

Abstract
Progressive Multifocal Leukoencephalopathy (PML) is caused by lytic replication of JC virus (JCV) in specific cells of the central nervous system. Like other polyomaviruses, JCV encodes a large T-antigen helicase needed for replication of the viral DNA. Here, we report the development of a luciferase-based, quantitative and high-throughput assay of JCV DNA replication in C33A cells, which, unlike the glial cell lines Hs 683 and U87, accumulate high levels of nuclear T-ag needed for robust replication. Using this assay, we investigated the requirement for different domains of T-ag, and for specific sequences within and flanking the viral origin, in JCV DNA replication. Beyond providing validation of the assay, these studies revealed an important stimulatory role of the transcription factor NF1 in JCV DNA replication. Finally, we show that the assay can be used for inhibitor testing, highlighting its value for the identification of antiviral drugs targeting JCV DNA replication.
Keywords: Replication of JCV DNA, C33A cells, Nuclear localization of T-ag, Stimulation of replication by the non-coding control region, NF-1, Screening for inhibitors of JCV replication
Introduction
There are presently 12 known human polyomaviruses (reviewed in (Ehlers and Wieland, 2013; Scuda et al., 2013)), many of which have been associated with important diseases (reviewed in (Ferenczy et al., 2012; Jiang et al., 2009; White et al., 2013)). As a result, there is increased interest in understanding the life cycles of these viruses and their tropism for different cells within the human body. Progress in deciphering the biology of polyomaviruses will help address the growing need for therapeutic interventions that can limit the propagation of these infectious agents.
JC virus (JCV) is an example of a pathogenic human polyomavirus (reviewed in (Jiang et al., 2009; Padgett and Walker, 1973; Shishido-Hara, 2010)). JCV infects 50–80% of the human population (Knowles et al., 2003; Padgett and Walker, 1973) and is generally asymptomatic. However, its reactivation in immuno-compromised individuals can lead to the development of Progressive Multifocal Leukoencephalopathy (PML), an often-fatal demyelinating disease (reviewed in (Bellizzi et al., 2013; Brew et al., 2010; Ferenczy et al., 2012)). Examples of susceptible individuals are those suffering from AIDS or patients undergoing immunosuppressive therapy, such as organ transplant recipients or multiple sclerosis patients treated with the monoclonal antibody Tysabri (reviewed in (Ferenczy et al., 2012; Koralnik, 2013)). PML results from the ability of JCV to infect cells of the central nervous system and in particular glial cells (reviewed in (Major et al., 1992)), although some recent reports indicate that it may also infect neurons (e.g., (DuPasquier et al., 2003; Wuthrich et al., 2009)).
A central event during the life cycle of all polyomaviruses is replication of their viral DNA genome. The prototypic primate polyomavirus, simian virus 40 (SV40), has long been used as a paradigm to investigate the mechanisms employed during viral DNA replication (reviewed in (Fanning and Zhao, 2009; Meinke and Bullock, 2012)). As with all polyomaviruses, replication of the SV40 genome is dependent on a viral protein, termed large T antigen (T-ag); the other factors needed for this process being supplied by the infected cell (reviewed in (Bell and Dutta, 2002; Fanning and Knippers, 1992; Hurwitz et al., 1990; Kelly, 1988)). T-antigen contains several functional regions, including the N-terminal J-domain, the origin binding domain (OBD) and the helicase domain (An et al., 2012; Simmons, 2000; Topalis et al., 2013). Upon binding to the viral origin, T-ag engages in multiple protein/origin and protein/protein interactions (reviewed in (Borowiec et al., 1990; Bullock, 1997; Fanning and Knippers, 1992)). Critical insights into these fundamental processes were provided by recent structural studies of SV40 T-ag (reviewed in (Meinke and Bullock, 2012)). For example, studies addressing how the OBD interacts with DNA revealed how a region spanning the A1 & B2 loops binds site-specifically to the central region of the viral origin (Bochkareva et al., 2006; Chang et al., 2013; Meinke et al., 2007). Similarly, structural and functional studies of the helicase domain led to a number of fundamental observations, such as helping to reveal how ssDNA is pumped through the central channel of the helicase domain (Gai et al., 2004; Li et al., 2003). They have also provided critical insights into the oligomerization process (Gai et al., 2004; Li et al., 2003).
The first structural study of JCV T-ag was recently reported (Meinke et al., 2014). Remarkably, it established that differences exist between the T-ag OBDs encoded by JCV and SV40. Of particular interest was the discovery that the JCV OBD contains a relatively large C-terminal pocket that is bound by residues from the A1 and B2 loops of an adjacent OBD (Meinke et al., 2014). This and other observations led to the conclusion that the T-ag’s encoded by SV40 and JCV, despite being highly homologous, bear subtle but important differences.
To help establish JCV DNA replication as a valid antiviral target for the treatment of PML, we have set out to extend our initial structural studies of JCV T-ag to provide a broader understanding of the regions of the molecule responsible for binding to DNA and interacting with cellular replication proteins. In addition, we have initiated investigation of the regulatory mechanisms that govern replication of the JCV genome in different cell types, as they may also provide opportunities for antiviral drug discovery. With these goals in mind, we elected to establish a T-ag dependent high-throughput assay for JCV DNA replication. The assay described herein should greatly facilitate the study of JCV DNA replication and the screening for inhibitors of this process.
Results
Development of a luciferase based assay for JCV DNA replication
We previously described cellular assays of SV40 and HPV31 DNA replication based on a dual-luciferase readout (Fradet-Turcotte et al., 2010). Here, we tested if this methodology could be applied to the study of JCV DNA replication. In our initial series of experiments, we aimed to construct the necessary T-ag and origin-containing plasmids needed for this assay and to identify cell lines capable of supporting robust levels of JCV DNA replication. An illustration of the luciferase based JCV replication assay is presented in Fig. 1A. In overview, a plasmid encoding JCV T-ag is co-transfected into permissive cells along with a second plasmid containing both the JCV origin of replication and the firefly luciferase reporter gene. As a control, a third plasmid encoding Renilla luciferase is included in the transfection. After suitable time intervals, the amount of T-ag dependent replication is determined by measuring the levels of firefly luciferase activity expressed from the ori-containing plasmid, using a luminometer (Fradet-Turcotte et al., 2010). The levels of Renilla luciferase activity measured in the same cells are also determined and used for normalization.
Fig. 1.

Diagrams of the luciferase based JCV replication assay and the Mad-1 NCCR. A. On the left are depictions of three plasmid types used in the assay. They include a plasmid encoding JCV T-ag (pJCVT-ag), a plasmid containing both the JCV origin of replication and the firefly Fluc gene (pFL-JCVori) and a plasmid encoding Renilla luciferase (pRL). These plasmids are transfected into human cell lines where T-ag dependent replication takes place. Following an adequate time (e.g., 48 or 72 h) the amount of light released by the firefly and renilla luciferases is measured using a luminometer. B. A diagram of the tandemly duplicated regulatory sequences adjacent to the JCV origin of replication in the Mad-1 strain (Frisque, 1983). Individual regions within the NCCR are indicated; they include the core origin (containing the central Site II region and flanking early palindrome (EP) and TATA regions), Site I and subdomains within the enhancers.
To fully introduce this assay, it is also necessary to provide a brief description of the viral regulatory region. In the JCV genome, a region termed the non-coding control region (NCCR) contains the viral origin of replication and it separates the early and late genes (reviewed in (Bellizzi et al., 2013; Ferenczy et al., 2012)). The hyper-variable promoter/enhancer region within the NCCR is of particular interest because it is a significant modulator of JCV activity (e.g.,(Ault, 1997; Feigenbaum et al., 1992; Jensen and Major, 2001; Johnson et al., 2013)). In JCV isolates from the urine of healthy individuals, the single promoter/enhancer region is highly conserved (e.g., (Agostini et al., 1996; Jensen and Major, 2001; Yogo et al., 1990)) and is termed the archetype. Upon immuno-suppression and reactivation of the virus, rearrangements within the hyper-variable region occur (reviewed in (Jensen and Major, 2001; Koralnik, 2013; White et al., 2009)) that correlate with viral spread and development of PML. As a result, patients with poor clinical outcomes have a high proportion of tandem repeats and insertions in the enhancer sequences (e.g. (Ault, 1997; Pfister et al., 2001; Vaz et al., 2000). It is thought that such changes in the hyper-variable enhancer affect levels of viral transcription and replication which in turn, govern JCV cellular tropism (e.g., (Ault, 1997; Feigenbaum et al., 1987)). However, the exact mechanism(s) by which the enhancer/promoter regions stimulate JCV replication has not been fully established. A depiction of the Mad-1 derived tandem duplication present in pFL-JCVori-TD, the pFL-JCVori derivative used in our initial studies, is presented in Fig. 1B (note, the enhancers are subdivided into sections a–f (Jensen and Major, 2001); reviewed in (Ferenczy et al., 2012)).
Identifying a suitable cellular host for characterization of JCV DNA replication
In initial studies, we selected several cell lines to test for JCV DNA replication. Given the tropism of JCV for glial cells, we tested the human glioma cell line Hs 683 and the human primary glioblastoma cell line U87. The Hs 683 and U87 cells were of particular interest because they have been used in a number of previous studies of JCV DNA replication (e.g., (Ault, 1997; Bollag et al., 2010; Chang et al., 1996; Chang et al., 1994; Gosert et al., 2010)). The cervical carcinoma cell line C33A was also included for historical reasons, as it was the cell line used to implement our previous SV40 and HPV31 DNA replication assays (Fradet-Turcotte et al., 2010).
