particles were prepared as described elsewhere.30 PDMS stamps were treated with piranha solution (H2SO4:H2O2 = 1:1 v:v) to render them hydrophilic. A previous study has shown that this wet chemistry treatment is effective in removing the hydrophobic methyl groups from the PDMS surface.31 The 1:1 (v:v) piranha solution was used instead of the traditionally 3:1 (v:v) one to avoid the etch-induced cracking of the PDMS. To print the first protein patch, an etched stamp was incubated with 200 μL of fluorescently labeled proteins (20 μg mL−1), which were either BSA or BSA-biotin, for 1 hour at room temperature. The first stamp was dried under a stream of filtered air and immediately placed on top of a monolayer of silica particles with a pressure of 1.5 × 104 Pa. The stamp was peeled off 3 minutes later and placed onto a flat surface, with the particle-embedded side facing up. A second stamp that was “inked” with proteins was pressed against the first stamp to generate the second protein patch on particles. Particles were then sonicated off the stamps and collected in 1× PBS buffer containing 0.064 mg mL−1 BSA or BSA-biotin, depending on the type of particle made. Biotinylated IgG molecules were attached onto the particle surfaces via streptavidin–biotin conjugation. See the ESI† for details on the microcontact printing method.
2.3 Live cell imaging
RAW 264.7 macrophages were seeded in sterilized imaging chambers for 24 hours and serum starved for 3 hours before imaging. Cells were imaged at 37 °C on a Nikon Eclipse epi-fluorescence microscope that is equipped with an Andor iXon3 EMCCD camera and a Nikon Plan Apo 100x/1.49 N.A TIRF objective.
2.4 Single-particle rotational tracking and analysis
The tracking algorithm had three main steps. The first step was to reduce noise. The images were smoothed using a Gaussian surface function as previously reported by Crocker and Grier.32 The particles displayed no obvious rotation within 5 consecutive images (a total of 10 seconds with 2 s interval time between frames), so every 3 consecutive images of the same patch were averaged for further noise reduction. The second step was to identify the center of each protein patch. Because the fluorescence image of each protein patch had a non-circular cross section, conventional methods of identifying the particle center based on the Gaussian distribution of intensity were not applicable. Instead, we adopted a “polynomial-fit Gaussian weight” method, as reported by Rogers et al., which identifies the center of single particles of unknown shapes.33 Briefly, the position of each patch was first estimated based on the local maximum of the fluorescence intensity. The identified intensity peaks were kept if their brightness was more than one standard deviation above the average of the pixel brightness of the entire image; otherwise they were discarded as noise. The center position of each patch was refined by applying a two-dimensional polynomial fitting of the intensity around each intensity peak, weighted by a Gaussian function of the distance from the center. After identifying the center of each patch, a few criteria, including brightness and distance between two patches, were used to recognize the pair of patches corresponding to each single particle. The third and final step was to calculate the orientation of each vector drawn between the centers of two patches that were identified as belonging to a single particle, and also to obtain the midpoint of each of these vectors as correspondingly roughly to the center of the particle. See the ESI† for details on the single-particle tracking algorithm.
3. Results and discussion
3.1 Creating triblock patchy particles
We designed the microparticles to be functionally uniform but optically anisotropic. For surface functionality, the particles were uniformly coated with IgG molecules, which bind to macrophage Fc receptors to trigger phagocytosis. The purpose of the optical anisotropy was to allow direct imaging of the orientation and rotation of each particle. We generated triblock patchy particles that display protein patches of distinct fluorescence on either pole by “sandwiching” a monolayer of silica microparticles in between two PDMS stamps that were pre-inked with solutions of fluorescently labeled BSA-biotin (Fig. 1). Although similar procedures have been used previously to create trivalent particles,34–36 this was the first time that this technique had been used to print protein patches on particles. This fabrication process is applicable for both 3 and 1.6 μm particles. We observed that the first-printed protein patch is slightly larger than the second one, likely due to the longer contact time of particles with the first stamp. After the microcontact printing, we filled the non-printed area of the particles with BSA-biotin via incubation, and attached IgG molecules to the particle surface with streptavidin–biotin linkers. After these functionalization steps, particles displayed two patches of fluorescent BSA-biotin, labeled with either Alexa 488 or 568 dyes, on opposite poles (Fig. 1b). Meanwhile, these triblock particles were coated uniformly with IgG, which is indicated by the homogeneous intensity of fluorescently labeled IgG molecules on the particle surface (Fig. S1, ESI†). For brevity, the particles uniformly coated with IgG are referred to as the all-IgG particles in the following context. The two patches on the triblock particles are referred to as the green or the red patch according to their respective fluorescence emission color.
