

Abstract
Juvenile hormones have attracted attention as safe and selective targets for the design and development of environmentally friendly and biorational insecticides. In the juvenile hormone III biosynthetic pathway, the enzyme farnesol dehydrogenase catalyzes the oxidation of farnesol to farnesal. In this study, farnesol dehydrogenase was extracted from Polygonum minus leaves and purified 204-fold to apparent homogeneity by ion-exchange chromatography using DEAE-Toyopearl, SP-Toyopearl, and Super-Q Toyopearl, followed by three successive purifications by gel filtration chromatography on a TSK-gel GS3000SW. The enzyme is a heterodimer comprised of subunits with molecular masses of 65 kDa and 70 kDa. The optimum temperature and pH were 35°C and pH 9.5, respectively. Activity was inhibited by sulfhydryl reagents, metal-chelating agents and heavy metal ions. The enzyme utilized both NAD+ and NADP+ as coenzymes with K m values of 0.74 mM and 40 mM, respectively. Trans, trans-farnesol was the preferred substrate for the P. minus farnesol dehydrogenase. Geometrical isomers of trans, trans-farnesol, cis, trans-farnesol and cis, cis-farnesol were also oxidized by the enzyme with lower activity. The K m values for trans, trans-farnesol, cis, trans-farnesol and cis, cis-farnesol appeared to be 0.17 mM, 0.33 mM and 0.42 mM, respectively. The amino acid sequences of 4 tryptic peptides of the enzyme were analyzed by MALDI-TOF/TOF-MS spectrometry, and showed no significant similarity to those of previously reported farnesol dehydrogenases. These results suggest that the purified enzyme is a novel NAD(P)+-dependent farnesol dehydrogenase. The purification and characterization established in the current study will serve as a basis to provide new information for recombinant production of the enzyme. Therefore, recombinant farnesol dehydrogenase may provide a useful molecular tool in manipulating juvenile hormone biosynthesis to generate transgenic plants for pest control.
Introduction
Juvenile hormones (JHs) are a family of sesquiterpenes that play important roles in the development, metamorphosis, reproduction, polyphenism, and behavioral changes of insects [1]. Due to these properties, JHs have attracted attention as safe and selective targets for the design and development of environmentally friendly and biorational insecticides [2]. Several JH analogs have been reported, such as ethyl 4-[2-(tert-butylcarbonyloxy)butoxy]benzoate, fluoromevalonate, ethyl (E)-3-methyl-2-dodecenoate, fluvastatin and methoprene though none of these compounds has proven to be sufficiently active for practical use in pest control [3–6].
The biosynthetic pathway of juvenile hormone III (JH III, methyl-10R,11-epoxy-3,7,11-trimethyl-2E, 6E-dodecadienoate) is well conserved in insects [7]. The early steps in the biosynthetic pathway of insect JH III include the mevalonate pathway from acetyl-CoA to farnesyl pyrophosphate, a conserved pathway in both vertebrates and invertebrates [8]. Farnesyl pyrophosphate synthase catalyzes the synthesis of farnesyl pyrophosphate from dimethylallyl diphosphate and isopentenyl pyrophosphate [9]. Farnesol is synthesized by farnesyl pyrophosphatase, [10] and then oxidized by farnesol dehydrogenase to produce farnesal [11, 12]. Subsequently, farnesal is oxidized most probably by farnesal dehydrogenase to form farnesoic acid [13]. The last two steps of the JH III biosynthetic pathway depend on the particular order of insect. In Lepidoptera, a P450 monooxygenase converts farnesoic acid to JH acid which is subsequently methylated by JH acid O-methyltransferase to form JH III [14]. However, in orthopteran and dictyopteran insects, the methylation reaction is preceded by an epoxidation reaction [8]. In comparison to the number of enzymatically well characterized enzymes from insect JH III biosynthetic pathway [12, 15–18], enzymes involve in plant JH III biosynthetic pathway are not well elucidated. JH III and its biosynthetic precursor in insects, methyl farnesoate, were also identified in the sedges, Cyperus iria L. and C. aromaticus [19]. Inhibitor and precursor feeding studies suggest that the later steps of the JH III biosynthesis in C. iria are similar to those in the insect pathway [20]. In addition, enzyme activities of farnesyl pyrophosphate synthase, farnesyl pyrophatase, farnesol dehydrogenase and methyltransferase which involved in JH III biosynthesis were detected in several plants [11, 21–27]. High concentration of JH III in C. iria roots and its present throughout development indicated that this compound plays important roles in plant mechanism through plant-insect, plant-plant, or other interaction [13]. Furthermore, the crude extract of C. aromaticus cultured cells which contained JH III showed growth inhibitory against Aedes aegypti and A. albopictus. The economic importance of insect hormones in plants was highlighted by their ability to inhibit the development and reproduction of insect herbivores [28]. Therefore, more detailed understanding of the enzymes in JH III biosynthetic and metabolic pathways in plant will be useful for the development of new approaches towards integrated pest management using recombinant DNA technology [29] by deployment of the genetically transformed plants for pest control [30].
To elucidate the JH III biosynthetic pathway in plant, we investigated enzymes participating in this sesquiterpene metabolism pathway in Polygonum minus. This species belongs to the Polygonaceae family and possesses a wide range of medicinal properties [31–35]. Farnesol and farnesal have been identified in the essential oils of Polygonum sp. [36, 37]. Moreover, enzyme activities of farnesyl pyrophosphate synthase, farnesol dehydrogenase and farnesal dehydrogenase have also been detected in cell-free extracts of P. minus. Farnesol dehydrogenase is an enzyme that catalyzes the oxidation of farnesol to farnesal [38]. Farnesol dehydrogenase activity has been reported to be present in Arabidopsis thaliana [26], Ipomoea batatas [11], and in the corpora allata glands of Aedes aegypti [12]. However, only farnesol dehydrogenase from A. aegypti has been purified to homogeneity. In addition, the existing papers lack of the information on the enzyme recognition of substrate specificity. Based on the current state of research, the nature of substrates that are specifically oxidized by farnesol dehydrogenase has remained poorly understood. This paper reports the purification and characterization of farnesol dehydrogenase enzyme from P. minus leaves. Purification was achieved using ion exchange and gel filtration chromatographies. To the best of our knowledge, this is the first report that demonstrates the utilization of both NAD+ and NADP+ as coenzymes by a farnesol dehydrogenase enzyme. The deployment of transgenic plant with farnesol dehydrogenase enzyme will be beneficial for use in manipulating juvenile hormone biosynthesis in plants. Thus, offers an alternative method for controlling population of insect pest by means of non-toxic, selectively acting and biorationally safe practice [28].
Materials and Methods
Plant materials and chemicals
The P. minus leaves were obtained from plants growing in an experimental plot at the Institute of Systems Biology of Universiti Kebangsaan Malaysia (UKM). Trans, trans-farnesol (trans,trans-3,7,11-trimethyl-2,6,10-dodecatrien-1-ol) was obtained from Alfa Aesar (Ward Hill, MA). Cis, trans-farnesol was purchased from Tokyo Chemical Industry (TCI) (Tokyo, Japan). Cis, cis-farnesol was obtained from Echelon Bioscience, Inc. (Salt Lake City, UT). DEAE-Toyopearl 650M, SuperQ Toyopearl 650M, SP Toyopearl 650M and TSK-gel GS3000SW were purchased from Tosoh (Tokyo, Japan), whilst standard proteins for gel filtration were obtained from Bio-Rad (Hercules, CA). All other reagents were analytical-grade commercial products. Water-insoluble chemicals were dissolved in absolute dimethyl sulfoxide or acetone, and subsequent dilutions were conducted in water. The presence of dimethyl sulfoxide or acetone in the reaction mixture had no effect on the enzyme activity.
Extraction of farnesol dehydrogenase
Preparation of the cell-free extract was performed according to the method described by Hassan et al. [39] with slight modifications. Approximately 200 g (fresh weight) of P. minus leaves were frozen in liquid nitrogen and ground to a fine powder with a Waring blender. The frozen powder was immediately slurried with cold extraction buffer (100 mM tricine-NaOH buffer (pH 7.5) containing 2.5 mM of 2-mercaptoethanol (2-ME), 15% (v/v) of glycerol, 5 mM of thiourea, 1 mM of phenylmethylsulfonylfluoride (PMSF), 50% (w/w) Amberlite XAD-4 and 10% (w/v) polyvinylpolypyrrolidone (PVPP) for 15 min before being squeezed through four layers of cheesecloth. The homogenate was centrifuged at 20,000 × g at 4°C for 30 min to remove cell debris. The supernatant which was determined to contain farnesol dehydrogenase activity was used as the enzyme source.