Results from representative assays, designed to measure levels of JCV replication in different cell types and conducted with the tandem duplication containing pFL-JCVori-TD plasmid, are presented in Fig. 2A. Inspection of this figure establishes that reactions conducted with C33A cells had robust levels of replication. In contrast, the identical reagents did not support JCV DNA replication in Hs 683 cells (as negative controls, the reactions presented in columns 1 and 3 were performed in the absence of T-ag). Importantly, the failure to detect replication in Hs 683 cells was not due to problems related to transfection efficiencies as similar levels of Renilla luciferase were detected in C33A and Hs 683 cells (Fig. 2B. In other assays, the levels were equal (data not shown)). To compare the expression levels of JCV T-ag in C33A and Hs 683 cells, Western blots were performed. Inspection of Fig. 2C establishes that while JCV T-ag is readily detected in C33A cells, T-ag is present in Hs 683 cells only at very low levels. Selective proteo-somal degradation of T-ag in Hs 683 cells is unlikely since no changes in T-ag levels were detected upon treatment of cells with MG132 at concentrations as high as 0.32 mM (data not shown). Finally, in a similar series of experiments, we also failed to detect JCV DNA replication in U87 cells (data not shown).
Fig. 2.
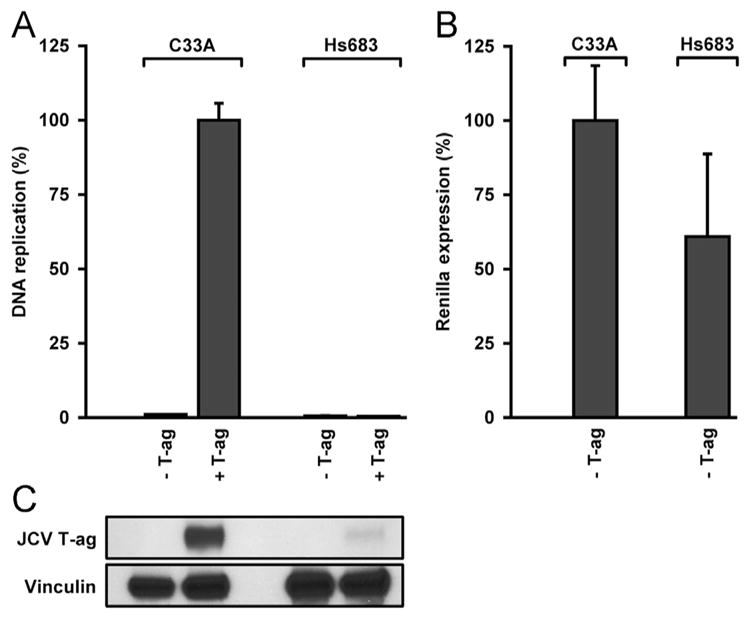
Examples of representative assays establishing that JCV DNA replication takes place in only certain cell lines. A. Results of JCV DNA replication assays conducted in the cervical carcinoma cell line C33A (left two columns) and the glioma cell line Hs 683 (right two columns). B. A plot of the Renilla luciferase levels in the “-T-ag” controls presented in Fig. 2A. C. A Western blot showing the relative levels of T-ag present in equal aliquots (10 μg protein) of whole cell extracts from C33A and Hs 683 cells. As a loading control, Vinculin levels were also determined.
JCV T-ag fails to accumulate in the nuclei of Hs 683 and U87 cells
The localization of T-ag to the nuclei of cells is essential for polyomavirus DNA replication (reviewed (An et al., 2012; Borowiec et al., 1990; Topalis et al., 2013)). Therefore, we investigated, via immunofluorescence microscopy ((Boichuk et al., 2010) and references therein), the localization of JCV T-ag in C33A cells, as well as in the glioma cell lines Hs 683 and U87. Inspection of Fig. 3A (rows 2–4, and the accompanying bar graphs) establishes that in replication competent C33A cells, JCV T-ag is generally found within the nucleus. This was true of nearly 75% of the T-ag positive cells examined. In contrast, in Hs 683 cells, JCV T-ag is largely excluded from the nuclei ((Fig. 3B, rows 2–4); only ~7% of Hs 683 cells had T-ag in their nuclei; most of the cells (~89%) had T-ag in the cytoplasm). Furthermore, the same distribution of T-ag was seen when these experiments were performed with vectors encoding additional viral factors (e.g., small t antigen (Bollag et al., 2010) and the entire Mad-1 genome (Dang et al., 2012): (data not shown)). Therefore, it is concluded that the failure to see JCV T-ag in the nuclei of glioma cells is not due to the absence of essential viral factors (e.g., the T′ proteins (Frisque, 2001; Prins and Frisque, 2001) or agnoprotein (Akan et al., 2006; Khalili et al., 2005; Okada et al., 2001; Safak et al., 2001; Saribas et al., 2012)).
Fig. 3.

Determination, via immunofluorescence, of the subcellular localization of JCV T-ag in C33A cells and two glial cell lines (Hs 683 and U87). A. Representative images of JCV T-ag (green) within C33A cells obtained using the previously described “standard” technique (Boichuk et al., 2010). The cell nuclei were stained with DAPI. The histograms present the percentage distribution of T-ag in the nuclei, both nuclei and cytoplasm or just cytoplasm of the two different cell types. B. Representative images of JCV T-ag within Hs 683 cells using the identical technique; the cell nuclei are indicated. (In both instances, the percentages were established after counting ~ 300 cells). C. Demonstration, using a modification of the immunofluorescence technique that involves pre-extraction of T-ag, of the “punctate distribution” of JCV T-ag in the nuclei of C33A, Hs 683 and U87 cells.
An additional set of immunofluorescence experiments was conducted using a method designed to “pre-extract” unbound T-ag from within the cell (Mirzoeva and Petrini, 2001). This method was employed in order to better visualize the distribution of JCV T-ag within the nuclei of these cells. Fig. 3C presents representative images that depict the distribution of T-ag in the nuclei of C33A, Hs 683 and U87 cells (images of the latter two cell types were derived from the minor population of cells with T-ag in the nuclei). Inspection of Fig. 3C (top line) provides additional evidence that in C33A cells, JCV T-ag is in the nuclei where it clusters in discrete foci. An analogous punctate distribution of T-ag in the nuclei of replication competent cells was seen in previous studies of SV40 DNA Zhao et al., 2008 and JCV replication ((Gasparovic et al., 2009; Orba et al., 2010; Shishido-Hara et al., 2008); reviewed in (Shishido-Hara, 2010)). Inspection of Fig. 3C (middle and bottom rows) establishes that a similar punctate distribution of JCV T-ag occurs in the Hs 683 and U87 cells. The significance of this distribution of JCV T-ag in the nuclei of cells will require additional studies. Nevertheless, the general absence of T-ag from the nuclei of glial derived cells lines, and relatively low levels of T-ag expression, are obvious reasons why these glioma cells fail to support JCV DNA replication. In contrast, it is apparent that C33A cells appear very promising for subsequent studies of JCV DNA replication. Therefore, we elected to further validate the replication assay in C33A cells.
Validation of the JCV DNA replication assay conducted in C33A cells
Time- and origin-dependence of the assay
The time- and origin-dependence of the assay were tested using a number of different NCCR configurations with similar results; for simplicity, only those obtained with the pFL-JCVori plasmid that contains the “archetype” single enhancer are presented in Figs. 4 and 5. Inspection of the left side of the graph presented in Fig. 4A establishes that in the presence of a plasmid containing the JCV origin of replication (Ori+), there is a time dependent increase in luciferase signal. Moreover, it is apparent from the right side of the figure that this increase in luciferase signal does not occur when the assay is conducted with a plasmid lacking the JCV origin (Ori−). Given the importance of T-ag to viral replication, a Western blot was used to monitor the levels of T-ag expression (Fig. 4B). It is apparent that T-ag is stably expressed in these cells and that the failure to detect replication of JCV DNA in the Ori− reactions is not due to aberrant T-ag levels.
Fig. 4.

A time course of JCV DNA replication conducted in C33A cells. A. The replication reactions on the left were conducted with the single enhancer containing pFL-JCVori plasmid; those on the right were conducted with pFL; a derivative of pFL-JCVori lacking the JCV ori. The times at which the assays were conducted are indicated, as are the Fluc/Rluc ratios for the indicated reactions. B. Results of a Western blot used to determine the amount of JCV T-ag present in C33A cells following the indicated amounts of time. As a loading control, Vinculin levels were also determined. C. Levels of JCV replication measured via quantitative real-time PCR (qPCR). Column 1 presents the results of a reaction conducted in the presence of pFL-JCVori, but in the absence of the plasmid encoding JCV T-ag (-T-ag). Column 2 presents the results of a reaction conducted in the presence of plasmids containing both the JCV origin and T-ag (+T-ag). The third column presents the results of a reaction conducted only in the presence of plasmid encoding T-ag (Ori−).
Fig. 5.

JCV DNA replication assays conducted with mutant forms of T-ag. A. A linear depiction of JCV large T-ag showing its major domains (i.e., J, OBD, ATPase/helicase and host range (HR)). The relative locations of the T-ag residues that were mutated to alanines (see section ‘Materials and methods’) are indicated on the top of the figure. B1. A surface representation of the JCV OBD (Meinke et al., 2014) showing the relative position of R155 and H204 (shown in red). B2. A surface representation of the JCV helicase domain, prepared using the program Phyre 2 (Kelley and Sternberg, 2009). This model shows the location of a residue at the base of the beta-hairpin (H514) and a residue that, based on studies of SV40 T-ag (Li et al., 2003), is likely involved in ATP binding (R499). C. JCV replication assays conducted in C33A cells for 72 h. with T-ag molecules containing mutations at residues that are predicted to be essential for DNA replication. The extreme left column presents the result of a replication reaction conducted in the absence of T-ag, while the amount of replication obtained with wt JCV T-ag is shown in the second column. The following four columns present the amount of replication obtained with the T125A, R155A, R499A and H514A mutants. D. Results from a Western blot demonstrating that levels of “mutant T-ag” are not different from those of wt JCV T-ag. As a loading control, levels of Vinculin were also determined.