Fig. 1.
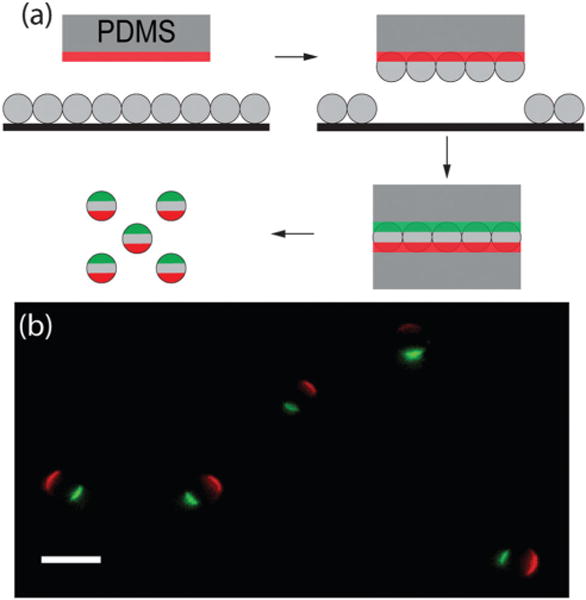
Fabrication and characterization of the triblock patchy particles. (a) Schematic illustration of the “sandwich” micro-contact printing method for creating the trivalence on spherical silica microparticles. The proteins to be printed on the particles are illustrated in red and green. A protein incubation step follows the microcontact printing to coat the particles either with BSA for passivation or with BSA-biotin for uniform ligand coating. (b) Overlaid epifluorescence images of 3 μm triblock Janus particles. Scale bar: 5 μm.
3.2 Macrophage uptake of the triblock patchy particles
Macrophage uptake of the all-IgG particles (3 μm) was imaged using epi-fluorescence microscopy. In addition to fluorescently labeling the triblock particles, we also sought to label the macrophage cells in order to identify the different stages of the internalization process. The choice of dyes for labeling macrophages is significantly limited because the phagocytic function of these cells leads to rapid degradation of dyes. The Vivotrack dye, though photobleached easily and having low quantum yield, stains the cell membrane most efficiently among many that we have tested. Labeling the RAW264.7 cells with Vivotrack 680 allowed us to roughly estimate the onset and completion of the particle internalization process. 3 μm particles were typically engulfed within 10 minutes after the initial particle–cell contact. A representative particle internalization event is shown in Fig. 2a. We roughly divided the entire process into three stages: initial cell–particle binding, cell entry, and completion of particle engulfment. We observed that the triblock particle rotated at different times as it entered the cell. The two patches stayed in focus of the epi-fluorescence imaging, meaning that the rotation of the particle occurred mostly within the imaging plane.
Fig. 2.
