

Abstract
Squaramides constitute a novel class of RNA polymerase inhibitors of which genetic evidence and computational modeling previously have suggested an inhibitory mechanism mediated by binding to the RNA polymerase switch region. An iterative chemistry program increased the fraction unbound to human plasma protein from below minimum detection levels, i.e. <1%, to 4~6%, while retaining biochemical potency. Since in vitro antimicrobial activity against an efflux-negative strain of Haemophilus influenzae was 4~8-fold higher, the combined improvement was at least 20~60-fold. Co-crystal structures of Escherichia coli RNA polymerase with two key squaramides showed displacement of the switch 2, predicted to interfere with the conformational change of clamp domain and/or with binding of non-template DNA, a mechanism akin to that of natural product myxopyronin. Furthermore, the structures confirmed the chemical features required for biochemical potency. The terminal isoxazole and benzyl rings bind into distinct relatively narrow, hydrophobic pockets and both are required for biochemical potency. In contrast, the linker composed of squarate and piperidine accesses different conformations in their respective co-crystal structures with RNA polymerase, reflecting its main role of proper orientation of the aforementioned terminal rings. These observations further explain the tolerance of hydrophilic substitutions in the linker region that was exploited to improve the fraction unbound to human plasma protein while retaining biochemical potency.
Keywords: Escherichia coli, RNA polymerase, X-ray crystallography, antibacterial, switch region, squaramide, myxopyronin
INTRODUCTION
There is a growing interest in identification of new compounds with antibacterial therapeutic potential, driven by a heightened incidence of clinical failures due to drug-resistant bacterial pathogens 1. The mounting number of macromolecular structures determined by X-ray crystallography, cryo-electron microscopy and NMR increasingly enables structure-based drug design of inhibitors of large antibacterial targets such as the ribosome and RNA polymerase (RNAP) 2–4. The (near-) atomic-resolution structures of macromolecules in complex with inhibitors identify their binding sites and elucidate their mechanism of inhibition, thus providing a structural framework for an iterative chemistry program aimed at rationally designing new compounds with higher affinity and efficacy.
Bacterial RNAP is essential for gene expression, cell growth and viability 5. The bacterial RNAP core enzyme, consisting of α (two copies), β, β′ and ω subunits, is responsible for binding to DNA template and subsequent RNA synthesis, and is complemented by a σ factor to start promoter-specific transcription 6. The molecular shape of a bacterial core RNAP resembles a crab claw, with two pincers that clamp around an RNA/DNA assembly during transcription 7. The larger pincer of RNAP, the DNA binding clamp, is mainly formed by the β′ subunit and connects to the rest of RNAP via a flexible hinge at the clamp’s base, composed of so-called “switches”. These switches allow a swinging motion of the clamp to different conformational states, transitioning from closed to open (with a maximum of 30°) 8. The open clamp configuration has been proposed to be a prerequisite for entry of DNA into the main channel during the DNA unwinding, whereas the subsequent reverse transition to the closed state clamp is required for stabilization of the open complex, followed by processive transcription elongation. Amino acid sequence conservation of RNAPs among bacterial species and structural dissimilarities of some regions including RNA binding site and switches with eukaryotic RNAPs make bacterial RNAP an attractive target for developing broad-spectrum, yet selective antibiotics 9–11.
Proof of principle is provided by rifampin and fidaxomicin, two bactericidal RNAP inhibitors currently in clinical use 12, 13. Rifampin binds to the RNA binding site of RNAP, thus sterically blocking RNA extension 14, 15, and has been a cornerstone of antituberculosis combination therapy for over a half-century 12. However, the clinical importance of rifampin has eroded in the last decade due to the rise of rifampin-resistant strains of Mycobacterium tuberculosis 16, which poses a serious threat to public health, leading to ~1.3 million tuberculosis-related deaths worldwide in 2012 including 170,000 from multidrug-resistant tuberculosis 17. Fidaxomicin is a recently approved narrow-spectrum antibiotic for the treatment of Clostridium difficile infections, responsible for 15,000~20,000 deaths in the United States each year 18. Unlike rifampin, fidaxomicin binds to the RNAP switches, thus preventing formation of the transcription ready promoter DNA open complex 19.