Further validation of JCV DNA replication via qPCR
To test whether the increase in luciferase activity was the result of a higher copy number of DNA, we used qPCR to directly measure the amount of Fluc containing plasmid DNA (see section ‘Materials and methods’). The qPCR reactions in Fig. 4C, columns 1–3, all contained genomic DNA extracted from C33A cells following transfection with the indicated plasmids. Inspection of column 1 demonstrates that in the absence of the plasmid encoding JCV T-ag (i.e., pCMV-JCVT-ag), the primers specific for the firefly luciferase gene amplified very little DNA. In contrast, in the presence of JCV T-ag and the origin containing pFL-JCVori plasmid (column 2), there is a marked increase in the amount of luciferase encoding DNA that is amplified. Finally, it is apparent from the data presented in column 3, the product of a reaction conducted with pCMV-JCVT-ag and pFL (the plasmid that lacks the JCV origin) that replication is dependent on the JCV origin of replication.
Using mutant T-ag’s to further validate the assay and for initial structure function studies
Previous SV40 based studies identified a number of residues in T-ag that are essential for viral DNA replication. For example, they established that residues in the A1 and B2 loops are critical for DNA binding (e.g., (Luo et al., 1996; Simmons et al., 1990; Simmons et al., 1990)). They also revealed that residues in the helicase domain at the tip of the “beta hairpin” are needed for melting of the origin (Kumar et al., 2007; Reese et al., 2004; Shen et al., 2005) and for interactions with the sugar-phosphate backbone of ssDNA (Gai et al., 2004). Therefore, given the extensive homology between SV40 and JCV T-ag’s, mutations at the corresponding residues in JCV T-ag should, in principle, inactivate the C33A based JCV DNA replication assay.
The relative locations of the residues in the JCV T-ag OBD and helicase domains that were selected for mutation are indicated in Fig. 5A. Structural models of the JCV OBD (Meinke et al., 2014) and helicase domains (see section ‘Materials and methods’) are presented in Fig. 5-B.1 and B.2. Regarding mutations in the OBD (Fig. 5B.1), the locations of R155 and H204, situated in the A1 and B2 loops, respectively, are shown on the surface of the molecule. Within the JCV T-ag helicase domain (Fig. 5B.2), the location of a residue at the tip of the beta-hairpin (i.e., H514) is shown. Also presented is the location of a residue (i.e., R499) that was predicted, based on studies of SV40 T-ag (Gai et al., 2004; Li et al., 2003), to play a critical role in ATP binding. Finally, phosphorylation of JCV T-ag residue Thr 125 has been demonstrated to be essential for the replication of JC virus ((Swenson et al., 1996; Tyagarajan and Frisque, 2006). Unfortunately, a structure does not exist for this region of any polyomavirus T-ag).
Inspection of Fig. 5C establishes that JCV T-ag molecules, containing alanine substitutions at the predicted “essential” residues, were all unable to support JCV DNA replication (four columns on the right). Moreover, the failure of these mutant T-ag molecules to increase the levels of firefly luciferase activity provides further validation that this readout provides an accurate measure of ori-plasmid replication or, stated otherwise, that the signal obtained with wt T-ag (second column) reflects replication of the viral DNA and not other processes (e.g., transactivation of the luciferase gene). In addition, inspection of the Western blot presented in Fig. 5D establishes that the failure of the individual T-ag mutant proteins to support replication is not due to changes in their levels of expression. (It is noted that the H204A T-ag mutation was also constructed and its ability to support DNA replication analyzed; however, much lower protein levels were associated with this mutant (data not shown). Therefore, replication data obtained with this mutant T-ag are not presented).
Systematic characterization of the JCV regulatory sequences in terms of their contributions to viral DNA replication
Having validated the JCV DNA replication assay in C33A cells, it was of interest to conduct a systematic analysis of the origin sequences needed for this process. Initially, we determined the extent to which the enhancer sequences modulate the levels of DNA replication within C33A cells. The DNA constructs containing different derivatives of the “archetype” NCCR used in this study, are presented in Fig. 6A. The single Mad 1 derived 98 bp enhancer (Frisque, 1983) present in pFL-JCVori, the construct used in certain previous replication assays (i.e., Figs. 4,5), is presented on line 1. Variants of this construct, in which the enhancer sequences were deleted, include one in which all of sub-region e and part of c were deleted ((18 bp and 31 bp, respectively; this represents ~51% of the enhancer). The plasmid containing this truncation was termed pFL-JCVoriΔ1/2 enh; the NCCR is depicted on line 2. In a second variant, the entire enhancer was deleted; the plasmid containing this derivative was termed pFL-JCVoriΔenh and the NCCR is presented on line 3. Finally, the Mad 1 strain has a tandem duplication of the enhancer sequences, as well as the addition of the F sub-region (Frisque et al., 1984). Therefore, as previously noted, the Mad 1 sequences were engineered into an additional derivative of the pFL-JCVori plasmid that was termed pFL-JCVori-TD (a depiction of the NCCR in this plasmid is shown in Fig. 1B).
Fig. 6.
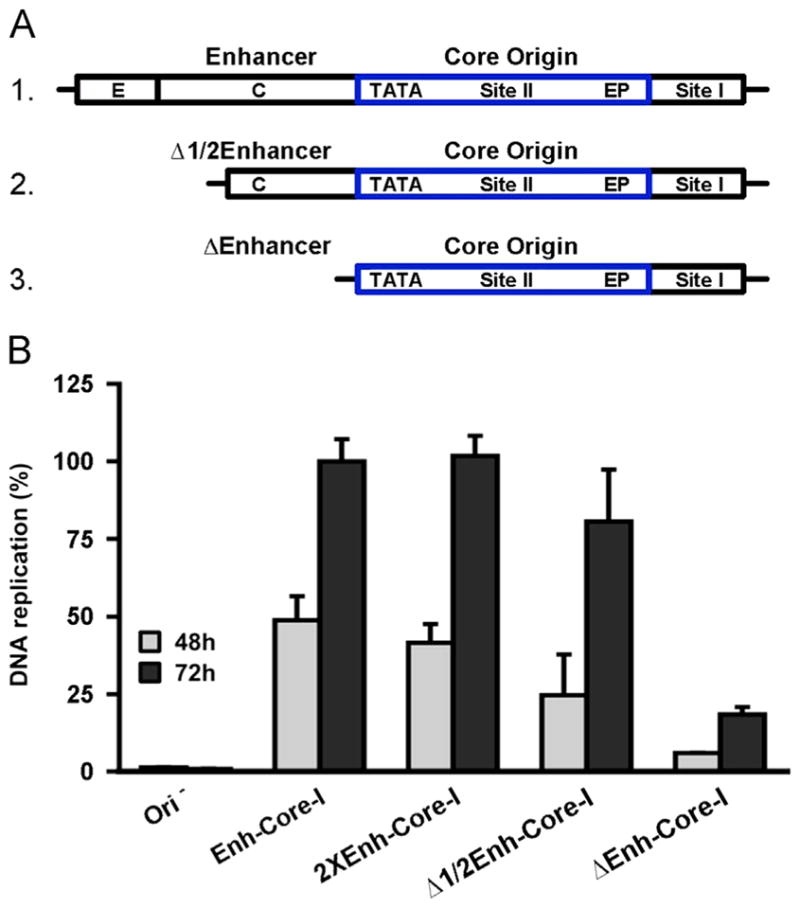
Determining the extent to which the enhancer/promoter regions stimulate JCV DNA replication. A. Alternative forms of the enhancer/promoter region analyzed in these studies. The monomeric enhancer present in plasmid pFL-JCVori is presented on the first line (abbreviated “enh-core-I”). The 98 bp enhancer is subdivided into subregions e, c and a (the TATA region includes subregion a). Removal of approximately half of the enhancer created the partial enhancer deletion (line 2; abbreviated “Δ1/2enh-core-I”). Removal of the entire enhancer created the enhancer deletion (line 3; abbreviated “Δenh-core-I”). B. The levels of JCV DNA replication supported by plasmids containing the different configurations of the NCCR depicted in Fig. 6A, and Fig. 1B, after 48 and 72 h. As a control, the first two columns depict the amount of DNA replication obtained with pFL, the construct lacking the JCV origin. The levels of replication obtained after 48 and 72 h, with the standard pFL-JCVori plasmid containing a single enhancer (Fig. 6A, line 1), and the duplicated enhancer found in the Mad-1 strain (Fig. 1B) are shown in the following four columns. Also shown are the relative levels of replication obtained with truncated forms of the single enhancer (i.e., the “Δ1/2 enh” (Fig. 6A; line 2) and the “Δenh” configurations (Fig. 6A; line 3)).
Levels of replication obtained by different promoter/enhancer elements
The data presented in Fig. 6B compare the relative levels of JCV DNA replication supported by plasmids containing different enhancer elements (i.e., those presented in Fig. 6A and Fig. 1B. Note, in Fig. 6B plasmids pFL-JCVori, pFL-JCVoriΔ1/2 enh, pFL-JCVoriΔenh and pFL-JCVori-TD are abbreviated Enh-core-I, Δ1/2 Enh-core-I, ΔEnh-core-I and 2XEnh-core-I, respectively. Furthermore, the pFL plasmid, lacking the JCV ori, was abbreviated as (Δ ori)). As expected, no replication was detected with the ori minus control plasmid (Δ ori) at either 48 or 72 h post transfection (left two columns). Of interest, the presence of the second enhancer did not increase JCV DNA replication (compare the levels of replication supported by the single enhancer (enh-core-I) versus the two enhancer (2X-enh-core-I) constructs). Therefore, the previously reported tandem duplication dependent stimulation of JCV transcription/replication in glial cells (e.g., (Jensen and Major, 2001; Pfister et al., 2001; Vaz et al., 2000); reviewed in (Gheuens et al., 2012)) is not a feature of JCV DNA replication in C33A cells. The extent to which sequences within the single 98-bp enhancer (i.e., the archetype) contribute to JCV replication was also addressed. The results of these studies reveal that relative to the single enhancer containing plasmid (Fig. 6A; line 2), plasmid Δ1/2 enh (Fig. 6A; line 3) had nearly an equivalent amount of replication activity (compare columns labeled enh-core-I with those labeled Δ1/2 enh-core-I). However, replication was greatly reduced by the complete removal of the remaining enhancer sequences (last pair of columns labeled Δenh-core-I).