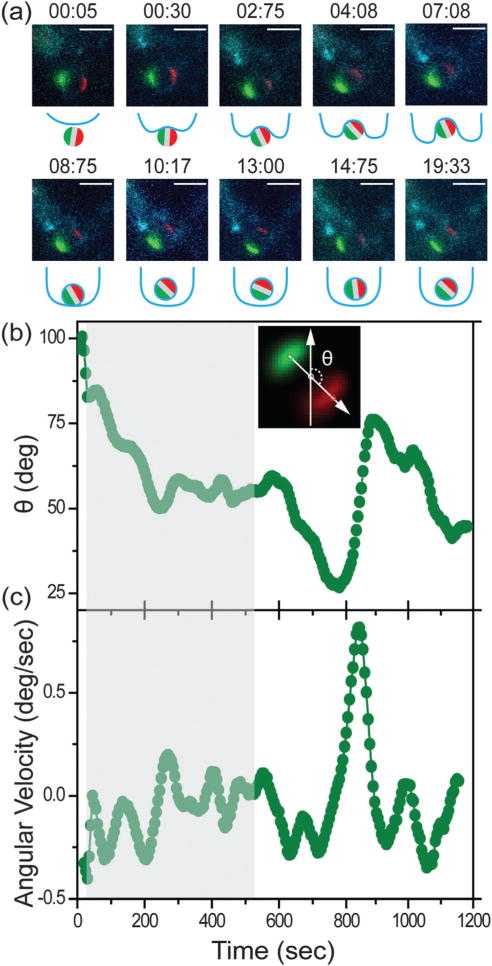
Fluorescence imaging and single-particle tracking of a representative 3 μm all-IgG particle during macrophage uptake. (a) Overlaid epi-fluorescence images and schematic illustrations showing the particle uptake at various times as indicated. The two protein patches, labeled with either Alexa 568 or Alexa 488 dye, are shown in red and green, respectively. Scale bars: 5 μm. (b) Angle θ of the triblock particle is plotted as a function of time for the particle shown in (a). The inset shows the definition of angle θ, which is the angle between the y-axis and the vector pointing from the green to the red patch. (c) Angular velocity of the triblock particle is plotted a function of time. The grey shades in both (b) and (c) indicate the period from the initial cell–particle contact to the time when the particle is visibly engulfed by the cell membrane. Both time points were estimated from the fluorescence images. Plots are representative of N = 25 particles.
3.3 Single-particle rotational tracking and analysis
We next analyzed the orientation and rotation of single triblock patchy particles using a single-particle tracking algorithm (see ESI† for details). The center of each patch was identified in a manner similar to the conventional center-of-mass particle localization procedure. To obtain the orientation of each particle, we drew a vector from the center of its green patch to that of its red one. The midpoint of each vector was identified as the center of the particle, assuming that the two protein patches are positioned exactly on the opposite poles of a particle. The angle between each vector and the y-axis vector is defined as angle θ and taken as the orientation of the particle (Fig. 2b inset and Fig. S2, ESI†). Out of three rotational angles, in this study we quantified only the rotational angle within the imaging plane. No particles were observed to undergo rotations larger than 180 degrees during the entire course of the internalization. The tracking inaccuracy was estimated to be 1 degree (see ESI† for details).
The in-plane angle θ for the particle shown in Fig. 2a is plotted as a function of time in Fig. 2b, and more in Fig. S3 (ESI†). We observed two dynamically different phases during the entire internalization process. In one phase, particles mostly rotated in one direction, as shown between 0–200 s and 600–900 s in Fig. 2b. In between the directional rotational movements, however, particle motion appeared relatively random and rotated in small steps around a certain orientation, as shown from 300–600 s. The mixture of rotational motion was observed in all the all-IgG particles. To correlate the particle rotation with specific steps in the internalization process (cell–particle binding, entry, and completion of engulfment), we used gray shades in all plots to indicate the period from the initial particle–cell contact to the point when the particles were visibly engulfed by the cell membrane. We found that all particles exhibited the directional rotation during the cell entry step. However, some particles also rotated directionally after they were completely inside the cell (Fig. S3, ESI†). The one-directional rotation of particles during cell entry is possibly associated with the membrane protrusion around the particles, but we could not clearly resolve the membrane structures in fluorescence images due to the aforementioned technical challenge of efficiently labeling macrophage cell membranes.