Protein measurement
Protein concentration was measured using the Lowry method [40] with bovine serum albumin as a standard. The proteins eluted from column chromatography were monitored by measuring absorbance at 280 nm.
Enzyme assay
Farnesol dehydrogenase activity was measured by observing the increase in absorbance at 340 nm at 35°C. The standard reaction mixture (1.5 ml) contained 100 mM of glycine-NaOH buffer (pH 9.5), 1.0 mM of trans, trans-farnesol, 1.0 mM of NAD+ and an appropriate amount of enzyme. The reaction was initiated by addition of the enzyme. Enzyme activity was calculated using an extinction coefficient of 6,200 M-1 cm-1 for NADH. One unit of enzyme activity was defined as the amount of enzyme required to catalyze the formation of 1 μmol of NADH per min under the described assay conditions. Specific activity was defined as the units of enzyme activity per mg of protein.
Purification of farnesol dehydrogenase
Purification of farnesol dehydrogenase was performed at 4°C. Throughout the purification procedure, 100 mM tricine-NaOH buffer (pH 7.5) containing 2.5 mM of 2-ME was used, unless otherwise stated. The flow rate during loading, washing, and elution was maintained at 1.5 ml/min unless stated. The cell-free extract (6600 mg protein) was put on a DEAE-Toyopearl 650M column (2.6 × 65 cm) equilibrated with the buffer. The column was washed with five column volumes of the buffer (15ml/fraction) and the unbound proteins with enzyme activity (300 ml) were collected. The fractions with enzyme activity from DEAE-Toyopearl were combined and applied to a SP-Toyopearl 650M column (1.6 × 37 cm) which had been equilibrated and washed with the buffer. Farnesol dehydrogenase activity was found in the unbound proteins. Fractions with farnesol dehydrogenase activity (15ml/fraction) from SP-Toyopearl were pooled (135 ml) and applied to a Super-Q Toyopearl 650M column (2.6 × 20 cm) that had been equilibrated with the buffer. The unbound proteins with enzyme activity were collected, concentrated with a Macrosep 10K Omega centrifugal devices (Pall Life Sciences, Ann Arbor, MI) and applied to a TSK-gel GS3000SW column (0.78 × 30 cm) equilibrated with the buffer. The protein was eluted with the buffer at a flow rate of 0.5 ml/min. Fractions with enzyme activity were pooled, concentrated using Macrosep 10K Omega centrifugal device and subjected to the same gel filtration procedure. Fractions showing farnesol dehydrogenase activity were pooled together, concentrated, and applied to a TSK-gel GS3000SW column for the third times. Finally, fractions containing enzyme activity were pooled and stored at -80°C until further use. The proteins eluted from column chromatography were monitored by measuring absorbance at 280 nm. The purity of the enzyme was determined by polyacrylamide gel electrophoresis (native-PAGE).
Polyacrylamide gel electrophoresis
Native-PAGE electrophoresis was performed using a 12.5% polyacrylamide gel at pH 8.8, with the Laemmli buffer system without SDS [41]. The protein was silver-stained using the PlusOne silver staining kit (GE Healthcare, Uppsala, Sweden). Enzyme activity staining was performed according to the method described by Hassan et al. [39] with slight modifications. The activity staining of the gel was performed in the presence of 1.0 mM of trans, trans-farnesol in 100 mM of glycine-NaOH buffer (pH 9.5) containing 54 μM of 1-methoxy phenazine methosulphate, 0.3 mM of nitroblue tetrazolium, and 1.0 mM of NAD+ at 35°C for 2 h.
Measurement of molecular mass and isoelectric point
The molecular mass of the enzyme was estimated by gel filtration on a TSK-gel GS3000SW column (0.75 × 30 cm) equilibrated with 0.1 M Tris-HCl buffer (pH 7.5) containing 2.5 mM of 2-ME. The purified enzyme (33 μg) was dialyzed against 3.0 L of 0.1 M Tris-HCl buffer (pH 7.5) containing 2.5 mM of 2-ME for 24 h and applied to the TSK-gel GS3000SW column. The proteins used for molecular-weight standards were thyroglobulin, γ-globulin, ovalbumin, myoglobin, and vitamin B12. SDS polyacrylamide gel electrophoresis (SDS-PAGE) was performed using a 12.5% polyacrylamide gel using the Laemmli method [41]. The PageRuler™ Prestained Protein Ladder, which has a standard-protein molecular-weight range of approximately 10–170 kDa (Product SM0671, Fermentas, St. Leon-Rot, Germany), was used as molecular marker. The protein was silver-stained as described previously.
Isoelectric focusing was performed with an 18 cm ReadyStrip IPG strip (pH 3–10) (GE Healthcare Bioscience, Uppsala, Sweden). The strip was passively rehydrated with 0.2 μg of purified farnesol dehydrogenase in rehydration buffer (8.0 M urea, 4% (w/v) CHAPS, 0.5% (v/v) of pH 3–10 ampholites, 30 mM 2-ME, and 0.002% bromophenol blue) for 12 h. The isoelectric focusing was carried out using Ethan IPGphor (GE Healthcare Bioscience, Uppsala, Sweden) according to the manufacturer’s instructions. The strip was silver stained.
Protein identification by MALDI-TOF/TOF mass spectrometry (MALDI-TOF/TOF-MS)
Identification and analysis of the purified protein was carried out by peptide mass fingerprinting using MALDI-TOF/TOF mass spectrometry (MALDI-TOF/TOF-MS). The purified farnesol dehydrogenase was dialyzed against 100 mM Tris-HCl buffer (pH 7.5) overnight before the protein solution was sent to the Proteomics Facility, Medical Biotechnology Laboratory, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia for mass spectrometry analysis. In-solution trypsin digestion, protein extraction and mass spectrometry analysis by MALDI-TOF/TOF-MS were carried out according to protocols described by Sarah et al. [42] with slight modification. Protein was identified by the set of its proteolytic peptide masses using Peptide Mass Fingerprint option of Mascot software (Matrix Science, USA, http://www.matrixscience.com). Peptide mass profiles were searched and the peptides sequences were blasted against NCBI non-redundant (nr) database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The following combined parameters were used in NCBI searches: Viridiplantae was set as the organism, and the search was applied to other known full-length sequences of terpene alcohol dehydrogenases and benzyl alcohol dehydrogenases from Persicaria minor (nerol dehydrogenase; AFQ59973.1), Castellaniella defragrans (geraniol dehydrogenase; CCF55024.1), Carpoglyphus lactis (geraniol dehydrogenase; BAG32342.1), Ocimum basilicum (geraniol dehydrogenase; AAX83107.1), A. thaliana (Rossmann-fold NAD(P)-binding domain-containing protein; AEE86213.1), A. aegypti (NADP+-dependent farnesol dehydrogenase; ADB03640.1), Lavandula x intermedia (borneol dehydrogenase; AFV30207.1), Picea abies (cinnamyl alcohol dehydrogenase; CAA0597.1), Fragaria x ananassa (cinnamyl alcohol dehydrogenase; AAD10327.1), Artemisia annua (cinnamyl alcohol dehydrogenase; ACB54931.1), Pseudomonas putida (p-cumic alcohol dehydrogenase; AAB62297.1), Mentha x piperita ((-)-isopiperitenol dehydrogenase; AAU20370.1), Streptomyces sp. NL15-2K (coniferyl alcohol dehydrogenase; BAN09098.1), A. thaliana (allyl alcohol dehydrogenase; AAG50689.1), Nicotiana tabacum (allyl alcohol dehydrogenase; BAA89423.1), P. putida (aryl alcohol dehydrogenase; P39849.1), and P. putida (benzyl alcohol dehydrogenase; AAC32671.1).