Evidence that NF-1 family members are needed for JCV DNA replication in C33A cells
The experiments presented in Fig. 6B established that the level of JCV DNA replication obtained with the partial enhancer (Δ1/2 enh-core-I) was nearly the same as with the entire enhancer. In contrast, very little replication was detected upon the complete removal of the enhancer (Δenh-core-I; last pair of histograms). This is an interesting observation given that the NF-1A binding site (Amemiya et al., 1989) in sub-region c (reviewed in (Ferenczy et al., 2012)) is intact in the partial, but not in the full, deletion. Given that NF-1 family members have been previously implicated in JCV replication (Amemiya et al., 1992; Monaco et al., 2001; Ravichandran and Major, 2008; Sock et al., 1991), it was of interest to test whether the NF-1A binding site is required for JCV replication in the C33A based system. Therefore, the NF-1A binding site (Amemiya et al., 1989) in the NCCR derivatives presented in Fig. 7A were mutated (see section ‘Materials and methods’) and the plasmids containing these mutations were used in additional JCV replication assays. Inspection of Fig. 7B establishes that relative to the parental sequences in which the NF-1A site was intact, derivatives containing the mutation at the NF-1A binding site had reduced levels of JCV DNA replication (compare shaded columns to striped columns). As previously noted, the luciferase based JCV replication assay expresses T-ag from a separate plasmid. Therefore, the results presented in Fig. 7B indicated that NF-1 family members directly promote JCV DNA replication in C33A cells, rather than playing an indirect role via up-regulating the expression of T-ag.
Fig. 7.

Determining the levels of JCV DNA replication supported by plasmids containing mutations within the NF-1A binding sites. A. The locations (in red) of the NF-1A binding site within the archetype (line 1) and “Δ1/2enhancer” (line 2) enhancer sequences (Frisque et al., 1984). B. The relative levels of JCV DNA replication obtained at 48 and 72 h with the parental pFL-JCVori and pFL-JCVori (Δ1/2enhancer) plasmids and the derivatives containing mutations in the NF-1A binding sites (Fig. 7A).
Is Site I required for JCV replication in C33A cells?
The Site I region of the SV40 origin is required for viral DNA replication in vivo ((Guo et al., 1991) and references therein). Likewise, it has been reported that the Site I region of the JCV origin stimulates replication in the U138 glial cell line (Sock et al., 1993). To determine if Site I stimulates JCV DNA replication in the C33A based luciferase assay, additional derivatives of the NCCR in plasmid pFL-JCVori were made (Fig. 8A). In one, the sequences encompassing the “late side” enhancer were kept intact and only Site I deleted (line 1; the NCCR in this derivative is abbreviated “enh-core-ΔI”). In the second construct, both Site I and the enhancer sequences are deleted; as a result, this pFL-JCVori derivative contained just the JCV core origin (line 2; the NCCR in this construct was abbreviated “Δenh-core-ΔI”).
Fig. 8.
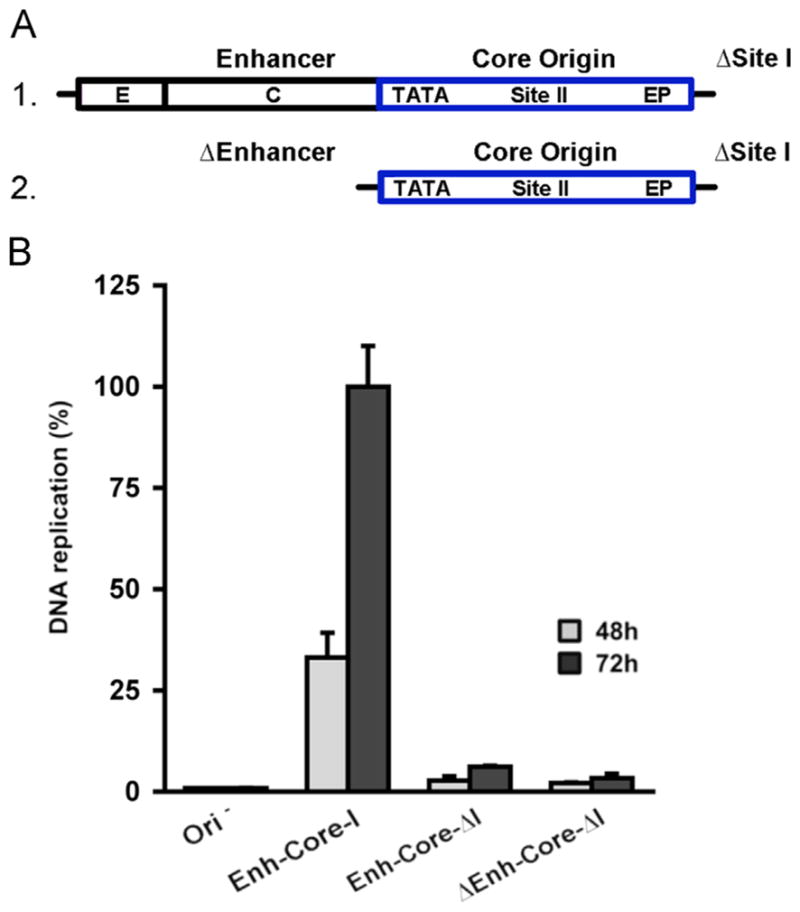
Measuring the levels of JCV DNA replication obtained with plasmids having deletions of Site I. A. Depictions of the NCCR regions in the wild-type “enh-core-I” derivative containing a deletion in Site I (termed “enh-core-ΔI”; Fig. 8A; line1) and in a second plasmid in which both the enhancer and Site I were deleted (the core origin derivative that was termed “Δenh-core-ΔI”; Fig. 8A; line 2). B. The relative levels of JCV DNA replication obtained at 48 and 72 h with the plasmids containing deletions in Site I and a wild type control (i.e., Enh-Core-I).
The plasmids containing these deletions are then used in additional JCV DNA replication assays (Fig. 8B). Inspection of this figure reveals that both plasmids containing Site I deletions were significantly reduced in terms of their ability to support JCV DNA replication. (Compare the data obtained with the positive control (i.e., pFL-JCVori, abbreviated as enh-core-I) to the data obtained with plasmids containing deletions in Site I (i.e., enh-core-ΔI and Δenh-core-ΔI)). Control reactions, conducted with plasmids lacking the JCV origin, are presented in the left two columns). Collectively, these assays establish that Site I is required for the replication of JCV DNA in C33A cells.
Establishing the pentanucleotide requirements for JCV DNA replication
All four pentanucleotides in the SV40 core origin are needed for DNA replication (Dean et al., 1987; Joo et al., 1998). In contrast, it has been reported that only three pentanucleotides in the analogous “core origin” region are needed for the replication of murine polyomavirus (Harrison et al., 2013) and Merkel cell polyomavirus (Harrison et al., 2011). Thus, the literature contains differences in terms of the pentanucleotide requirements for polyomavirus DNA replication. Therefore, we elected to determine if all four penta-nucleotides in the JCV core origin are required for the replication of JCV DNA in C33A cells.
A diagram of the sequences comprising the JCV core origin of replication is presented in Fig. 9A. As illustrated by the two pairs of sequences above and below the central sequence, four derivatives of the pFL-JCVori plasmid were constructed in which transition mutations replaced the individual pentanucleotides in the core origin. These plasmids were then used in an additional series of JCV replication assays (Fig. 9B). As a negative control, replication reactions were conducted with plasmid pFL (Ori−; containing the fluc gene but lacking the JCV origin (column 1)). As a positive control, a reaction was conducted with the pFL-JCVori plasmid (column 2). As expected, there was no detectable signal with the negative control, but clear evidence for replication with plasmid pFL-JCVori (compare columns 1 and 2). The results obtained in reactions conducted with plasmids containing transition mutations in P1, P2, P3 and P4 are shown in the following four columns. It is apparent that none of the mutated origins supported replication.
Fig. 9.

Determining whether all four pentanucleotides in the Site II region of the core origin are required for JCV replication. A. Sequences of the JCV core origin, including the four pentanucleotides in the Site II regions. The transition mutations that are substituted for the individual pentanucleotides (P1-P4) are shaded in gray. B. Results of JCV replication assays conducted with the pFL-JCVori derivatives containing transition mutations of the individual pentanucleotides. As a negative control, a reaction was conducted with a plasmid lacking an origin (i.e., pFL; far left column). As a positive control, an additional reaction was conducted with plasmid pFL-JCVori (second column from the left). The levels of replication obtained with the plasmids containing the transition mutations in the individual pentanucleotides are shown in the following four columns.
The JCV replication assay can be used to screen for inhibitors of DNA replication
The studies presented above provided evidence that the luciferase-based assay faithfully recapitulates many of the known features of JCV DNA replication. This, coupled with the high-throughput and quantitative nature of the dual-luciferase readout, makes this assay an attractive one for the screening of small molecule inhibitors of JCV DNA replication and/or of human DNA replication. To assess the potential of this assay for compound screening, we first determined that the C33A cells could tolerate up to 0.5% DMSO (data not shown) and then tested the effect of three well-characterized DNA replication inhibitors: aphidicolin, hydroxyurea and gemcitabine. The results presented in Fig. 10 demonstrate that the luciferase signal could be reduced, in a dose dependent manner, by all three compounds. Furthermore, their measured EC50 values in the JCV assay are in good agreement with those obtained in our related SV40 assay (data not shown). An advantage of the dual luciferase readout is that the Renilla luciferase internal control can be used to identify compounds that are toxic or inhibit viral DNA replication by an undesired mechanism. In the case of aphidicolin, hydroxyurea and gemcitabine, neither one of them decreased the Renilla luciferase levels by more than 20% at the highest concentration tested (10 μM, 10 mM and 2 μM, respectively), thus attesting to their specificity. Collectively, these results highlight the potential of this assay for future screening of small-molecule libraries for inhibitors of JCV DNA replication. Finally, since at high concentrations the replication inhibitors greatly reduce levels of JCV DNA replication (Fig. 10), these studies further establish that the luciferase signal is largely dependent on replication of the ori-containing plasmid.