To demonstrate that the observed mixture of rotations is indeed non-Brownian, we first calculated the angular velocity of the particles as a function of time (Fig. 2c). Counterclockwise in-plane rotation results in negative angular velocity, based on our definition of the angle θ. One can see that the all-IgG particles exhibit a heterogeneous distribution of angular velocities when sampled over time. To confirm this dynamical heterogeneity, we analyzed the probability distribution of particle rotational displacements P(Δθ, Δt) for the all-IgG particles (N = 25). This distribution, also known as the van Hove function, plots the probability that particles undergo a rotational displacement of Δθ during a time interval of duration Δt. It is commonly used to reveal dynamical heterogeneity in colloidal systems.37–39 P(Δθ, Δt) is a single Gaussian distribution for particles undergoing simple random rotation, but the function deviates from a Gaussian in the presence of heterogeneous step sizes, providing a quantitative measure of the uniformity of the particle dynamics. We tested the reliability of this analysis by imaging and analyzing the rotation of triblock particles dispersed in water containing 5% glycerol (to slow down the particle dynamics). In such a solution particles should rotate at random. P(Δθ, Δt = 10 s) of the in-plane rotational steps of these particles shows a single Gaussian distribution, as expected for Brownian motion (Fig. S4, ESI†). However, P(Δθ, Δt = 40 s) for the 3 μm all-IgG particles is much different from that for the Brownian particles (Fig. 3a). The distribution develops substantial weight in the large-displacement wings, indicating a mixture of slow and fast rotation, confirming our observation from the plot of angular velocity with time. The fraction of large rotational steps, however, is low. Δt = 40 s was used for the analysis shown, but the deviation from a single Gaussian was consistently observed at other interval times.
Fig. 3.
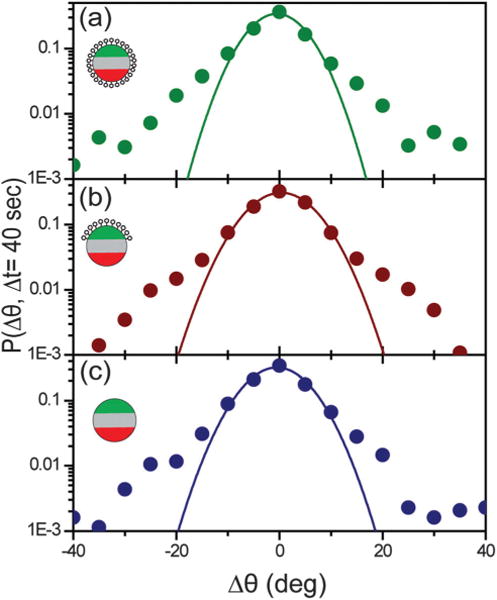
The probability distribution of particle rotational displacements, P(Δθ, Δt = 40 s), for 3 μm particles with different surface functionalities: (a) all-IgG (N = 25), (b) IgG–BSA (N = 41), and (c) all-BSA (N = 12). Each plot is fitted with a single Gaussian function indicated by the solid lines. Approximately 4000–5000 steps were averaged for each P(Δθ, Δt = 40 s). Insets are schematic illustrations of the three types of particles: (a) all-IgG, (b) IgG–BSA, and (c) all-BSA.
Could the observed heterogeneity in the distribution of particle rotation be due to steps when particles rotated outside the plane of imaging? In our single-particle tracking analysis, particles that rotated out-of-plane by more than 40° were screened out by a patch-to-patch distance threshold and not analyzed. By performing a computer simulation of Brownian particles (see ESI† for details), we found that the 40° out-of-plane rotation has negligible effect on the single-Gaussian shape of the displacement probability distribution of all in-plane angles (Fig. S5, ESI†). The results confirm that the non-Gaussian tails in all P(Δθ, Δt = 40 s) plots of the triblock patchy particles are caused by the dynamical heterogeneity of those particles, and not by their out-of-plane rotation.
3.4 Effect of IgG presentation on particle rotational dynamics
We next investigated if the heterogeneous rotational dynamics of particles are affected by the surface presentation of the ligand, IgG antibody. To answer this question, we designed two additional types of triblock patchy particles that were both optically anisotropic but had different surface presentations of ligands: one type was coated with IgG on one side and BSA on the other (referred to as the IgG–BSA particles), and the other type was uniformly passivated with BSA (referred to as the all-BSA particles). RAW264.7 macrophages have receptors for IgG, but no known receptors for BSA. Our previous study has shown that a smaller fraction of BSA-coated particles are internalized than IgG-coated ones due to the non-specific uptake pathways.40 Despite the different surface functionalities, both the IgG–BSA and all-BSA particles have the same optical anisotropy as the all-IgG ones: a green and a red patch on two poles.