Effects of pH and temperature on farnesol dehydrogenase activity
The effect of pH on the activity of the purified farnesol dehydrogenase (0.2 μg) was studied with 1.0 mM of trans, trans-farnesol and 1.0 mM NAD+ as substrates at 35°C. Farnesol dehydrogenase activity was assayed at pH values ranging from 5.0 to 11.0 in 0.5 of increment. The following buffers were used at final concentration of 100 mM in the incubation mixture: citrate buffers (pH 5.0–6.0), potassium phosphate buffers (pH 6.5–7.5), glycine-NaOH buffers (pH 9.0–10.5), and carbonate buffers (pH 10.5–11.0). The enzyme activity was expressed as the percentage of the maximum activity. For the optimum temperature, the activity of farnesol dehydrogenase (0.2 μg) was measured under the standard assay conditions, except with the reaction temperature varied between 25–70°C. The enzyme activity was expressed as the percentage of the maximum activity. The temperature stability was determined by incubating the purified enzyme (0.2 μg) at temperature ranging from 25–70°C for 10 min at pH 7.5 (100 mM of tricine-NaOH buffer containing 2.5 mM of 2-ME). The residual farnesol dehydrogenase activity was assayed under the standard enzyme assay conditions. The enzyme activity was defined as the percentage of the maximum activity level.
Effects of metal ions and inhibitors on the farnesol dehydrogenase activity
To determine the effects of inhibitors and metal ions on farnesol dehydrogenase activity, the purified enzyme (0.2 μg) was preincubated with various metal ions and inhibitors at a final concentration of 1.0 mM for 10 min at 35°C, followed by the standard enzyme assay as described before. The effect of inhibitors tested include 2,2-dipyridil, iodoacetamide, sodium azide, 5,5’-dithiobis (2-nitrobenzoic acid), p-chloromercuribenzoate, EDTA, 1, 10-phenanthroline and the metal ions tested include Li+, Ca+, Ag+, Zn2+, Cu2+, Mg2+ and Fe3+. These inhibitors and metal ions were widely used in many previous reports of alcohol dehydrogenase [11, 39, 43–45], and thus were selected for this study. The enzyme activity obtained from the reaction mixture without any extra ion or inhibitor was taken as a control, corresponding to 100% relative activity.
Substrate specificity and Michaelis-Menten constants
The ability of P. minus farnesol dehydrogenase (0.2 μg) to oxidize a range of substrates was determined at pH 9.5 (Fig 1). Enzyme activities were evaluated with different alcohol as substrates namely allylic alcohols (trans, trans-farnesol, cis, trans-farnesol, cis, cis-farnesol, nerolidol, geraniol, nerol, linalool, 2,4-octadien-1-ol, and 2,5-dimethyl-1,5-hexadien-3-ol), non-allylic alcohols (β-citronellol, and 3,7-dihydrolinalool), aromatic alcohols (carveol, (S)-perillyl alcohol, cinnamyl alcohol, p-cumic alcohol, borneol, and menthol), and aliphatic alcohols (tetrahydrogeraniol, tetrahydrolinalool, tetrahydrolavandulol, ethanol, and methanol) at concentrations of 1.0 mM, respectively. The enzyme activity was measured as described before. The effect of different substrates concentration, ranging from 0.5 mM to 1.5 mM with 0.25 increment on enzyme activity was estimated under optimal assay conditions (35°C, pH 9.5 and 5 min). The relative rate of oxidation for each substrate was determined as the percent of the enzyme activity measured with trans, trans-farnesol which was considered to correspond to 100%. The kinetic parameters (Michaelis-Menten constant (K m) and maximal reaction velocity (V max) were determined by linear regression from double-reciprocal plots according to Lineweaver-Burk [46]. The K m and V max were expressed in mM and μmol·min−1, respectively.
Fig 1. List of substrates tested for substrate specificity of P. minus farnesol dehydrogenase.

Results and Discussion
Purification of farnesol dehydrogenase from P. minus leaves
In plants, farnesol dehydrogenase activity has been identified in A. thaliana [26], chicory [47], sweet potato root tissue [11], and orange flavedo [48]. Recently, a gene on chromosome 4 of the Arabidopsis genome (At4g33360), called FLDH was shown to encode a NAD+-dependent dehydrogenase that oxidizes farnesol more efficiently than other prenyl alcohol substrates [26]. In this study, NAD(P)+-dependent farnesol dehydrogenase from P. minus leaves has been purified and characterized. P. minus farnesol dehydrogenase was purified with a high yield (3.2%) by 6 chromatographic steps, including three successive runs on a gel filtration chromatography. All farnesol dehydrogenase activities were recovered in the flow-through fractions of the DEAE-Toyopearl 650M, SP-Toyopearl 650M and Super-Q Toyopearl 650M with increased specific activity. The last step of purification was performed with a TSK-gel GS3000SW gel filtration chromatography. The purification scheme and their results are summarized in Table 1. The purification procedures purified farnesol dehydrogenase about 234-fold with about 3.2% recovery of the enzyme activity. The third run on the TSK-gel GS3000SW resulted in fractions containing farnesol dehydrogenase activity that gave a single protein band by native-PAGE at the same position on the gel where the enzyme activity was detected (Fig 2), strongly suggesting that the farnesol dehydrogenase enzyme from P. minus was purified to homogeneity. The three successive run on TSK-gel GS3000SW was crucial to maintain the homogeneity of the enzyme where elimination of these step will resulted in impurity of the protein sample.
Table 1. Summary of purification of farnesol dehydrogenase from P. minus leaves.
| Purification step | Total activity (U) | Total protein (mg) | Specific activity (U·mg-1) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| Cell-free extract | 7.81 | 6600 | 1.2 × 10−3 | 1.0 | 100.0 |
| DEAE Toyopearl 650M | 7.22 | 946 | 7.6 × 10−3 | 6.5 | 92.5 |
| SP Toyopearl 650M | 6.44 | 349 | 1.7 × 10−2 | 14.0 | 82.5 |
| Super-Q Toyopearl 650M | 2.70 | 67 | 4.0 × 10−2 | 34.0 | 34.6 |
| 1st TSK-Gel GW3000SW | 0.65 | 5 | 1.3 × 10−1 | 107.0 | 8.4 |
| 2nd TSK-Gel GW3000SW | 0.48 | 2 | 2.4 × 10−1 | 204.0 | 6.2 |
| 3rd TSK-Gel GW3000SW | 0.25 | 0.9 | 2.7 × 10−1 | 234.0 | 3.2 |
Fig 2. Native-PAGE of the purified farnesol dehydrogenase from P. minus.
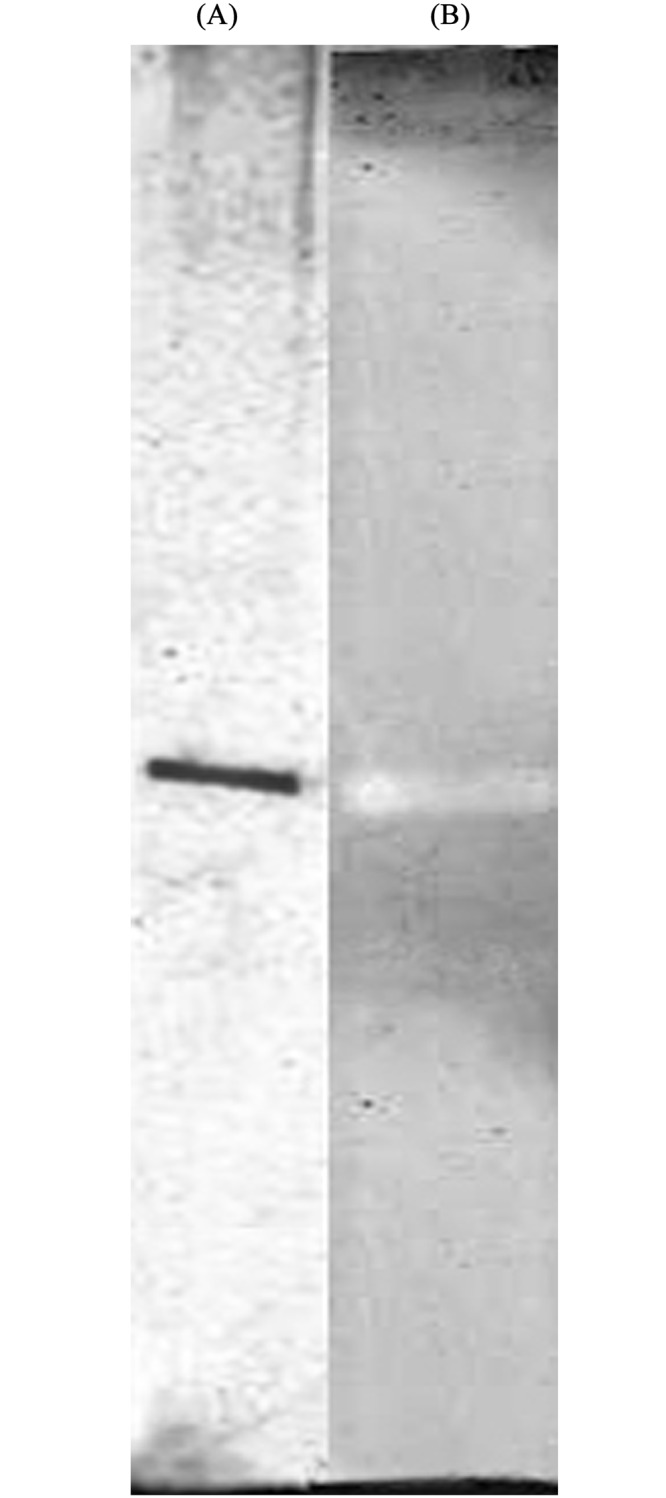
Purified enzyme (33 μg) was subjected to electrophoresis in the absence of SDS with 12.5% gel at pH 8.8. Protein gel were stained by silver stain (A) and activity stain (B).