Rifampin-resistant mutants were not cross-resistant to the switch region binding RNAP inhibitors 20–23. Furthermore, the importance of mobility of the clamp during the transcription-competent open complex formation and identification of a number of natural and synthetic antimicrobial molecules that bind the switches have raised the importance of studying and subsequent exploitation of the molecular basis of their action, and these compounds could be promising antibiotic candidates for treatment of multi-drug resistant tuberculosis. These include chemically diverse natural compounds such as the α-pyrones, myxopyronin (Myx) (Fig. 1A) and corallopyronin, and the macrocycles ripostatin and fidaxomicin 19, 23–25. More recently, synthetic scaffolds including ureidothiophene-2-carboxylic acid-based inhibitors 21, 26, and squaramide-based inhibitors (Fig. 1B) 27 have been added. Structural and biochemical studies of Myx-mediated inhibition of RNAP culminated into two possible hypotheses regarding its mechanism. In the hinge-jamming model, Myx prevents conformational changes of the RNAP clamp required for the promoter DNA melting 8, 25. Alternatively, in the steric clash model, Myx alters the conformation of switch 2, not preventing the nucleation of the promoter DNA melting but interfering with its further accommodation into the active-center cleft 24.
Figure 1.

Chemical structures of (A) Myxopyronin and (B) Squaramide (compound 14). The chemical moieties are indicated. The stereocenter at the C7-position on Myx is indicated by an asterisk.
In a previous study, a series of antimicrobial squaramides were synthesized of which a subset acts by binding to the switches, as concluded from mapping of resistance mutations to the β and β′ subunits 27. Using the crystal structure of Thermus thermophilus RNAP – Myx complex 25, a computational model of the RNAP and squaramide complex was constructed to predict possible squaramide binding modes around the switches. Here, in an extension of these studies, crystal structures of E. coli RNAP holoenzyme in complex with squaramides were determined, structurally confirming that the squaramides bind the RNAP switches. This revealed that Myx and squaramides, although chemically distinct, bind to the same region of RNAP and identified new binding crevices within the switches that could be explored for the design of a new class of antibiotics.
RESULTS AND DISCUSSION
Determination of structure activity relationships of squaramides
A high-throughput screen of a corporate compound library using S30 extracts of E. coli in a coupled Transcription-Translation assay (TT assay) identified a series of squaramides of which its most active member, compound 1 (Table 1), had a sub-micromolar half maximal inhibitory concentration (IC50)27. Since this assay was 10–100-fold more sensitive than an RNAP assay using purified components27, the TT assay was used to monitor biochemical potency. In addition, compound 1 was selective as determined in an eukaryotic TT assay, using rabbit reticulocytes, with an IC50 of >100 μM (data not shown). Although hydrophobicity and solubility of the compound were reasonable for a chemical starting point for development of an antibacterial lead, the fraction unbound to human plasma protein (fu) was below the minimum detection level of 1% (Table 1). Animal efficacy of a compound is determined by its antibacterial activity and exposure in tissues to its unbound, or “free” fraction 28. Since compound 1 was highly plasma protein bound (Table 1) as reported for Myx 29, an iterative chemistry program was started to improve human plasma protein binding of the compound while retaining its biochemical activity and solubility.
Table 1.