Fig. 10.

Evidence that the C33A based JCV DNA replication assay can be used to test inhibitors of JCV DNA replication. JCV DNA replication assays were conducted with the indicated amounts of the replication inhibitors aphidicolin, hydroxyurea and gemcitabine. The EC50 values are indicated.
Discussion
Using the cervical carcinoma line C33A, we have developed a quantitative and versatile luciferase based HTS assay for studying JCV DNA replication. The two virally encoded factors that are essential for DNA replication in this cell line are T-ag and the origin containing NCCR region. Previous studies, including those employing a cell free replication assay (Nesper et al., 1997), also led to the conclusion that JCV large T-ag is the only viral protein required to mediate the replication of an origin containing plasmid.
The reason(s) why C33A cells are so efficient at supporting JCV DNA replication has yet to be determined. Perhaps relevant to this issue, it was previously established that the accumulation of SV40 T-ag in nuclear foci is essential for viral replication (Tang et al., 2000; Zhao et al., 2008). Therefore, one reason why C33A cells may be efficient at supporting JCV DNA replication is that JCV T-ag accumulates in analogous nuclear foci. This finding may indicate that in C33A cells, ND10 domains are involved in JCV replication; a possibility supported by previous studies ((Gasparovic et al., 2009; Shishido-Hara et al., 2008); reviewed in (Shishido-Hara, 2010)). However, it should be noted that C33A cells do not contain papillomavirus DNA (Yee et al., 1985); therefore, papillomavirus encoded proteins are not involved in establishing the favorable conditions for JCV DNA replication.
Prior studies have also identified a number of proteins that bind to the JCV enhancer and promoter regions (reviewed in (Bellizzi et al., 2013; Ferenczy et al., 2012)). Therefore, a related issue is whether C33A cells are enriched in particular cellular proteins that bind to sequences within the NCCR and stimulate JCV replication. Regarding specific protein candidates, several reports indicate that the binding of NF-1 family members to the NF-1 binding sites stimulates viral replication (Amemiya et al., 1989; Marshall et al., 2010; Monaco et al., 2001; Ravichandran et al., 2006; Sock et al., 1993). Furthermore, it has been suggested that a particular NF-1 family member (i.e., NF1-X) is a molecular determinant of JCV susceptibility that functions throughout viral pathogenesis (Messam et al., 2003). Indeed, it has been reported that overexpression of NF1-X is sufficient to cause nonproductive cells to become permissive for JCV replication (Messam et al., 2003). Therefore, it is noteworthy that mutation of the NF-1A binding site reduces JCV DNA replication in C33A cells. It is stressed that since T-ag expression is dependent on regulatory sequences on a second plasmid, the stimulation observed by the presence of the NF-1 binding site has to reflect direct promotion of JCV DNA replication. The binding of NF-1 family members to NF-1 binding sites is also known to play a role in BKV DNA replication, but only in the presence of competitor templates (Liang et al., 2011). Thus, the C33A based luciferase assay, in which direct stimulation is observed, will be useful for subsequent studies designed to establish how JCV DNA replication is promoted by the binding of NF-1 family members. This assay might also play an important role in the identification of additional enhancer binding cellular factors needed for JCV DNA replication.
Our studies also establish that the Site I region of the JCV origin is essential for DNA replication in C33A cells, a finding consistent with previous observations (Sock et al., 1993). The Site I region of the SV40 origin is also necessary for DNA replication in vivo ((Guo et al., 1991) and references therein), but not in vitro (Bullock et al., 1997). These observations suggest that cellular factors, such as histones or other chromatin components may play important roles in the Site I dependent regulation of DNA replication. However, the precise role played by Site I in polyomavirus DNA replication has yet to be established.
It has been recently reported that the replication of certain polyomaviruses is dependent upon only a subset of the pentanucleotides found in Site II (Harrison et al., 2011, 2013). Therefore, the C33A based replication system was also used to investigate the pentanucleotide requirements for JCV DNA replication. The results presented herein establish, however, that all four pentanucleotides in the JCV core origin are needed for replication. Likewise, it was previously concluded that all four pentanucleotides in the Site II region of the SV40 core origin are needed for DNA replication (Dean et al., 1987; Joo et al., 1998). Collectively, these results suggest that within the polyomavirus family, there are significant differences in the pentanucleotide requirements for replication. Whether these findings reflect a fundamental difference in the mechanism(s) utilized during the initiation of viral DNA replication will require additional studies.
Replication of JC virus is, in general, limited to human cells of macroglial origin within the CNS (Ferenczy et al., 2012). The limited host range is largely a function of the NCCR region, which as previously discussed, is regulated by diverse transcription and replication factors (reviewed in (Bellizzi et al., 2013; Ferenczy et al., 2013)). Therefore, it is of interest that the reagents that support DNA replication in C33A cells do not support JCV DNA replication in established glioma cell lines (e.g., Hs 683 and U87). The reason(s) why these glioma cell lines fail to support JCV DNA replication is not understood. However, our Western blot experiments demonstrate that relative to C33A cells, JCV T-ag accumulates at relatively low levels in Hs 683 and U87 cells. Moreover, our immunofluorescence studies have established that JCV T-ag accumulates in only a small percentage of the glial cell nuclei. These limitations in terms of the amounts and cellular distribution of JCV T-ag may reflect that there are cellular based restrictions (e.g. Windl and Dorries (1995)) on JCV replication in the glioma cells used herein. The identity of the missing cellular factor(s) is unclear; however, they may include regulatory proteins such as NF2 (Beltrami et al., 2013), p53 (Staib et al., 1996) or cyclins A and B (Radhakrishnan et al., 2003). In addition, low levels of JCV T-ag in glial cells may reflect autophagy (Sariyer et al., 2012), interactions with the cellular degradation complex SCFβTrCP1/2 (Reviriego-Mendoza and Frisque, 2011) or metabolic changes, such as glucose deprivation (Noch et al., 2012).
Curiously, while JCV T-ag was absent from the nuclei of approximately 90% of the Hs 683 or U87 cells, it was present in ~7% of the cells. Why this distribution exists within the glioma cell population is also unknown. Detecting JCV DNA replication in Hs 683 and U87 cells will likely depend upon increasing the percentage of glioma cells that have JCV T-ag in their nuclei (Broekema and Imperiale, 2012). Nevertheless, since JCV T-ag is known to accumulate in the nuclei of glial cells derived from PML patients (Stoner et al., 1986), it is concluded that Hs 683 and U87 cells are not ideal models for additional studies of this disease.
That the C33A based JCV DNA replication assay will be used for many additional studies is suggested by its cost-effectiveness, speed at which replication experiments can be performed and the high-throughput format of the reactions. Indeed, our studies with known DNA replication inhibitors establish that it is an excellent assay for subsequent screening for additional inhibitors of JCV DNA replication. The C33A based JCV DNA replication assay will also be useful for further refining our understanding of the protein/DNA interactions that regulate JCV DNA replication. It will also serve as a very valuable control during subsequent studies designed to detect JCV DNA replication in glial cells. Finally, given that JCV DNA is efficiently replicated in C33A cells, it is likely that C33A cells will also be very useful for studies of the propagation of JC Virus.
Materials and methods
Plasmids
The construction of a plasmid encoding full-length JCV T-ag, that was termed pCMV-JCVT-ag, was previously described (Meinke et al., 2014). A plasmid containing the JCV origin of replication was obtained from Dr. H.P. Nasheuer. The origin regions present in this construct include Site II, Site I and one copy of the 98 bp enhancer sequence present in the Mad-1 strain of JCV (Frisque, 1983; Frisque et al., 1984). This origin containing fragment was obtained by PCR and then subcloned, via Gibson Assembly (New England Biolabs), into plasmid pFL (Fradet-Turcotte et al., 2010) at the Dra III site; the resulting plasmid was termed pFL-JCVori. Subsequent deletions of the 98 bp enhancer and Site I regions were generated using the QuikChange II kit (Agilent Technologies). Additional Gibson Assembly reactions were used to insert a second copy of the enhancer into pFL-JCVori, this re-created the tandem duplication found in the Mad-1 strain (Frisque et al., 1984). The resulting tandem duplication containing plasmid was termed pFL-JCVori-TD.
Point mutations at selected residues were introduced into pCMV-JCVT-ag using the QuikChange II kit (Agilent Technologies). The oligonucleotides used in these procedures are as follows (the complements to the individual oligonucleotides are not listed): for T125A (5′–GGATCCCAACACTCTGCCCCACCTAAGAAG-3′), for R155A (5′–GTCAAGCTGTGTTTAGTAATGCAACTGTTGCTTCTTTTGCTG-3′), for H204A (5′–GGGGGTCATAATATTTTGTTTTTCTTAACACCACATA-GAGCTAGAGTTTCAGCAATTAATAATTACTGTCAAAAACTATG-3′), for R499A (5′ GCAACCTTGATTGCCTAGCAGATTACTTAGATGGAAG-3′) and for H514A (5′ GTGTGAAAGTTAATTTAGAGAGAAAAGCCCAAAA-CAAAAGAACACAGGTGTTTC-3′). The bases that were inserted in order to introduce the indicated alanines are underlined. Additional QuikChange reactions were used to generate the mutation of the NF-1A binding sites within the enhancer. The oligonucleotide used to mutate the NF-1A binding site (Amemiya et al., 1992) was 5′–CCCTAGGTATGAGCTCATGCTCAGCTGGCAGTAATCCCTTCCC-3 (the complement is not listed); the mutated bases in the NF1 binding site are underlined (Ravichandran et al., 2006). Finally, DNA sequencing at the Tufts University Core Facility was used to verify the DNA sequences present in all plasmid constructs.
Cell culture and transfections
The C33A cervical carcinoma cell line was grown in EMEM medium (ATCC) supplemented with 10% FBS (Atlanta Biologicals) in 5% CO2. The Hs 683 cells (ATCC) were grown in DMEM supplemented with 15% FBS. The U87 cells were grown in EMEM supplemented with 10% FBS. Transfections of cells were conducted in bulk using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s recommendation.