Angle θ of single particles for both types of particles shows a mixture of directional and random movements at various times of the particle uptake process (Fig. 4). The mixture of fast and slow rotation is evident in the displacement probability distribution P(Δθ, Δt = 40 s), which deviates from a single Gaussian distribution in the large displacement wings, similarly to the distribution of the all-IgG particles (Fig. 3). We also investigated particles of different sizes. 1.6 μm particles overall exhibited faster angular velocities than 3 μm particles, which is likely due to the smaller size (Fig. S6, ESI†). But similar heterogeneous rotational dynamics were observed with all three types of 1.6 μm particles, confirming the generality of our findings (Fig. S6 and S7, ESI†). The results indicate that all particles, regardless of the ligand display, exhibited similar rotational dynamics as they were taken up by the macrophage cells, suggesting that the surface presentation of IgG on the particles has negligible effect on the rotational heterogeneity of particles. However, it remains to be explored if this conclusion can be applied to other ligands or other cells, because particle uptake is known to be highly specific to cell types and receptors involved.5
Fig. 4.
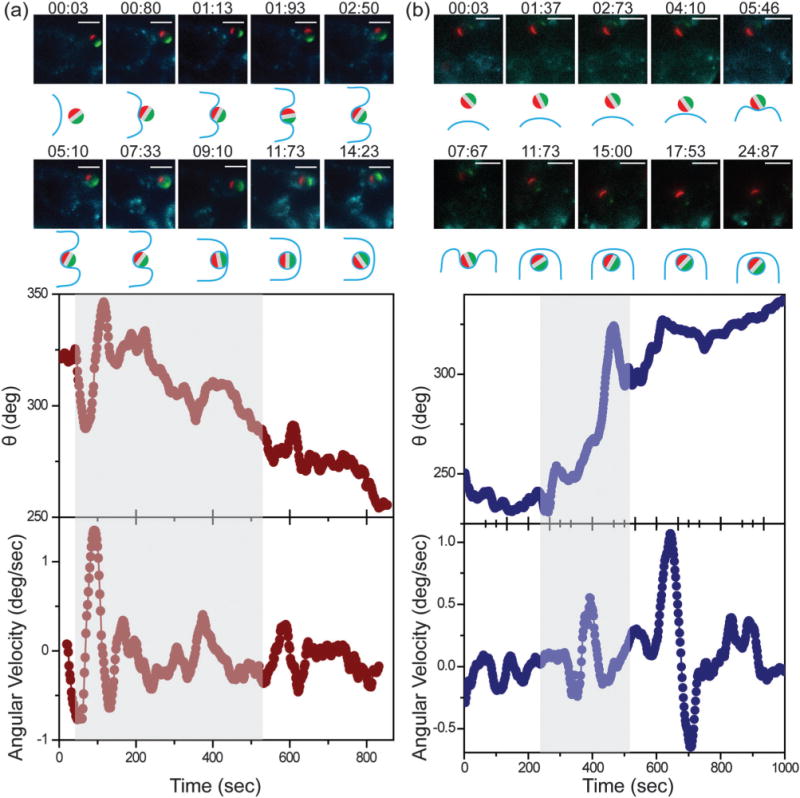
Fluorescence imaging and single-particle tracking of representative 3 μm (a) IgG–BSA and (b) all-BSA particles during macrophage uptake. In both (a) and (b), overlaid epi-fluorescence images and schematic illustrations show particle uptake at various times as indicated. Angle θ and angular velocity of the triblock particles are plotted as a function of time. Grey shades indicate the period from the initial cell–particle contact to the time when the particle is visibly engulfed by the cell membrane. Both time points were estimated from the fluorescence images. Scale bars: 5 μm. Plots are representative of N = 41 IgG–BSA and N = 12 all-BSA particles, respectively.