Determination of molecular mass and isoelectric point
The native molecular mass of the purified enzyme was determined by gel filtration (TSK-gel GS3000SW). The farnesol dehydrogenase showed a relative molecular mass of 130 kDa (Fig 3A). The molecular weight of the monomers of farnesol dehydrogenase were 65 kDa and 70 kDa (Fig 3B), suggesting that P. minus farnesol dehydrogenase appears to be a heterodimeric enzyme. In contrast, both of farnesol dehydrogenase from I. batatas and A. aegypti are homodimers with molecular masses of 60 and 90 kDa, respectively [11, 12]. In addition, several plant alcohol dehydrogenases have been reported to be heterodimeric [39, 49, 50] or homodimeric enzymes [51–53]. Rauwolfia serpentina acyclic monoterpene primary alcohol dehydrogenase [54] and the C. lactis geraniol dehydrogenase [55] are both reported to function as monomers.
Fig 3. Determination of molecular mass of the farnesol dehydrogenase from P. minus.

(A) Estimation of native molecular mass of farnesol dehydrogenase by TSK-gel GS3000SW column. Experimental conditions are described in “Materials and Methods”. Standard protein marker (■): thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1350 Da). Farnesol dehydrogenase (●). (B) SDS-PAGE analysis of purified farnesol dehydrogenase. Purified enzyme and standard proteins were subjected to electrophoresis in the presence of SDS with a 12.5% polyacrylamide gel. The PageRuler™ Prestained Protein Ladder, ~10–170 kDa (SM0671) (Fermentas) was used as the molecular marker.
The purified farnesol dehydrogenase was subjected to isoelectric focusing and developed by silver staining. One band was visualized, that showed a pI value of 6.8 (S1 Fig). The pI value for P. minus farnesol dehydrogenase is different from other reported farnesol dehydrogenases. The calculated pI values for A. thaliana and A. aegypti farnesol dehydrogenases are 8.27 and 7.06, respectively [11, 12]. The lower pI suggests that farnesol dehydrogenase from P. minus is rich in acidic amino acid residues.
Analysis of the protein sequence by MALDI-TOF/TOF-MS
Dialyzed farnesol dehydrogenase was subjected to in-solution digestion with trypsin. Four peptides with m/z values of 2223.04, 2435.06, 3652.21 and 3685.77 were further analyzed with MALDI-TOF/TOF-MS spectrometry and the amino acid sequence of each peptide was determined (Table 2). All 4 amino acid sequences (A,B,C and D) showed similarity (30–70%) to several predicted oxidoreductases and terpene alcohol dehydrogenases, including P. putida p-cumic alcohol dehydrogenase, M. piperita (-)-isopiperitenol dehydrogenase, C. defragrans geraniol dehydrogenase and S. indicum (+)-neomenthol dehydrogenase. The homology comparison of the peptide sequence (A) and (B) showed a putative conserved domain for Rossmann-fold NAD(P)-binding domain-containing protein. Database searches showed that the peptides of the purified P. minus farnesol dehydrogenase shared no significant similarity with any reported farnesol dehydrogenases enzymes; however, peptide fragment (A) and (D) showed less than 45% identities to NADP+-dependent farnesol dehydrogenase 2 of Aedes aegypti and partial farnesol dehydrogenase from Diploptera punctata. These results suggested that the purified enzyme might be a novel farnesol dehydrogenase.
Table 2. Identification of tryptic peptides from P. minus farnesol dehydrogenase.
| Species | Peptide | E value | Identity (%) |
|---|---|---|---|
| (A) The homology comparison of the peptide sequence from P. minus farnesol dehydrogenase showed a putative conserved domain for NADB_Rossmann superfamily by a BLASTp search. | |||
| P. minus | KVWLITGCSTGFGKELTLAALKRGDKVIATARTPAKL | nd. | nd. |
| Ricinus communis 1 | KVWFITGASRGFGRIWTEAALARGDKVAATARQLASL | 2.0×10- 12 | 68 |
| Brassica rapa 2 | KVVLITGVSKGLGRALSLEMAKRGHTVIGCARTQEKL | 2.0×10- 9 | 57 |
| Musa acuminata 3 | KTVLVTGVSRGLGRALSLELARRGHAVIGCARSPDKV | 1.0×10- 8 | 46 |
| Nicotiana sylvestris 4 | KVVMVTGASSGIGREFSLDLAKSGCRIIAAARRVDRL | 2.0×10- 8 | 43 |
| Pseudomonas putida 5 | KVAIVTGAATGIGNAIVRSYLAEGAKVV | 3.0×10- 5 | 39 |
| Aedes aegypti 6 | KVAVVTGSSSGIGAAIAKDLAKAGMVVVGLAR | 2.0×10- 4 | 38 |
| Arabidopsis thaliana 7 | ---LVTGSTGYLGARLCHVLLRRGHSVRALVRRTSDL | 5.0×10- 4 | 35 |
| Mentha x piperita 8 | KVAIVTGGASGIGEVTARLFAERGARAVVIA | 5.0×10- 3 | 29 |
| (B) The homology comparison of the peptide sequence from P. Minus farnesol dehydrogenase showed a putative conserved domain for NADB_Rossmann superfamily by a BLASTp search. | |||
| P. minus | RAFLPHMRARRSGVIALIGS | nd. | nd. |
| Coccomyxa subellipsoidea 9 | QAVLPHMRAAQSGQIINITSLVGFSAIP | 5.0×10- 7 | 50 |
| Solanum lycopersicum 10 | ---VPHMASRRSGSIVNVGSVVGKVSTP | 5.0×10- 7 | 48 |
| Oryza sativa Japonica 11 | RAVAPHMASRRSGRVVNVGSVVGTAATP | 1.0×10- 6 | 46 |
| Castellaniella defragrans 12 | --RIAGVGVCHTDLVCRDGFP | 6.9 | 32 |
| (C) The homology comparison of the peptide sequence from P. minus farnesol dehydrogenase showed no putative conserved domain by a BLASTp search. | |||
| P. minus | KQAELLTRQLSEVHDEAETVIRL | nd. | nd. |
| Citrus sinensis 13 | KQSELLSKLTRQLSIHD | 5.0×10- 8 | 67 |
| Sesamun indicum 14 | -------EIHENSVAEAETVIR | 3.0×10- 6 | 60 |
| (D) The homology comparison of the peptide sequence from P. Minus farnesol dehydrogenase showed no putative conserved domain by a BLASTp search. | |||
| P. minus | KTPAAAAIAHHAAVDGKQPGDPVKA | nd. | Nd. |
| Rhodococcus opacus 15 | --------AINEAVAAQHRGDVVKA | 7.1×10- 2 | 47 |
| Diploptera punctata 16 | -----AAITQQLVKDG | 1.1 | 45 |
1 (XP_002534918.1) putative 3-oxoacyl-[acyl-carrier-proten] reductase of Ricinus communis
2 (XP_009144454.1) predicted carbonyl reductase family member 4 of Brassica rapa
3 (XP_009415001.1) predicted uncharacterized oxidoreductase YMR226C isoform X1 of Musa acuminata subsp. Malaccensis
4 (XP_009792301.1) Predicted dehydrogenase/reductase SDR family member 7-line of Nicotiana sylvestris
5 (AAB62297.1) p-cumic alcohol dehydrogenase of Pseudomonas putida
6 (ADB03640.1) NADP+-dependent farnesol dehydrogenase 2 of Aedes aegypti
7 (AEE86213.1) Rossmann-fold NAD(P)-binding domain-containing protein of Arabidopsis thaliana
8 (AAU20370.1) (-)-isopiperitenol dehydrogenase of Mentha x piperita
9 (XP_005650109.1) NAD(P)-binding protein from Coccomyxa subellipsoidea C-169