JCV replication assays
As with our previously described SV40 and HPV31 assays (Fradet-Turcotte et al., 2010), the JCV replication assay requires several plasmids. One contains the firefly luciferase gene, either in the presence of the JCV origin (e.g., pFL-JCVori), or in its absence (pFL). The reactions also require a plasmid encoding JCV T-ag (e.g., pCMV-JCVT-ag or, alternatively, mutant forms of pCMV-JCVT-ag). Moreover, as a transfection control, all reactions contained 0.5 ng of the Renilla luciferase (Rluc) containing plasmid pRL. Finally, the total amount of plasmid transfected was adjusted to 100 ng with plasmid pCI ((Fradet-Turcotte et al., 2010); used as an empty vector control). Following transfection for 4-h, with the requisite plasmids in the absence of serum, FBS was added (10% for C33A and U87 cells, 15% for Hs 683 cells). The transfected C33A cells were then plated in white flat-bottom 96-well plates (costar #3917) at a density of 25,000 cells/well. Firefly and Renilla luciferase activities were measured at the indicated times using a Dual-Glo Luciferase assay system (Promega).
Western blots
JCV large T-ag was detected using the Pab 416 antibody (Santa Cruz Biotechnology). As a loading control, vinculin levels were determined using the HVIN-1 monoclonal Ab (Sigma). To conduct the actual Western blots, proteins were transferred to a PVDF membrane (Millipore). After addition of the appropriate primary antibody, the protein bands were detected using a horseradish peroxidase-conjugated rat anti-mouse secondary antibody (BD Pharmingen) and enhanced chemiluminescence detection kit (Millipore).
qPCR
Six well plates containing C33A cells (~3 × 105) were trans-fected with 50 ng of pFL-JCVori and 400 ng of pCMV-JCVT-ag. To standardize the reactions, the Renilla encoding plasmid was added (10 ng) and the total amount of plasmid DNA was adjusted to 2 μg with pCI. Total genomic DNA was extracted 48 h. post-transfection using the DNeasy Blood and Tissue Kit from Qiagen. To measure the amount of replicated pFL-JCVori plasmid, an aliquot of the genomic DNA (25 μl) was digested with 10 units of Dpn I for 16 h, followed by digestion with exo III (100 units) for 30 min.. The primers used to amplify an 80 bp region of the firefly luciferase gene were 5′–TCCTTCGATAGGGACAAGACAATT-3′ and 5′–GGCAGAGCGACACCTTTAGG-3′. The site-specific probe used in these assays, derived from the luciferase gene, was 5′-/56-FAM/CACTGATCA/ZEN/TGAACTCCTCTGGATCTACTGGTC/3IABkFQ/-3′. All samples were run in triplicate using the TaqMan PerfeCTa qPCR ToughMix UNG ROX (Quanta).
Immunofluorescence
The “standard” immunofluorescence microscopy assays were conducted as previously described (Boichuk et al., 2010). After seeding, cells (~2 × 105) were allowed to attach to poly-L-Lysine-coated glass coverslips for approximately 24 h.. Using the Lipofectamine 2000 reagent, the cells were then transfected with the desired plasmids (2 μg total of DNA). After the addition of serum, the cells were incubated at 37 °C for an additional 48 h. Cells were then washed with phosphate-buffered saline (PBS) and then fixed in 4% paraformaldehyde in PBS for 15 min. The cells were then washed twice with PBS and permeabilized via the addition of 0.2% Triton X-100, dissolved in PBS, for 5 min.. The cells were washed again three times with PBS. To block nonspecific binding the cells were then incubated with 10% goat serum (Invitrogen) for 20 min. The permeabilized cells were then treated with a 1/1000-fold dilution of the Pab 416 anti T-ag monoclonal Ab (Santa Cruz Biotechnology) for 1 h, followed by three washes with PBS. The secondary goat anti-mouse antibody, conjugated with Alexa 488 (Life technologies) was diluted 1/1000 and applied for 1 h.. After three additional washes with PBS, the cells were stained briefly with DAPI (4′, 6′-diamidino-2-phenylindole) and covered with cover slips coated with “mounting medium” (Electron Microscopy Sciences). The cells were then visualized using a Zeiss Axiovert 200 M microscope employing the 63X objective. The OpenLab software package (Perkin-Elmer) was used to acquire and process images. A variation of this protocol involved “pre-extraction“ of unbound T-ag (Mirzoeva and Petrini, 2001). Cells were initially pre-extracted with CSK buffer (10 mM PIPES, pH 7, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA and 0.5% Triton X-100); the remaining steps are identical to the “Standard method”.
Molecular modeling
The molecular graphics images were generated using the program PyMOL (DeLano, 2002). The structures used in the molecular modeling discussed herein are the apo JCV T-ag OBD (RCSB PDB code 4LMD) and the JCV helicase domain threaded onto the SV40 T-ag helicase domain (PDB code 1SVM) using the program Phyre2 (Kelley and Sternberg, 2009).
Inhibitor testing
Approximately 25,000 C33A cells were seeded in a 96-well plate and co-transfected the day after with 20 ng of pCMV-JCVT-ag, 2.5 ng pFL-JCVori, 0.5 ng of pRL and 77 ng of carrier DNA (pCI). The culture medium was replaced 4 h post-transfection with fresh medium containing the inhibitor or with vehicle alone (0.1% DMSO) as a control. Cells were then incubated for 24 h and the culture medium replaced with fresh medium lacking inhibitor prior to measuring the levels of firefly and Renilla luciferase using the Dual-Glo Luciferase assay system (Promega). Inhibitors were tested in duplicate 12-point dose response curves, in two independent experiments. Dose response curves were generated by two-fold serial dilution of each compound starting at a maximal concentration of 10 μM for aphidicolin, 10 mM for hydroxyurea and 2 μM for gemcitabine. EC50 values were calculated with the GraphPad Prism 6 software by fitting the data to the following equation: % DNA replication=Low+(100–Low)/(1+10^((LogEC50 –[I]) × n)), in which Low represents the lowest percentage of DNA replication reached at saturating amounts of inhibitor, [I] is the concentration of the inhibitor (M), and n is the Hill coefficient.
Acknowledgments
We thank Alexei Degterev for U87 cells, Karl Munger for the C33A cells and H.P. Nasheuer for the original JCV plasmids. We also acknowledge Ralph Isberg for use of his Zeiss Axiovert microscope and Gretchen Meinke and Andrew Bohm for helpful discussions. This work was supported by a grant from the Canadian Institutes for Health Research (CIHR) to JA (MOP-126103) and institutional funding from Tufts University to P.A.B.
References
- Agostini HT, Ryschkewitsch CF, Stoner GL. Genotype profile of human polyomavirus JC excreted in urine of immunocompetent individuals. J Clin Microbiol. 1996;34:159–164. doi: 10.1128/jcm.34.1.159-164.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology. 2006;349:66–78. doi: 10.1016/j.virol.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Amemiya K, Traub RG, Durham L, Major EO. Interaction of a nuclear factor-1-like protein with the regulatory region of the human polyomavirus JC virus. J Biol Chem. 1989;264:7025–7032. [PubMed] [Google Scholar]
- Amemiya K, Traub RG, Durham L, Major EO. Adjacent nuclear factor-1 and activator protein binding sites in the enhancer of the neurotropic JC virus. The J Biol Chem. 1992;267:14204–14211. [PubMed] [Google Scholar]
- An P, Saenz Robles MT, Pipas JM. Large T antigens of polyomaviruses: amazing molecular machines. Annu Rev Microbiol. 2012;66:213–236. doi: 10.1146/annurev-micro-092611-150154. [DOI] [PubMed] [Google Scholar]
- Ault GS. Activity of JC virus archetype and PML-type regulatory regions in glial cells. J Gen Virol. 1997;78:163–169. doi: 10.1099/0022-1317-78-1-163. [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Bellizzi A, Anzivino E, Rodio DM, Palamara AT, Nencioni L, Pietropaolo V. New insights on human polyomavirus JC and pathogenesis of progressive multifocal leukoencephalopathy. Clin Dev Immunol. 2013;2013 doi: 10.1155/2013/839719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrami S, Brancheti E, Sariyer IK, Otte J, Weaver M, Gordon J. Neurobibromatosis type 2 tumor suppressor protein, NF2, induces proteasome-mediated degradation of JC virus T-antigen in human glioblastoma. PLOS One. 2013;8:e53447. doi: 10.1371/journal.pone.0053447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva E, Martynowski D, Seitova A, Bochkarev A. Structure of the origin-binding domain of simian virus 40 large T antigen bound to DNA. EMBO J. 2006;25:5961–5969. doi: 10.1038/sj.emboj.7601452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boichuk S, Hu L, Hein J, Gjoerup OV. Multiple DNA damage signaling and repair pathways deregulaed by simian virus 40 large T antigen. J Virol. 2010;84:8007–8020. doi: 10.1128/JVI.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag B, Hofstetter CA, Reviriego-Mendoza MM, Frisque RJ. JC virus small t antigne binds phosphatase PP2A and Rb family proteins and is required for efficient viral DNA replication activity. PLoS One. 2010;5:e10606. doi: 10.1371/journal.pone.0010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiec JA, Dean FB, Bullock PA, Hurwitz J. Binding and unwinding -how T antigen engages the SV40 origin of DNA replication. Cell. 1990;60:181–184. doi: 10.1016/0092-8674(90)90730-3. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Davies NWS, Cinque P, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol. 2010;6:667–679. doi: 10.1038/nrneurol.2010.164. [DOI] [PubMed] [Google Scholar]
- Broekema NM, Imperiale MJ. Efficient propagation of archetype BK and JC polyomaviruses. Virology. 2012;422:235–241. doi: 10.1016/j.virol.2011.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock PA. The initiation of Simian virus 40 DNA repliation in vitro. Crit Rev Biochem Mol Biol. 1997;32:503–568. doi: 10.3109/10409239709082001. [DOI] [PubMed] [Google Scholar]
- Bullock PA, Joo WS, Sreekumar KR, Mello C. Initiation of SV40 DNA replication in vitro: analysis of the role played by sequences flanking the core origin on initial synthesis events. Virology. 1997;227:460–473. doi: 10.1006/viro.1996.8347. [DOI] [PubMed] [Google Scholar]