Biochemical and antimicrobial potency, and physicochemical properties of RNA polymerase inhibitors mediating their effect via the switch domain.
| Structure | IC50 (μM) | MIC (μM) | Physicochemical properties | |||
|---|---|---|---|---|---|---|
| Eco-TT | H. influenzae acrB | logD | fu (%)§ | Solubility (μM) | ||
| 1 |
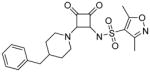
|
0.28 | 200 | 0.1 | <1 | >1000 |
| 2 |
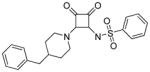
|
2.1 | 200 | 0.2 | <1 | >1000 |
| 3* |
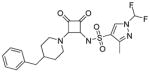
|
7.9 | 100 | −0.1 | <1 | >1000 |
| 4 |
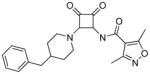
|
>100 | n.d. | 1.7 | <1 | 508 |
| 5 |
|
>100 | n.d. | −1.0 | 1.6 | 773 |
| 6 |
|
26 | n.d. | −0.6 | <1 | 763 |
| 7 |
|
71 | n.d. | −0.3 | <1 | 985 |
| 8* |
|
0.29 | 12.5 | 0.5 | <1 | >1000 |
| 9 |
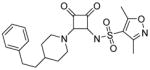
|
0.32 | n.d. | 0.0 | <1 | 636 |
| 10 |
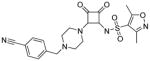
|
1.5 | 200 | −1.3 | 2.9 | >1000 |
| 11 |
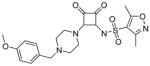
|
3.0 | >200 | −0.9 | 1.8 | 497 |
| 12 |
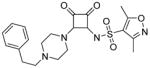
|
43 | n.d. | −0.4 | 4.9 | 560 |
| 13 |
|
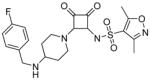
2.7 | >200 | 0.0 | 26 | 52 |
| 14* |
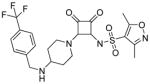
|
0.29 | 25 | 0.8 | 6.6 | >1000 |
| 15 |
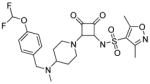
|
0.43 | 50 | 0.2 | 6.2 | 203 |
| 16* |
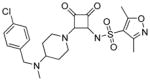
|
0.33 | 50 | 0.5 | 3.8 | >1000 |
| 17 |
|
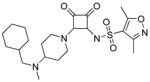
>100 | n.d. | −0.4 | 38 | 12.2 |
| 18 |
|
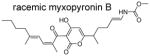
1.0 | n.d. | −0.3 | <1 | 679 |
| Linezolid | 2.4 | 12.5 | 0.7 | >70 | >1000 | |
| Rifampin | 0.014 | 0.025 | 1.3 | 16.5 | 751 | |
compounds described27; n.d. not determined
compound fraction unbound to human plasma protein
Squaramides can be divided into a left hand side (LHS) and a right hand side (RHS) by bisecting the squarate along the north south axis (Fig. 1B). As the final analogs are available in a few synthetic steps, compound synthesis was straightforward. The synthetic route as exemplified for compound 1 is as follows (Scheme 1): diethyl squarate was treated with ammonia to displace one ethoxy residue and afford 3-amino-4-ethoxycyclobut-3-ene-1,2-dione 1a cleanly in 74% isolated yield. Installation of the sulfonamide was accomplished using sodium hydride as base to afford key intermediate compound 1b in 47% yield after normal phase chromatography. Treatment of 1b with a second amine under more forcing conditions displaced the second ethoxy group to afford the final compound 1 in 53% yield. Other analogs were prepared using the same general synthetic route (see Experimental Section).
Scheme 1.

Synthesis of squaramide (1).
A wide variety of sulfonamide analogs were prepared and from that scan of the RHS a few alternatives to the dimethylisoxazole sulfonamide were identified. Compound 2 with a phenyl sulfonamide showed a 7-fold reduction in activity in the TT assay. Another sulfonamide substituent that was biochemically active was the substituted pyrazole 3. Changing the sulfonamide linker to an amide resulted in an inactive compound (compound 4). However, none of these modifications improved human serum protein binding to levels above minimum detection levels (Table 1). Furthermore, additional sulfonamide analogs not shown here were inactive; therefore, attention was then turned to the LHS while keeping the preferred dimethylisoxazole sulfonamide from compound 1 intact.