10 (XP_004231947.1) predicted NADPH-dependent -1-acylihydroxyacetone phosphate reductase-like of Solanum lycopersicum
11 (ABA99950.2) oxidoreductase, short chain dehydrogenase/reductase family protein, expressed from Oryza sativa Japonica group
12 (CCF55024.1) geraniol dehydrogenase of Castellaniella defragrans
13 (KDO39154.1) hypothetical protein CISIN_1g030018mg of Citrus sinensis
14 (XP_011091715.1) predicted (+)-neomenthol dehydrogenase of Sesamun indicum
15 (AII08455.1) geraniol dehydrogenase (Rhodococcus opacus)
16 (AHZ20737.1) farnesol dehydrogenase, partial (Diploptera punctata)
a nd.–not determined
Effects of temperature and pH
The effects of pH and temperature on the enzyme activity and stability of P. minus farnesol dehydrogenase was investigated. The optimal temperature of the enzyme was found to be 35°C (Fig 4A), which is comparable to the optimal temperature of farnesol dehydrogenases from I. batatas [11] and A. aegypti [12] (25°C-30°C). The residual activity of farnesol dehydrogenase was measured after heat treatment at various temperatures for 10 min in 2.5 mM 2-ME containing tricine-NaOH buffer (100 mM, pH 7.5) (Fig 4A). The enzyme showed the highest activity at 35°C, and remained stable at temperatures up to 50°C. The enzyme activity of P. minus farnesol dehydrogenase declined rapidly at temperatures greater than 50°C. Rapid inactivation of enzyme activity was observed when the enzyme activity was measured from 55 to 65°C. Approximately 70% of the activity at was observed at 55°C and only 16% of the activity left remained at 65°C. The enzyme activity was completely lost at 70°C.
Fig 4. Effects of temperature and pH.

(A) Effects of temperature on enzyme activities of farnesol dehydrogenase and stability of the enzyme. The temperature stability was determined by incubating the purified enzymes at a temperature in the range of 25–70°C for 10 min at pH 7.5 (100 mM tricine-NaOH containing 2.5 mM 2-ME). The residual farnesol dehydrogenase activity was assayed as described in “Materials and Method”. The optimal temperature was determined by performing the standard enzyme assay as described in “Materials and Methods,” except that the reaction temperature was varied. Thermo stability (●), optimal temperature (■). (B) Effect of pH on enzyme activity of farnesol dehydrogenase. Enzyme activity was assayed under the standard assay conditions, except that the following buffers were used at a final concentration of 100 mM in the incubation mixture: citrate buffers (■), potassium phosphate buffers (×), Tris-HCl buffers (○), glycine-NaOH buffers (▲), and carbonate buffers (●).
The optimal pH for P. minus farnesol dehydrogenase was found to be 9.5 (Fig 4B). At pH below 8.5 or above 10.0, the enzyme activity declined sharply, suggesting that P. minus farnesol dehydrogenase has a narrow alkaline pH optimum. This result is comparable to the optimal pH for farnesol dehydrogenases from I. batatas [11] and A. aegypti [12] (pH 9.5–11), and to the terpene alcohol dehydrogenases [39, 49, 54, 56, 57] (pH 9.0–9.5).
Effects of metal ions and inhibitors
Oxidation of trans, trans-farnesol by P. minus farnesol dehydrogenase was inhibited completely by heavy metal ions and sulfhydryl agents such as p-chloromercuribenzoate, 5,5’-dithiobis (2-nitrobenzoic acid), and iodoacetoamide (Table 3). The activity was also completely inhibited by EDTA and 1,10-phenanthroline, a major chelating agent of Zn ion. Chelating agents of the Fe ion, 2, 2’-dipyridyl and sodium azide caused approximately 25–50% inhibition in farnesol dehydrogenase activity. Terpene alcohol dehydrogenases were also inhibited by sulfhydryl group inhibitors and heavy metals [11, 39, 49, 54, 56, 58–60]. Inactivation of P. minus farnesol dehydrogenase by sulfhydryl reagents suggested that the sulfhydryl group of the enzyme is essential for catalytic activity. The inhibition of activity by EDTA and other metal chelators also suggests the participation of one or more metal ions in the enzyme activity.
Table 3. Effects of inhibitors on the farnesol dehydrogenase activity.
The enzyme was preincubated for 5 min at 35°C with the various reagents before addition of the substrate. Each reagent was added at the final concentration as indicated.
| Reagent (1.0 mM) | Relative activity (%) |
|---|---|
| None | 100 |
| 2,2-Dipyridil | 48 |
| Iodoacetamide | 34 |
| Sodium azide | 75 |
| 5,5’-Dithiobis (2-nitrobenzoic acid) | 0 |
| p-Chloromercuribenzoate | 0 |
| EDTA | 0 |
| 1, 10-Phenanthroline | 0 |
| Lithium chloride | 0 |
| Silver nitrate | 0 |
| Zinc chloride | 0 |
| Magnesium chloride | 27 |
| Calcium chloride | 76 |
| Iron (III) chloride | 0 |
| Cuprum sulphate | 0 |
Substrate specificity and Michaelis-Menten constants
P. minus farnesol dehydrogenase utilized both NAD+ and NADP+ as coenzymes. The utilization of NAD+ by the enzyme was 5 times more efficient than for NADP+, while the K m value for NADP+ was over 54-fold greater than the corresponding value for NAD+ (Table 4). Both NAD+ and NADP+ also can be utilized as coenzymes for several terpene alcohol dehydrogenases, such as M. piperita isopiperitenol dehydrogenase [59], N. tabacum allylic alcohol dehydrogenase [61], and S. officinalis borneol dehydrogenase [60]. On the other hand, farnesol dehydrogenases from I. batatas [11] and A. aegypti [12] and several plant terpene alcohol dehydrogenases [39, 56, 57, 58, 62]; acyclic monoterpene primary alcohol DH [49, 54] were highly specific for NADP+. Other terpene alcohol dehydrogenases such as C. lactis geraniol dehydrogenase[55], Pseudomonas perillyl alcohol dehydrogenase [63], Nepeta racemosa nepetalactol oxidoreductase[49], and L. x intermedia borneol dehydrogenase [64] were very specific to NAD+.
Table 4. Substrate specificity, coenzyme specificity, and kinetic parameters of P. minus farnesol dehydrogenase.
| Substrate | Relative activity (%) | K m value (mM) | V max value (μmol/ml) |
|---|---|---|---|
| Trans-trans-farnesol | 100 | 0.17 | 0.24 |
| Cis-trans-farnesol | 66 | 0.33 | 0.26 |
| Cis-cis-farnesol | 47 | 0.42 | 0.25 |
| Nerodilol | 36 | 1.00 | 0.29 |
| Geraniol | 37 | 12.50 | 0.63 |
| Nerol | 0 | n.d | n.d |
| Carveol | 32 | 0.71 | 0.45 |
| (S)-Perillyl alcohol | 32 | 0.77 | 0.20 |
| Cinnamyl alcohol | 35 | 1.04 | 0.15 |
| p-Cumic alcohol | 0 | n.d | n.d |
| NAD+ | 100 | 0.74 | 0.38 |
| NADP+ | 22 | 40.00 | 2.50 |
n.d- not determined.