- Chang CF, Tada H, Kahalili K. The role of a pentanucleotide repeat sequence, AGGGAAGGA, in the regulation of JC virus DNA replication. Gene. 1994;148:309–314. doi: 10.1016/0378-1119(94)90704-8. [DOI] [PubMed] [Google Scholar]
- Chang CF, Gallia GL, Muralidharan V, Chen NN, Zoltick P, Johnson EM, Khalili K. Evidence that replication of human neurotropic JC virus DNA in glial cells is regulated by the sequence-specific single-stranded DNA-binding protein Pur alpha. J Virol. 1996;70:4150–4156. doi: 10.1128/jvi.70.6.4150-4156.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YP, Xu M, Machado ACD, Yu XJ, Rohs R, Chen XS. Mechanism of origin DNA recognition and assembly of an initiator-helicase complex by SV40 large tumor antigen. Cell. 2013;3:1–11. doi: 10.1016/j.celrep.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang X, Wuthrich C, Gordon J, Sawa H, Koralnik IJ. JC virus encephalopathy is associated with a novel agnoprotein-deletion JCV variant. PLoS One. 2012;7:e35793. doi: 10.1371/journal.pone.0035793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. Delano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- Dean FB, Borowiec JA, Ishimi Y, Deb S, Tegtmeyer P, Hurwitz J. Simian virus 40 large tumor antigen requires three core replication origin domains for DNA unwinding and replication in vitro. Proc Natl Acad Sci USA. 1987;84:8267–8271. doi: 10.1073/pnas.84.23.8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPasquier RA, Corey S, Margolin DH, Williams K, Pfister LA, De Girolami U, MacKey JJ, Wurtich C, Joseph JT, Koralnik IJ. Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology. 2003;61:775–782. doi: 10.1212/01.wnl.0000081306.86961.33. [DOI] [PubMed] [Google Scholar]
- Ehlers B, Wieland U. The novel human polyomaviruses HPyV6, 7, 9 and beyond. APMIS. 2013 doi: 10.1111/apm.12104. doi:10.1111:1-13. [DOI] [PubMed] [Google Scholar]
- Fanning E, Knippers R. Structure and function of simian virus 40 large tumor antigen. Annu Rev Biochem. 1992;61:55–85. doi: 10.1146/annurev.bi.61.070192.000415. [DOI] [PubMed] [Google Scholar]
- Fanning E, Zhao K. SV40 DNA replication: from the A gene to a nanomachine. Virology. 2009;384:352–359. doi: 10.1016/j.virol.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenbaum L, Khalili K, Major E, Khoury G. Regulation of the host range of human papovavirus JCV. Proc Natl Acad Sci USA. 1987;84:3695–3698. doi: 10.1073/pnas.84.11.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenbaum L, Hinrichs SH, Jay G. JC virus and simian virus 40 enhancers and transforming proteins: role in determining tissue specificity and pathogenicity in transgenic mice. J Virol. 1992;66:1176–1182. doi: 10.1128/jvi.66.2.1176-1182.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, Marshall LJ, Nelson CDS, Atwood WJ, Nath A, Khalili K, Major EO. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced deymelinating disease of the human brain. Clin Microbiol Rev. 2012;25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenczy MW, Johnson KR, Marshall LJ, Monaco MC, Major EO. Differentiation of human fetal multipotential neural progenitor cells to astrocytes reveals susceptibility factors for JC virus. J Virol. 2013;87:6221–6231. doi: 10.1128/JVI.00396-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology. 2010;399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ. Nucleotide sequence of the region encompassing the JC virus origin of DNA replication. J Virol. 1983;46:170–176. doi: 10.1128/jvi.46.1.170-176.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisque RJ. Structure and function of JC virus T′ proteins. J NeuroVirol. 2001;7:293–297. doi: 10.1080/13550280152537120. [DOI] [PubMed] [Google Scholar]
- Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51:458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai D, Li D, Finkielstein CV, Ott RD, Taneja P, Fanning E, Chen XS. Insights into the oligomeric states, conformational changes and helicase activities of SV40 large tumor antigen. J Biol Chem. 2004;279:38952–38959. doi: 10.1074/jbc.M406160200. [DOI] [PubMed] [Google Scholar]
- Gai D, Zhao R, Li D, Finkielstein CV, Chen XS. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell. 2004;119:47–60. doi: 10.1016/j.cell.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Gasparovic M, Maginnis MS, O’Hara BA, Dugan AS, Atwood WJ. Modulation of PML protein expression regulates JCV infection. Virology. 2009;390:279–288. doi: 10.1016/j.virol.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheuens S, Wuthrich C, Koralnik IJ. Progressive multifocal leukoencephalopathy: why gray and white matter. Annu Rev Pathol Mech Dis. 2012;8:189–215. doi: 10.1146/annurev-pathol-020712-164018. [DOI] [PubMed] [Google Scholar]
- Gosert R, Kardas P, Major EO, Hirsch HH. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increases virus early gene expression and replication rates. J Virol. 2010;84:10448–10456. doi: 10.1128/JVI.00614-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZS, Heine U, DePamphilis ML. T-antigen binding to site I facilitates initiation of SV40 DNA replication but does not affect bidirectionality. Nucl Acids Res. 1991;19:7081–7088. doi: 10.1093/nar/19.25.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C, Jiang T, Banerjee P, Meinke G, D’Abramo CM, Schaffhausen B, Bohm A. Polyomavirus large T-antigen binds symmetrical repeats at the viral origin in an asymmetrical manner. J Virol. 2013;87:13751–13759. doi: 10.1128/JVI.01740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CJ, Meinke G, Kwun HJ, Rogalin H, Phelan PJ, Bullock PA, Chang Y, Moore PS, Bohm A. Asymmetric assembly of merkel cell polyomavirus large T-antigen origin binding domain at the viral origin. J Mol Biol. 2011;409:529–542. doi: 10.1016/j.jmb.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz J, Dean FB, Kwong AD, Lee SH. The in vitro replication of DNA containing the SV40 origin. J Biol Chem. 1990;265:18043–18046. [PubMed] [Google Scholar]
- Jensen PN, Major EO. A classification scheme for human polyomavirus JCV variants based on the nucleotide sequence of the noncoding regulatory region. J NeuroVirol. 2001;7:280–287. doi: 10.1080/13550280152537102. [DOI] [PubMed] [Google Scholar]
- Jiang M, Abend JR, Johson SF, Imperiale MJ. The role of polyomaviruses in human disease. Virology. 2009;384:266–273. doi: 10.1016/j.virol.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Wortman MJ, Dagdanova AV, Lundberg PS, Daniel DC. Polyomavirus jc in the context of immunosuppression: a series of adapative, dna replication-driven recombination events in the development of progressive multifocal leukoencephalopathy. Clin Dev Immunol. 2013;2013:1–10. doi: 10.1155/2013/197807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo WS, Kim HY, Purviance JD, Sreekumar KR, Bullock PA. Assembly of T-antigen double hexamers on the Simian virus 40 core origin requires only a subset of the available binding sites. Mol Cell Biol. 1998;18:2677–2687. doi: 10.1128/mcb.18.5.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJE. Protein structure prediction on the web: A case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kelly TJ. SV40 DNA replication. J Biol Chem. 1988;263:17889–17892. [PubMed] [Google Scholar]
- Khalili K, White MK, Sawa H, Nagashima K, Safak M. The agnoprotein of polyomaviruses: a multifunctional auxiliary protein. J Cell Physiol. 2005;204:1–7. doi: 10.1002/jcp.20266. [DOI] [PubMed] [Google Scholar]
- Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DWG, Miller E. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71:115–123. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: has the disease outgrown its name? Ann Neurol. 2013;60:162–173. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- Kumar A, Meinke G, Reese DK, Moine S, Phelan PJ, Fradet-Turcotte A, Archambault J, Bohm A, Bullock PA. Model for T-antigen-dependent melting of the simian virus 40 core origin based on studies of the interaction of the beta-hairpin with DNA. J Virol. 2007;81:4808–4818. doi: 10.1128/JVI.02451-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhao R, Lilyestrom W, Gai D, Zhang R, DeCaprio JA, Fanning E, Jochimiak A, Szakonyi G, Chen XS. Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature. 2003;423:512–518. doi: 10.1038/nature01691. [DOI] [PubMed] [Google Scholar]
- Liang B, Tikhanovich I, Nasheuer HP, Folk WR. Stimulation of BK virus DNA replication by NF1 family transcription factors. J Virol. 2012;86:3264–3275. doi: 10.1128/JVI.06369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Sanford DG, Bullock PA, Bachovchin WW. Structure of the origin specific DNA binding domain from simian virus 40 T-antigen. Nat Struct Biol. 1996;3:1034–1039. doi: 10.1038/nsb1296-1034. [DOI] [PubMed] [Google Scholar]
- Major EO, Amemiya K, Tornatore CS. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5:49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall LJ, Dunham L, Major EO. Transcription factor Spi-B binds unique sequences present in the tandem repeat promoter/enhancer of JC virus and supports viral activity. J Gen Virol. 2010;91:3042–3052. doi: 10.1099/vir.0.023184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke G, Bullock PA. Structural “snap-shots” of the initiation of SV40 replication. In: Gaston K, editor. Small DNA Tumor Viruses. Horizon Scientific Press; Norwich: 2012. pp. 195–215. [Google Scholar]