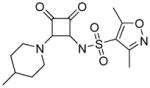
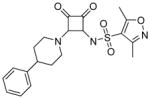
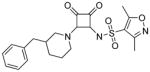
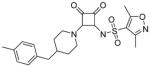
Modifications of the LHS squaramide delineated the minimal pharmacophore. Truncation of the LHS versus 1 resulted in either inactive (compound 5) or much less active (compound 6) analogs. In addition, moving the benzyl substituent to the 3-position of the piperidine also eroded the biochemical activity (compound 7) pointing to a requirement for substituents at the 4-position. Substitution off of the benzyl group appeared to be tolerated (compound 8). Hydroxylation of either C4 of the piperidine, or the benzylic carbon resulted in slight improvements in protein binding, but reduced TT activity three-fold (data not shown). Replacement of the lipophilic phenyl ring with a 3- or 4-pyridyl ring showed even larger effects of the introduction of polarity to this portion of the compound, making for nearly inactive compounds (data not shown). Homologation of the benzyl group to a phenethyl substituent (compound 9) was tolerated in regards to biochemical activity, however with no measurable improvements in plasma protein binding. Substituted piperazines, which add a basic amine to the LHS of the molecule, showed measurable free fractions (fu) of 2–5% while retaining single digit micromolar activity in the TT assay (compounds 10–12). However, the best analogs inserted a nitrogen off of the piperidine ring in the four position to afford the most active compounds in the series (compounds 13–16) with the best free fractions. The best aminopiperidine analogs had a benzylic substituent off of the 4-amino group, whereas placing a saturated ring in that position resulted in an inactive analog (compound 17). Compound 13 showed an unbound fraction of 26% but with a ten-fold reduction in biochemical activity compared to compound 1. Compounds 14–16 were biochemically equipotent to compound 1, and showed unbound fractions of 4–7%, a significant advancement versus starting compound 1.
Squaramides made to date have shown modest antibacterial activity in efflux-negative strains of H. influenzae 27 with, for instance, minimum inhibitory concentrations (MIC) for compound 1 of 200 μM (Table 1). Although compounds 14–16 are biochemically equipotent with compound 1, their antimicrobial activity has increased 4~8-fold while retaining a mode of action via RNAP (Table 1) 27. Combined with enhanced free fractions, this amounts to at least a 20~60-fold improvement.
Structural basis of transcription inhibition by squaramides
To elucidate the mechanism of transcription inhibition by squaramides, crystal structures of the E. coli RNAP holoenzyme in complex with compound 8 and 14 were determined (Table 2). Biochemically and microbiologically these squaramides are equivalent, but the free fraction (fu) of compound 14 was 7-fold better (Table 1). Crystals of these complexes were prepared by soaking 1 mM compounds into crystals of RNAP. The crystal structures contain two RNAP molecules per asymmetric unit, designated RNAPA and RNAPB 30 and these two RNAP-compound complexes have nearly identical structures. Therefore, the structural analysis throughout this study is based mainly on RNAPA.
Table 2.