We speculate that such directional yet transient rotation is a result of interactions between the particle and the cell membrane. A computer simulation study, aiming to understand the rotation of non-spherical particles during endocytosis, has suggested that particles can be rotated by asymmetric membrane deformation.41 The particles in our experiments are spherical. However, it is possible that the membrane wrapping around a particle is uneven at times, leading to transient directional rotation. In fact, we have demonstrated in a previous study with a different cell type that transient uneven membrane protrusion during particle uptake is possible.42 We intended to do the same analysis with macrophage cells in this study, but were limited by the lack of dyes to clearly label the macrophage cell membrane. We are currently exploring different strategies to overcome this challenge, so that we may simultaneously resolve the membrane protrusion and particle rotation to understand the mechanisms underlying the heterogeneous particle rotation. Of course, other mechanisms may also be possible. For example, a simulation study has suggested that particles that display mixed patches of hydrophobic and hydrophilic functional groups rotate during membrane penetration in order to maximize favorable particle–lipid interactions.43
Another interesting question but unclear at this moment is why the surface presentation of IgG and BSA has negligible effect on the particle rotation. Macrophage cells are capable of uptaking particles via two distinct pathways.44 One is the ligand-guided phagocytosis, in which the membrane protrusion tightly follows the presence of ligands on particle surfaces. IgG-coated particles typically enter macrophages via this mechanism.45 The other is macropinocytosis. It is independent of ligands and facilitates particle uptake through dramatic membrane ruffling.46 It is likely that all-IgG particles enter macrophages via ligand-guided phagocytosis, whereas all-BSA particles use the macropinocytosis pathway. Both the ligand-guided membrane protrusion and membrane ruffling are possible to lead to particle rotation, as observed in our experiments. Future studies such as correlating membrane dynamics with particle rotation, as mentioned above, may provide insights into distinguishing these two pathways.
4. Conclusions
Here we studied rotation of single particles during their uptake by macrophage cells. The particle probes were triblock microparticles that display two patches of different colors on opposite sides. These particles allowed precise measurement of the orientation and rotation of particles during particle internalization by macrophages. We found that all microparticles, regardless of the particle size or the surface presentation of IgG ligands, exhibit a mixture of fast and slow rotation. Particles also transiently undergo directional rotation during cell entry, which might be caused by transient particle–cell membrane interactions. While translational dynamics of particles during macrophage uptake has been previously studied,47 our study shows that particles also rotate in a non-Brownian fashion, which may provide new insights into understanding particle–cell interactions during this complex process.
This study demonstrates a new application of patchy particles that exploits their optical anisotropy. Unlike the previously reported approach for tracking half-coated MOON particles, our tracking method is largely built upon conventional single-particle algorithms that locate the particle positions. Therefore, researchers who currently use single-particle translational tracking can easily adopt this method to study particle rotation with only minimal modifications. We realize that a limitation of this study is that we only tracked the in-plane angle of particles. We are currently developing methods that enable tracking of other rotational angles of triblock particles. Nevertheless, this quantitative study showcases the potential of employing patchy particles to probe complex dynamics and interactions in biological systems.