The P. minus enzyme showed the highest activity toward trans, trans-farnesol, cis, trans-farnesol and cis, cis-farnesol. Nerolidol, geraniol, (S)-perillyl alcohol, cinnamyl alcohol, and carveol were also oxidized at rates less than 40% compared to trans, trans-farnesol (Table 4). On the other hand, P. minus farnesol dehydrogenase did not react with other terpene alcohols such as p-cumic alcohol, borneol, linalool, or menthol. No detectable activity was observed with β-citronellol, 3,7-dihydrolinalool, benzyl alcohol, 2,4-octadien-1-ol, or 2,5-dimethyl-1.5-hexadien-3-ol. Tetrahydrogeraniol, tetrahydrolinalool, tetrahydrolavandulol, ethanol, and methanol were inert as substrates. The kinetic parameters determined for farnesol dehydrogenase were shown in Table 4. No reduction reaction was observed.
P. minus farnesol dehydrogenase oxidizes not only trans, trans-farnesol, but also its geometrical and structural isomers. The geometrical isomers of trans, trans-farnesol, (cis, trans-farnesol and cis, cis-farnesol) were oxidized at 66% and 47% of the rate observed for trans, trans-farnesol. The K m values of cis, trans-farnesol and cis, cis-farnesol were approximately 2-fold and 2.5-fold higher than for trans, trans-farnesol, respectively. Nerolidol, a structural isomer of trans, trans-farnesol, was also oxidized at a lower rate compared to trans, trans-farnesol. The K m value for nerolidol was approximately 6-fold higher than trans, trans-farnesol. In addition, farnesol dehydrogenases from A. aegypti and I. batatas oxidize cis, cis-farnesol [12] and cis, trans-farnesol [11], respectively, at lower rates compared to trans, trans-farnesol. These results indicate that P. minus farnesol dehydrogenase is capable of distinctly recognizing the geometrical and structural isomers of substrates, similar with other farnesol dehydrogenases [11, 12] and geraniol dehydrogenases [39, 55, 58, 62].
The positions of the double bonds at carbon positions 2 and 6 in the substrate also play a significant role in the substrate selectivity of P. minus farnesol dehydrogenase. Besides trans, trans-farnesol and its isomers, P. minus farnesol dehydrogenase selectively oxidizes geraniol ((trans)-3,7-dimethyl-2,6-octadien-1-ol) but at a much lower rate. Moreover, linalool, a structural analog of geraniol, that has double bond in position 1 and 6 was not oxidized. Neither non-allylic nor aliphatic analogs of geraniol, β-citronellol (3, 7-dimethyl-6-octen-1-ol) and tetrahydrogeraniol (3, 7-dimethyl-1-octanol), were substrates for P. minus farnesol dehydrogenase. These results were consistent with previous reports of farnesol dehydrogenases from A. aegypti [12] and I. batatas [11]. The K m values for geraniol and β-citronellol of A. aegypti farnesol dehydrogenase [12] was about 2.3-fold and 1.3-fold higher than obtained with trans, trans-farnesol as substrate, respectively. The relative activities of geraniol and β-citronellol for I. batatas farnesol dehydrogenase were 57% and 37% of the rate observed with trans, trans-farnesol, respectively [11].
Interestingly, P. minus farnesol dehydrogenase also oxidized several aromatic alcohols such as carveol, (S)-perillyl alcohol, and cinnamyl alcohol. On the other hand, aromatic alcohols, such as benzyl alcohol and p-cumic alcohol (which does not have an alkenyl group near the carbinol) were not oxidized by P. minus farnesol dehydrogenase.
P. minus [39], O. basilicum [57], and C. defragrans [65] geraniol dehydrogenases, as well as P. putida benzyl alcohol dehydrogenase [66] and P. putida MB-1 allylic alcohol dehydrogenase [67] show broad substrate specificity with substrates containing an allylic double bond or with an aromatic ring attached to the carbinol carbon. Benzyl alcohol dehydrogenase from Acinetobacter calcoaceticus oxidized not only a range of aromatic alcohols related to benzyl alcohol but also the allylic alcohol moieties in perillyl, cinnamyl, and coniferyl alcohols [68]. However, 2-phenylethanol, which does not have an alkenyl group near the carbinol, was not a substrate for this enzyme. MacKintosh and Fewson suggested that for cinnamyl and coniferyl alcohols, the alkenyl group, which is located between the aromatic ring and the carbinol center may help correctly position the alcohol in the active site [68].
Conclusion
In this study, we purified the farnesol dehydrogenase enzyme from P. minus leaves and characterized its biochemical properties. This enzyme has several different biochemical characteristics from the previously identified farnesol dehydrogenases regarding the substrate profile, isoelectric point, molecular weight and utilization of both NAD+ and NADP+ as coenzymes. Furthermore, a MALDI-TOF/TOF-MS analysis of the purified enzyme revealed no homology to any known farnesol dehydrogenase. Altogether, these data suggest that the purified enzyme is a novel form of NAD(P)+-dependent farnesol dehydrogenase. Studies of the enzyme’s substrate specificity and kinetic parameter results suggest that the selectivity of substrate specificity of P. minus farnesol dehydrogenase depends on the cis or trans configuration, the number of the isoprene units, and the position of double bonds of the substrate. In addition, P. minus farnesol dehydrogenase exhibited no activity against aliphatic alcohols demonstrating that this enzyme was specific for substrates containing an allylic double bond. The complete sequence, structure and functional analysis of P. minus farnesol dehydrogenase may lead to the discovery of novel strategies for development of integrated pest management in plant by engineering the transgenic plants using insect-resistant genes to fight insect pests [69]. Nevertheless, further studies on the hormonally linked relationship between plants and insect pest could well indicate the method for utilizing the chemicoecological interaction in integrated pest management [28].
Supporting Information
The arrows indicate the protein bands approximately at pI 6.8. ReadyStrip IPG strips are preprinted to indicate anode end and pH range. A barcode is printed toward the pointed end of the strip holder.