- Meinke G, Phelan PJ, Moine S, Bochkareva E, Bochkarev A, Bullock PA, Bohm A. The crystal structure of the SV40 T-antigen origin binding domain in complex with DNA. PloS Biol. 2007;5:e23. doi: 10.1371/journal.pbio.0050023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke G, Phelan PJ, Kalekar R, Shin J, Bohm A, Bullock PA. Insights into the initiation of JC virus DNA replication derived from the crystal structure of the T-Antigen origin binding domain. PLOS Pathog. 2014;10:e1003966. doi: 10.1371/journal.ppat.1003966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messam CA, Hou J, Gronostajski RM, Major EO. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann Neurol. 2003;53:636–646. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol. 2001;21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco MC, Sabath BF, Durham LC, Major EO. JC virus multiplication in human hematopoietic progenitor cells requires the NF-1 classD transcription factor. J Virol. 2001;75:9687–9695. doi: 10.1128/JVI.75.20.9687-9695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesper J, Smith RWP, Kautz AR, Sock E, Wegner M, Grummt F, Nasheuer HP. A cell-free replication system for human polyomavirus JC DNA. J Virol. 1997;71:7421–7428. doi: 10.1128/jvi.71.10.7421-7428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noch E, Sariyer IK, Gordon J, Kahalili K. JC Virus T-antigen regulates glucose metabolic pathways in brain tumor cells. PLos One. 2012;7:e35054. doi: 10.1371/journal.pone.0035054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Endo S, Takahaski H, Sawa H, Umemura T, Nagashima K. Distribution and function of JCV agnoprotein. J NeuroVirol. 2001;7:302–306. doi: 10.1080/13550280152537148. [DOI] [PubMed] [Google Scholar]
- Orba Y, Suzuki T, Makino Y, Kubota K, Tanaka S, Kimura T, Sawa H. Large T antigen promotes JC virus replication in G2-arrested cells by inducing ATM-and ATR-mediated G2 checkpoint signaling. J Biol Chem. 2010;285:1544–1554s. doi: 10.1074/jbc.M109.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett BL, Walker DL. Prevalance of antibodies in human sera against JC virus, an isolate from a case of progressive multifocal leukoencephalopathy. J Infect Dis. 1973;127:467–470. doi: 10.1093/infdis/127.4.467. [DOI] [PubMed] [Google Scholar]
- Pfister LA, Letvin NL, Koralnik IJ. JC virus regulatory region tandem repeats in plasma and central nervous system isolates correlate with poor clinical outcome in patients with progressive multifocal leukoencephalopathy. J Virol. 2001;75:5672–5676. doi: 10.1128/JVI.75.12.5672-5676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins C, Frisque RJ. JC virus T′ proteins encoded by alternatively spliced early mRNAs enhance T antigen-mediated viral DNA replication in human cells. J NeuroVirol. 2001;7:250–264. doi: 10.1080/13550280152403290. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S, Otte J, Enam S, Del Valle L, Kahalili K, Gordon J. JC virus-induced changes in cellular gene expression in primary human astrocytes. J Virol. 2003;77:10638–10644. doi: 10.1128/JVI.77.19.10638-10644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran V, Major EO. DNA-binding transcription factor NF-1A negatively regulates JC virus multiplication. J Gen Virol. 2008;89:1396–1401. doi: 10.1099/vir.0.2008/000059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran V, Sabath BF, Jensen PN, Houff SA, Major EO. Interactions between c-Jun, nuclear factor 1, and JC virus promoter sequences: implications for viral tropism. J Virol. 2006;80:10506–10513. doi: 10.1128/JVI.01355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese DK, Sreekumar KR, Bullock PA. Interactions required for binding of simian virus 40 T antigen to the viral origin and molecular modeling of initial assembly events. J Virol. 2004;78:2921–2934. doi: 10.1128/JVI.78.6.2921-2934.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviriego-Mendoza MM, Frisque RJ. Interaction and co-localization of JC virus large T antigen and the F-box protein B-transducin-repeat containing protein. Virology. 2011;5:119–128. doi: 10.1016/j.virol.2010.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Kahalili K. Interactions of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75:1476–1486. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: evidence for its involvement in viral DNA replication. Virology. 2012;433:12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Merabova N, Patel PK, Knezevic T, Rosati A, Turco MC, Khalili K. Bag3-induced autophagy is associated with degradation of JCV oncopro-tein,T-Ag. PLOS One. 2012;7:e45000. doi: 10.1371/journal.pone.0045000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scuda N, Madinda NF, Akoua-Koffi C, Adjogous EV, Wevers D, Hoffman J, Cameron KN, Leendertz SAJ, Couacy-Hymann E, Robbins M, Boesch C, Jarvis MA, Moens U, Mugisha L, Calvignac-Spencer S, Leendertz FH, Ehlers B. Novel polyomaviruses of nonhuman primates: genetic and serological predictors for the existence of multiple unknown polyomaviruses within the human population. PLoS Pathog. 2013;9:e1003429. doi: 10.1371/journal.ppat.1003429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Gai D, Patrick A, Greenleaf WB, Chen XS. The roles of the residues on the channel β-hairpin and loop structures of simian virus 40 hexameric helicase. Proc Natl Acad Sci USA. 2005;102:11248–11253. doi: 10.1073/pnas.0409646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y. Progressive multifocal leukoencephalopathy and promyelocytic leukemia nuclear bodies: a review of clinical, neuropathological, and virolgical aspects of JC virus-induced demyelinating disease. Acta Neuropathol. 2010;120:403–417. doi: 10.1007/s00401-010-0694-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y, Higuchi K, Ohara S, Duyckaerts C, Hauw JJ, Uchihara T. Promyelocytic leukemia nuclear bodies provide a scaffold for human polyomavirus JC replication and are disrupted after development of viral inclusions in progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol. 2008;67:299–308. doi: 10.1097/NEN.0b013e31816a1dd3. [DOI] [PubMed] [Google Scholar]
- Simmons DT. SV40 large T antigen functions in DNA replication and transformation. Adv Virus Res. 2000;55:75–134. doi: 10.1016/s0065-3527(00)55002-7. [DOI] [PubMed] [Google Scholar]
- Simmons DT, Loeber G, Tegtmeyer P. Four major sequence elements of simian virus 40 large T antigen coordinate its specific and nonspecific DNA binding. J Virol. 1990;64:1973–1983. doi: 10.1128/jvi.64.5.1973-1983.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DT, Wun-Kim K, Young W. Identification of simian virus 40 T-antigen residues important for specific and nonspecific binding to DNA and for helicase activity. J Virol. 1990;64:4858–4865. doi: 10.1128/jvi.64.10.4858-4865.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sock E, Wegner M, Grummt F. DNA replication of human polyomavirus JC is stimulated by NF-1 in vivo. Virology. 1991;182:298–308. doi: 10.1016/0042-6822(91)90673-y. [DOI] [PubMed] [Google Scholar]
- Sock E, Wegner M, Grummt F. Large T-antigen and sequences within the regulatory region of JC virus both contribute to the features of JC virus DNA replication. Virology. 1993;197:537–548. doi: 10.1006/viro.1993.1627. [DOI] [PubMed] [Google Scholar]
- Staib C, Pesch J, Gerwig R, Gerber JK, Brehm U, Stangl A, Grummt F. p53 inhibits JC virus DNA replication in vivo and interacts with JC virus large T-antigen. Virology. 1996;219:237–246. doi: 10.1006/viro.1996.0241. [DOI] [PubMed] [Google Scholar]
- Stoner GL, Ryschkewitsch CF, Walker DL, Webster HD. JC papovavirus large tumor (T)-antigen expression in brain tissue of acquired immune deficiency syndrome (AIDS) and non-AIDS patients with progressive multifocal leukoencephalopathy. Proc Natl Acad Sci USA. 1986;83:2271–2275. doi: 10.1073/pnas.83.7.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson JJ, Trowbridge PW, Frisque RJ. Replication activity of JC virus large T antigen phosphorylation and zinc finger domain mutants. J NeuroVirol. 1996;2:78–86. doi: 10.3109/13550289609146541. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bell P, Tegtmeyer P, Maul GG. Replication but not transcription of Simian virus 40 DNA is dependent on Nnuclear domain 10. J Virol. 2000;74:9694–9700. doi: 10.1128/jvi.74.20.9694-9700.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalis D, Andrei G, Snoeck R. The large tumor antigen: a Swiss army knife protein possessing the functions required for the polyomavirus life cycle. Antiviral Res. 2013;97:122–136. doi: 10.1016/j.antiviral.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Tyagarajan SK, Frisque RJ. Stability and function of JC virus large T antigen and T′ proteins are altered by mutation of their phosphorylated threonine 125 residues. J Virol. 2006;80:2083–2091. doi: 10.1128/JVI.80.5.2083-2091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, Pickhardt M, Weber T. Analysis of the transcription control region in progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:398–409. doi: 10.3109/13550280009018304. [DOI] [PubMed] [Google Scholar]
- White MK, Safak M, Khalili K. Regulation of gene expression in primate polyomaviruses. J Virol. 2009;83:10846–10856. doi: 10.1128/JVI.00542-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MK, Gordon J, Khalili K. The rapidly expanding family of human polyomaviruses: recent developments in understanding their life cycle and role in human pathology. PLOS Pathog. 2013;9:e1003206. doi: 10.1371/journal.ppat.1003206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windl O, Dorries K. Expression of human polyomavirus JC T antigen by an adenovirus hybrid vector and its binding to DNA sequences encompassing the JC virus origin of DNA replication. J Gen Virol. 1995;76:83–92. doi: 10.1099/0022-1317-76-1-83. [DOI] [PubMed] [Google Scholar]
- Wuthrich C, Dang X, Westmoreland S, McKay J, Maheshwari A, Anderson MP, Ropper AH, Viscidi RP, Koralnik IJ. Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol. 2009;65:742–748. doi: 10.1002/ana.21619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequeces in human cervical carcinoma cell lines. Am J Pathol. 1985;119:361–366. [PMC free article] [PubMed] [Google Scholar]
- Yogo Y, Kitamura T, Sugimoto C. Isolation of a possible archtypal JC virus DNA sequence from nonimmunocompromised individuals. J Virol. 1990;64:3139–3143. doi: 10.1128/jvi.64.6.3139-3143.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E. Ataxia Telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of MRE11-rad50-NBS1 subunits in simian virus 40-infected primate cells. J Virol. 2008;82:5316–5328. doi: 10.1128/JVI.02677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]