Supplementary Material
Acknowledgments
We thank Mr. Jim Powers of the IUB Light Microscopy Imaging Center for fluorescence imaging assistance, and Indiana University for funding. L.S. was supported by the Graduate Training Program in Quantitative and Chemical Biology (T32GM109825) and Indiana University. S.M.A. is currently a postdoctoral researcher supported by Sandia National Laboratories. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy under DOE contract DE-ACO4-94AL85000.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm00893j
References
- 1.Pacheco P, White D, Sulchek T. PLoS One. 2013;8:e60989. doi: 10.1371/journal.pone.0060989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Champion JA, Mitragotri S. Proc Natl Acad Sci U S A. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paul D, Achouri S, Yoon YZ, Herre J, Bryant CE, Cicuta P. Biophys J. 2013;105:1143–1150. doi: 10.1016/j.bpj.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci U S A. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conner SD, Schmid SL. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 6.Suh J, Dawson M, Hanes J. Adv Drug Delivery Rev. 2005;57:63–78. doi: 10.1016/j.addr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Schulz A, McDonagh C. Soft Matter. 2012;8:2579–2585. [Google Scholar]
- 8.van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. PLoS Pathog. 2008;4:e1000244. doi: 10.1371/journal.ppat.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin H, Heller DA, Sharma R, Strano MS. ACS Nano. 2009;3:149–158. doi: 10.1021/nn800532m. [DOI] [PubMed] [Google Scholar]
- 10.Bálint Š, Verdeny Vilanova I, Sandoval Álvarez Á, Lakadamyali M. Proc Natl Acad Sci U S A. 2013;110:3375–3380. doi: 10.1073/pnas.1219206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levi V, Gratton E. Cell Biochem Biophys. 2007;48:1–15. doi: 10.1007/s12013-007-0010-0. [DOI] [PubMed] [Google Scholar]
- 12.Rothenberg E, Sepúlveda LA, Skinner SO, Zeng L, Selvin PR, Golding I. Biophys J. 2011;100:2875–2882. doi: 10.1016/j.bpj.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang YH, Nam JM. Anal Chem. 2009;81:2564–2568. doi: 10.1021/ac802477h. [DOI] [PubMed] [Google Scholar]
- 14.Ritchie K, Shan XY, Kondo J, Iwasawa K, Fujiwara T, Kusumi A. Biophys J. 2005;88:2266–2277. doi: 10.1529/biophysj.104.054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murcia MJ, Minner DE, Mustata GM, Ritchie K, Naumann CA. J Am Chem Soc. 2008;130:15054–15062. doi: 10.1021/ja803325b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bruin K, Ruthardt N, von Gersdorff K, Bausinger R, Wagner E, Ogris M, Brauchle C. Mol Ther. 2007;15:1297–1305. doi: 10.1038/sj.mt.6300176. [DOI] [PubMed] [Google Scholar]
- 17.Welsher K, Yang H. Nat Nanotechnol. 2014;9:198–203. doi: 10.1038/nnano.2014.12. [DOI] [PubMed] [Google Scholar]
- 18.Xiao L, Ha JW, Wei L, Wang G, Fang N. Angew Chem, Int Ed. 2012;51:7734–7738. doi: 10.1002/anie.201202340. [DOI] [PubMed] [Google Scholar]
- 19.Gu Y, Sun W, Wang G, Zimmermann MT, Jernigan RL, Fang N. Small. 2013;9:785–792. doi: 10.1002/smll.201201808. [DOI] [PubMed] [Google Scholar]
- 20.Gu Y, Sun W, Wang G, Jeftinija K, Jeftinija S, Fang N. Nat Commun. 2012;3:1030. doi: 10.1038/ncomms2037. [DOI] [PubMed] [Google Scholar]
- 21.Brokmann X, Ehrensperger MV, Hermier JP, Triller A, Dahan M. Chem Phys Lett. 2005;406:210–214. [Google Scholar]