(TIF)
Acknowledgments
We wish to thank Mohd Fauzi Abd Razak and Saidi Adha Suhaimi for technical assistance.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by the Ministry of Science, Technology & Innovation (Grant number: 02-01-02-SF0852, URL: http://www.mosti.gov.my/), and International Foundation for Science (Grant number: RB-IFS-001-2012, URL: http://www.ifs.se/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kamita SG, Hammock BD. Juvenile hormone esterase: biochemistry and structure. J Pestic Sci. 2010; 35: 265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lopez O, Jose GF, Maria VG. New trends in pest control: the search for greener insecticides. Green Chem. 2005; 7: 431–42. [Google Scholar]
- 3. Hoffmann KH, Lorenz MW. Recent advances in hormones in insect pest control. Phytoparasitica. 1998; 26 (4): 323–30. [Google Scholar]
- 4. Furuta K, Kuwano E, Fujita N, Yamada N. Synthesis and biological activity of novel anti-juvenile hormone agents. J Pestic Sci. 2008; 33 (1): 14–6. [Google Scholar]
- 5. Pedigo LP, Rice ME. Entomology and pest management. 6th ed New Jersey: Pearson Prentice Hall; 2009. [Google Scholar]
- 6. Athanassiou CG, Arthur FH, Kavallieratos NG, Throne JE. Efficacy of spinosad and methoprene, applied alone or in combination, against six stored-product insect species. J Pest Sci. 2010; 84: 61–7. [Google Scholar]
- 7. Belles X, Martin D, Piulach M-D. The mevalonate pathway and the synthesis of juvenile hormone in insects. Annu Rev Entomol. 2004. August 5; 50: 181–199. [DOI] [PubMed] [Google Scholar]
- 8. Marchal E, Zhang JR, Badisco L, Verlinden H, Hult EF, Wielendaele PV, et al. Final steps in juvenile hormone biosynthesis in the desert locust, Schistocerca gregaria . Insect Biochem Molec Biol. 2011; 41: 219–27. [DOI] [PubMed] [Google Scholar]
- 9. Castillo-Gracia M, Couillaud F. Molecular cloning and tissue expression of an insect farnesyl diphosphate synthase. Eur J Biochem. 1999; 262: 365–70. [DOI] [PubMed] [Google Scholar]
- 10. Nah J, Song SJ, Back K. Partial characterization of farnesyl and geranylgeranyl diphosphatases induced in rice seedlings by UV-C irradiation. Plant Cell Physiol. 2001; 42 (8): 864–7. [DOI] [PubMed] [Google Scholar]
- 11. Inoue H, Tsuji H, Uritani I. Characterization and activity change of farnesol dehydrogenase in black rot fungus-infected sweet potato. Agric Biol Chem. 1984; 48 (3): 733–8. [Google Scholar]
- 12. Mayoral JG, Nouzova M, Navare A, Noriega FG. NADP+-dependent farnesol dehydrogenase, a corpora allata enzyme involved in juvenile hormone synthesis. P Natl Acad Sci. 2009; 106 (50): 21091–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bede JC, Goodman WG, Tobe SS. Development distribution of insect juvenile hormone III in the sedge, Cyperus iria L. Phytochem. 1999; 52: 1269–74. [Google Scholar]
- 14. Shinoda T, Itoyama K. Juvenile hormone acid methyltransferase: a key regulatory enzyme for insect metamorphosis. Proc Natl Acad Sci. 2003; 100 (21): 11986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sen SE, Trobaugh C, Beliveau C, Richard T, Cusson M. Cloning, expression and characterization of a dipteran farnesyl diphosphate synthase. Insect Biochem Mol Biol. 2007. November; 37(11): 1198–206. [DOI] [PubMed] [Google Scholar]
- 16. Nyati P, Nouzova M, Rivera-Perez C, Clifton ME, Mayoral JG, Noriega FG. Farnesyl phosphatase, a corpora allata enzyme involved in juvenile hormone biosynthesis in Aedes aegypti . PLoS One. 2013. July 8; 8(8): e71967 10.1371/journal.pone.0071967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayoral JG, Nouzova M, Yoshiyama M, Shinoda T, Hernandez-Martinez S, Dolghih E, et al. Molecular and functional characterization of a juvenile hormone acid methyltransferase expressed in the corpora allata of mosquitoes. Insect Biochem Mol Biol. 2009. January; 39(1): 31–7. 10.1016/j.ibmb.2008.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Helvig C, Koener JF, Unnithan GC, Feyereisen R. CYP15A1, the cytochrome P450 that catalyzes epoxidation of methyl farnesoate to juvenile hormone III in cockroach corpora allata. PNAS. 2004. March 23; 101(12) 4024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toong YC, Schooley DA, Baker FC. Isolation of insect juvenile hormone III from a plant. Nature. 1988; 333:170–1. [Google Scholar]
- 20. Bede JC, Teal PE, Goodman WG, Tobe SS. Biosynthetic pathway of insect juvenile hormone III in cell suspension cultures of the sedge Cyperus iria . Plant Physiology. 2001; 127 (2): 584–93. [PMC free article] [PubMed] [Google Scholar]
- 21. Cunillera N, Boronat A, Ferrer A. The Arabidopsis thaliana FPS1 gene generates a novel mRNA that encodes a mitochondrial farnesyl-diphosphate synthase isoform. J Biol Chem. 1997. June 13; 272(24): 15381–8. [DOI] [PubMed] [Google Scholar]
- 22. Gaffe J, Bru J-P, Causse M, Vidal A, Stamitti-Bert L, Carde J-P, et al. LEFPS1, a tomato farnesyl pyrophosphate gene highly expressed during early fruit development. Plant Physiology. 2000. August; 123: 1351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y-J, Chen X, Zhang M, Su P, Liu Y-J, Tong Y-R, et al. Molecular cloning and characterisation of farnesyl pyrophosphate synthase from Tripterygium wilfordii . PLoS One. 2015. May 4; 10(5): e0125415 10.1371/journal.pone.0125415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Flores-Sánchez IJ, Ortega-López J, Montes-Horcasitas MDC, Ramos-Valdivia AC. Biosynthesis of sterols and triterpenes in cell suspension cultures of Uncaria tomentosa . Plant Cell Physiol. 2002. September 20; 43(12): 1502–9. [DOI] [PubMed] [Google Scholar]
- 25. Ha SB, Lee DE, Lee HJ, Song SJ, Back K. Activities of soluble and microsomal farnesyl diphosphatases in Datura stramonium . Biol Plant. 2003. November; 47(3): 477–9. [Google Scholar]
- 26. Bhandari J, Fitzpatrick AH, Crowell DN. Identification of novel abscisic acid-regilated farnesol dehydrogenase from Arabidopsis . J Plant Physiol. 2010. August 31; 154:1116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y, Yuan JS, Ross J, Noel JP, Pichersky E, Chen F. An Arabidopsis thaliana methyltransferase capable of methylating farnesoic acid. Arch Biochem Biophys. 2006. April 15; 448(1–2): 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Slama K. Animal hormones and antihormones in plants In: Rosenthal GA, Janzen DH, Applebaum SW. Herbivores, their interaction with secondary plant metabolites. New York: Academic Press; 1979. p. 683–700. [Google Scholar]
- 29. Minakuchi C, Riddiford LM. Insect juvenile hormone as a potential target of pest management. J Pestic Sci. 2006; 31: 77–84. [Google Scholar]
- 30. Sharma HC, Sharma KK, Seetharama N, Ortiz R. Prospects for using transgenic resistance to insects in crop improvement. Electron J Biotechnol. 2000. August 15; 3(2): 1–20. [Google Scholar]
- 31. Qader SW, Mahmood AA, Chua LS, Hamdan S. Potential bioactive property of Polygonum minus Huds (kesum) review. Sci Res Essays. 2012. January 16; 7(2): 90–3. [Google Scholar]
- 32. Qader SW, Mahmood AA, Chua LS, Najim N, Zain MM, Hamdan S. Antioxidant, total phenolic content and cytotoxicity evaluation of selected Malaysian plants. Molecules. 2011; 16: 3433–43. 10.3390/molecules16043433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wasman SQ, Mahmood AA, Salehhuddin H, Zahra AA, Salmah I. Cytoprotective activities of Polygonum minus aqueous leaf extract on ethanol-induced gastric ulcer in rats. J Med Plants Res. 2010. December 18; 4(24): 2658–65. [Google Scholar]
- 34. Wiart C. Medicinal plants of Asia and the Pacific. USA: CRC Press; 2006. [Google Scholar]
- 35. Suhaila M, Suzana S, El-Sharkawy SH, Abdul Manaf A, Sepiah M. Antimycotic screening of 58 Malaysian plants against plant pathogens. Pesticide Sci.1999. March 26; 47: 259–64. [Google Scholar]
- 36. Baharum SN, Bunawan H, Ghani MA, Mustapha WAW, Noor NM. Analysis of the chemical composition of the essential oil of Polygonum minus Huds. using two-dimensional Gas Chromatography-Time-of-Flight Mass Spectrometry (GC-TOF MS). Molecules. 2010. October 12; 15: 7006–15. 10.3390/molecules15107006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyazawa M, Tamura N. Components of the essential oil from sprouts of Polygonum hydropiper L. (‘Benitade’). Flavour Frag J. 2007. January 17; 22(3): 188–90. [Google Scholar]
- 38. Baker FC, Mauchamp B, Tsai LW, Schooley DA. Farnesol and farnesal dehydrogenase (s) in corpora allata of the tobacco hornworm moth, Manduca sexta . J Lipid Res. 1983; 24(12):1586–94. [PubMed] [Google Scholar]
- 39. Hassan M, Maarof ND, Noor NM, Othman R, Mori N. Monoterpene alcohol metabolism: identification, purification, and characterization of two geraniol dehydrogenase isoenzymes from Polygonum minus leaves. Biosci Biotechnol Biochem. 2012. May 17; 76(8): 1463–70. [DOI] [PubMed] [Google Scholar]
- 40. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951. November; 193(1):265–75. [PubMed] [Google Scholar]
- 41. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970. August 15; 227(5259):680–5. [DOI] [PubMed] [Google Scholar]
- 42. Sarah SA, Karsani SA, Amin I, Mokhtar NFK, Sazili AQ. Differences in thermostable actin profile of goat meat as observed in two-dimensional gel electrophoresis (2DE). Int Food Res J. 2013; 20 (2): 897–901. [Google Scholar]
- 43. Sperry AE, Sen SE. Farnesol oxidation in insects: evidence that the biosynthesis of insect juvenile hormone is mediated by a specific alcohol oxidase. Insect Biochem Mol Biol. 2001; 31(2): 171–178. [DOI] [PubMed] [Google Scholar]
- 44. Koutsompogeras P, Kyriacou A, Zabetakis I. Characterizing NAD-dependent alcohol dehydrogenase enzymes of Methylobacterium extorquens and strawberry (Fragaria × ananassa cv. elsanta). J Agric Food Chem. 2006. January 11; 54(1): 235–42. [DOI] [PubMed] [Google Scholar]
- 45. Takeda M, Muranushi T, Inagaki S, Nakao T, Motomatsu S, Suzuki I, et al. Identification and characterization of a mycobacterial (2R, 3R)-2, 3-butanediol dehydrogenase. Biosci Biotechnol Biochem. 2011. December 7; 75(12): 2384–2389. [DOI] [PubMed] [Google Scholar]
- 46. Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc. 1934; 56 (3): 658–66. [Google Scholar]
- 47. de Kraker JW, Schurink M, Franssen MCR, König WA, de Groot A, Bouwmeester HJ. Hydroxylation of sesquiterpenes by enzymes from chicory (Cichorium intybus L.) roots. Tetrahedron. 2003. January 13; 59(3): 409–18. [Google Scholar]
- 48. Chayet L, Pont-Lezica R, George-Nascimento C, Cori O. Biosynthesis of sesquiterpene alcohols and aldehydes by cell free extracts from orange flavedo. Phytochemistry. 1972. July 26; 12(1): 95–101. [Google Scholar]
- 49. Hallahan DL, West JM, Wallsgrove RM, Smiley DW, Dawson GW, Pickett JA, Hamilton JG. Purification and characterization of an acyclic monoterpene primary alcohol: NADP+ oxidoreductase from catmint (Nepeta racemosa). Arch Biochem Biophys. 1995. May 10; 318(1): 105–112. [DOI] [PubMed] [Google Scholar]
- 50. Halpin C, Knight ME, Grima-Pettenati J, Goffner D, Boudet A, Schuch W. Purification and characterization of cinnamyl alcohol dehydrogenase from tobacco stems. Plant physiol. 1992. January; 98(1):12–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Malley DM, Porter S, Sederoff RR. Purification, characterization, and cloning of cinnamyl alcohol dehydrogenase in loblolly pine (Pinus taeda L.). Plant Physiol. 1992. April; 98(4): 1364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Igaue I, Yagi M. Alcohol dehydrogenase from cultured rice cells. Plant Cell Physiol. 1982; 23(2): 213–25. [Google Scholar]
- 53. Tong WF, Lin SW. Purification and biochemical-properties of rice alcohol-dehydrogenase. Bot Bull Acad Sinica. 1988; 29(4): 245–253. [Google Scholar]
- 54. Ikeda H, Esaki N, Nakai S, Hashimoto K, Uesato S, Soda K et al. Acyclic monoterpene primary alcohol:NADP+ oxidoreductase of Rauwolfia serpentina cells: the key enzyme in biosynthesis of monoterpene alcohols. J Biochem. 1991; 109 (2): 341–7. [PubMed] [Google Scholar]
- 55. Noge K, Kato M, Mori N, Kataoka M, Tanaka C, Yamasue Y, et al. Geraniol dehydrogenase, the key enzyme in biosynthesis of the alarm pheromone, from the astigmatid mite C. lactis (Acari: Carpoglyphidae). FEBS J. 2008. June; 275(11): 2807–17. 10.1111/j.1742-4658.2008.06421.x [DOI] [PubMed] [Google Scholar]
- 56. Potty VH, Bruemmer JH. Oxidation of geraniol by an enzyme system from orange. Phytochemistry. 1970. May; 9(5): 1003–7. [Google Scholar]
- 57. Iijima Y, Wang G, Fridman E, Pichersky E. Analysis of the enzymatic formation of citral in the glands of sweet basil. Arch Biochem Biophys. 2006. April 15; 448(1–2): 141–9. [DOI] [PubMed] [Google Scholar]
- 58. Sangwan RS, Singh-Sangwan N, Luthra R. Metabolism of acyclic monoterpenes: partial purification and properties of geraniol dehydrogenase from lemongrass (Cymbopogon flexuosus Stapf.) leaves. J Plant Physiol. 1993. August; 142(2): 129–34. [Google Scholar]
- 59. Kjonaas RB, Venkatachalam KV, Croteau R. Metabolism of monoterpenes: oxidation of isopiperitenol to isopiperitenone, and subsequent isomerization to piperitenone by soluble enzyme preparations from peppermint (Mentha piperita) leaves. Arch Biochem Biophys. 1985. April 1; 238(1):49–60. [DOI] [PubMed] [Google Scholar]
- 60. Croteau R, Hooper CL, Felton M. Biosynthesis of monoterpenes: Partial purification and characterization of a bicyclic monoterpenol dehydrogenase from sage (Salvia officinalis). Arch Biochem Biophys. 1978. May; 188(1): 182–93. [DOI] [PubMed] [Google Scholar]
- 61. Hirata T, Tamura Y, Yokobatake N, Shimoda K, Ashida Y. A 38 kDa allylic alcohol dehydrogenase from the cultured cells of Nicotiana tabacum . Phytochemistry. 2000. October; 55(4): 297–303. [DOI] [PubMed] [Google Scholar]
- 62. Sekiwa-Iijima Y, Aizawa Y, Kubota K. Geraniol dehydrogenase activity related to aroma formation in ginger (Zingiber officinale Roscoe). J Agric Food Chem. 2001. November 21; 49(12): 5902–6. [DOI] [PubMed] [Google Scholar]
- 63. Ballal NR, Bhattacharyya PK, Rangachari PN. Microbiological transformations of terpenes. XIV. Purification & properties of perillyl alcohol dehydrogenase. Indian J Chem. 1968. March; 5(1):1–6. [PubMed] [Google Scholar]
- 64. Sarker LS, Galata M, Demissie ZA, Mahmoud SS. Molecular cloning and functional characterization of borneol dehydrogenase from the glandular trichomes of Lavandula x intermedia . Arch Biochem Biophys. 2012. December 15; 528(2): 163–70. 10.1016/j.abb.2012.09.013 [DOI] [PubMed] [Google Scholar]
- 65. Lüddeke F, Wülfing A, Timke M, Germer F, Weber J, Dikfidan A, et al. Geraniol and geranial dehydrogenases induced in anaerobic monoterpene degradation by Castellaniella defragrans . Appl Environ Microbiol. 2012. April; 78(7): 2128–36. 10.1128/AEM.07226-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Curtis AJ, Shirk MC, Fall R. Allylic or benzylic stabilization is essential for catalysis by bacterial benzyl alcohol dehydrogenases. Biochem Biophys Res Commun. 1999. May 27; 259(1): 220–3. [DOI] [PubMed] [Google Scholar]
- 67. Malone VF, Chastain AJ, Ohlsson JT, Poneleit LS, Nemecek-Marshall M, Fall R. Characterization of a Pseudomonas putida allylic alcohol dehydrogenase induced by growth on 2-methyl-3-buten-2-ol. Appl Environ Microbiol. 1999. June; 65(6): 2622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. MacKintosh RW, Fewson CA. Benzyl alcohol dehydrogenase and benzaldehyde dehydrogenase II from Acinetobacter calcoaceticus. Substrate specificities and inhibition studies. Biochem J. 1988; 255: 653–61. [PMC free article] [PubMed] [Google Scholar]
- 69. Yu X, Wang G, Huang S, Ma Y, Xia L. Engineering plants for aphid resistance: current status and future perspectives. Theor Appl Genet. 2014. July 25; 127:2065–83. 10.1007/s00122-014-2371-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The arrows indicate the protein bands approximately at pI 6.8. ReadyStrip IPG strips are preprinted to indicate anode end and pH range. A barcode is printed toward the pointed end of the strip holder.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.