- 22.Watanabe TM, Fujii F, Jin T, Umemoto E, Miyasaka M, Fujita H, Yanagida T. Biophys J. 2013;105:555–564. doi: 10.1016/j.bpj.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuinness LP, Yan Y, Stacey A, Simpson DA, Hall LT, Maclaurin D, Prawer S, Mulvaney P, Wrachtrup J, Caruso F, Scholten RE, Hollenberg LCL. Nat Nanotechnol. 2011;6:358–363. doi: 10.1038/nnano.2011.64. [DOI] [PubMed] [Google Scholar]
- 24.Kukura P, Ewers H, Muller C, Renn A, Helenius A, Sandoghdar V. Nat Methods. 2009;6:923–927. doi: 10.1038/nmeth.1395. [DOI] [PubMed] [Google Scholar]
- 25.Robbins JR, Theriot JA. Curr Biol. 2003;13:R754–R756. doi: 10.1016/j.cub.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Behrend CJ, Anker JN, McNaughton BH, Kopelman R. J Magn Magn Mater. 2005;293:663–670. [Google Scholar]
- 27.Behrend CJ, Anker JN, McNaughton BH, Brasuel M, Philbert MA, Kopelman R. J Phys Chem B. 2004;108:10408–10414. [Google Scholar]
- 28.Anthony SM, Hong L, Kim M, Granick S. Langmuir. 2006;22:9812–9815. doi: 10.1021/la062094h. [DOI] [PubMed] [Google Scholar]
- 29.Kim M, Anthony SM, Bae SC, Granick S. J Chem Phys. 2011;135:054905. doi: 10.1063/1.3623489. [DOI] [PubMed] [Google Scholar]
- 30.Chen B, Jia Y, Gao Y, Sanchez L, Anthony SM, Yu Y. ACS Appl Mater Interfaces. 2014;6:18435–18439. doi: 10.1021/am505510m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maji D, Lahiri SK, Das S. Surf Interface Anal. 2012;44:62–69. [Google Scholar]
- 32.Crocker JC, Grier DG. J Colloid Interface Sci. 1996;179:298–310. [Google Scholar]
- 33.Rogers SS, Waigh TA, Zhao X, Lu JR. Phys Biol. 2007;4:220–227. doi: 10.1088/1478-3975/4/3/008. [DOI] [PubMed] [Google Scholar]
- 34.Kaufmann T, Gokmen MT, Wendeln C, Schneiders M, Rinnen S, Arlinghaus HF, Bon SAF, Du Prez FE, Ravoo BJ. Adv Mater. 2011;23:79–83. doi: 10.1002/adma.201003564. [DOI] [PubMed] [Google Scholar]
- 35.Kaufmann T, Wendeln C, Gokmen MT, Rinnen S, Becker MM, Arlinghaus HF, Du Prez F, Ravoo BJ. Chem Commun. 2013;49:63–65. doi: 10.1039/c2cc36483b. [DOI] [PubMed] [Google Scholar]
- 36.Jiang S, Granick S. Langmuir. 2009;25:8915–8918. doi: 10.1021/la902152n. [DOI] [PubMed] [Google Scholar]
- 37.Weeks ER, Crocker JC, Levitt AC, Schofield A, Weitz DA. Science. 2000;287:627–631. doi: 10.1126/science.287.5453.627. [DOI] [PubMed] [Google Scholar]
- 38.Yu Y, Anthony SM, Bae SC, Granick S. J Phys Chem B. 2011;115:2748–2753. doi: 10.1021/jp109146s. [DOI] [PubMed] [Google Scholar]
- 39.Wang B, Anthony SM, Bae SC, Granick S. Proc Natl Acad Sci U S A. 2009;106:15160–15164. doi: 10.1073/pnas.0903554106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Y, Yu Y. Langmuir. 2015;31:2833–2838. doi: 10.1021/la504668c. [DOI] [PubMed] [Google Scholar]
- 41.Yang K, Yuan B, Ma Y-q. Nanoscale. 2013;5:7998–8006. doi: 10.1039/c3nr01561k. [DOI] [PubMed] [Google Scholar]
- 42.Gao Y, Yu Y. J Am Chem Soc. 2013;135:19091–19094. doi: 10.1021/ja410687z. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Zhang X, Cao D. Soft Matter. 2014;10:6844–6856. doi: 10.1039/c4sm00236a. [DOI] [PubMed] [Google Scholar]
- 44.Swanson JA. Nat Rev Mol Cell Biol. 2008;9:639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swanson JA, Hoppe AD. J Leukocyte Biol. 2004;76:1093–1103. doi: 10.1189/jlb.0804439. [DOI] [PubMed] [Google Scholar]
- 46.Welliver TP, Chang SL, Linderman JJ, Swanson JA. J Cell Sci. 2011;124:4106–4114. doi: 10.1242/jcs.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishikawa J, Okano J, Ohki K, Amagai A, Maeda Y, Miyata H. Exp Cell Res. 2003;288:268–276. doi: 10.1016/s0014-4827(03)00212-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.