Data collection and refinement statistics
| Complex PDB code | Compound81 4YFK | Compound141 4YFN | MyxB2 4YFX | Apo2 4YG2 |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||
| a (Å) | 186.105 | 191.247 | 188.520 | 187.308 |
| b (Å) | 206.438 | 206.569 | 205.177 | 205.901 |
| c (Å) | 307.993 | 312.146 | 310.140 | 309.185 |
| Resolution (Å) | 30 – 3.50 | 30 – 3.80 | 50 – 3.80 | 30–3.60 |
| Total reflections | 679,098 | 704,430 | 488,892 | 592,860 |
| Unique reflections | 133,199 | 107,176 | 106,246 | 123,448 |
| Redundancy | 5.1 (3.5) | 6.6 (6.2) | 4.6 (4.3) | 4.5 (2.9) |
| Completeness (%) | 96.8 (89.9) | 89.7 (83.8) | 93.8 (95.0) | 96.8 (93.7) |
| I/σ | 12.8 (1.8) | 15.8 (1.1) | 9.0 (1.0) | 10.1 (2.0) |
| Rsym (%) | 10.5 (64.2) | 10.4 (100) | 13.9 (100) | 9.1 (46.9) |
| Refinement | ||||
| Resolution (Å) | 30 – 3.57 | 30 – 3.82 | 30 – 3.84 | 30 – 3.70 |
| Rwork | 0.2168 | 0.2215 | 0.2356 | 0.1914 |
| Rfree | 0.2521 | 0.2695 | 0.2999 | 0.2601 |
| R.m.s deviations | ||||
| Bond length (Å) | 0.018 | 0.018 | 0.014 | 0.013 |
| Bond angles (°) | 1.81 | 1.82 | 2.146 | 1.70 |
Data sets were collected at MacCHESS F1 line, Ithaca, NY
Data sets were collected at MacCHESS A1 line, Ithaca, NY
Highest resolution shells are shown in parentheses
The structures show unambiguous densities for both compounds located in the switches, that form the hinge part of the DNA binding clamp domain connecting to the main body of RNAP (Fig. 2A; Supplemental Fig. 1A–B). The trifluoro extension at the benzyl ring of compound 14, causes larger density compared with that of compound 8, thus unequivocally confirming the squaramide binding orientation. Both the position and orientation of the squaramides in their binding pocket are similar to those described in the computational docking model 27.
Figure 2.

RNAP – squaramide complex structures. (A) Overall structure of the E. coli RNAP in complex with compound 14. RNAP is depicted as a partially transparent molecular surface model (α subunits: white, β subunit: cyan, β′ subunit: pink, σ70: orange, ω subunit is not visible from this orientation). The DNA binding clamp is shown as a gray oval. Right panel shows a magnified view of the interface between β and β′ subunits upon removal of σ70 to visualize the squaramide binding site. Compound 14 is depicted as a stick model (carbon, yellow; oxygen, red; nitrogen, blue; fluorine, pale cyan; sulfur, light brown). β (cyan) and β′ (pink) subunits are depicted as a ribbon models, respectively. Five switches are denoted by a unique color and labeled correspondingly. (B) Surface representation of the squaramide-binding pocket. Surface properties are: white, neutral; green, hydrophobic; red, hydrogen bond acceptor potential; blue, hydrogen bond donor potential. Compound 14 is depicted as a stick model and its labeling is identical to that in (A). (C) Schematic summary of contacts between RNAP and compound 14. Amino acids belonging to the switches are labeled with those identified by resistance mutations highlighted in blue. Chemical groups of the squaramide are indicated.
The squaramide binding site is a deep crescent-shaped pocket of ~22 Å in length (Fig. 2B–C), which is overlaid by the σ factor region 3.2 (also known as σ finger) 31. The molecular surfaces of compound 8 and compound 14 are 531 Å2 and 590 Å2, respectively and only 366 Å2 and 398 Å2, respectively, are involved in the RNAP binding, largely due to exposure of the squarate and piperidine linker to solvent. These numbers are smaller than the molecular surface (646 Å2) and the contact area (486 Å2) of Myx in complex with the E. coli RNAP described below and the T. thermophilus RNAP (638 Å2 and 427 Å2, respectively) 25. Among the five switches of RNAP 25, 32, the squaramides mainly interact with switch 1 (β′ subunit residues from 1307 to 1329) and switch 2 (β′ subunit residues from 331 to 348) (Fig. 2A–C). However, a few interactions with switch 3 (β subunit residues from 1248 to 1270) and switch 4 (β subunit residues from 1291 to 1300) are also apparent, but there is no interaction with switch 5 (β′ subunit residues from 1356 to 1358). In addition to the switches, the squaramides also contact the α helices near the C-terminal ends of both β (residues from 1320 to 1330) and β′ subunits (residues from 1348 to 1353), which surround the switches but are not part of them. The squaramide-binding pocket is largely hydrophobic, which complements the hydrophobic nature of the preferred terminal substituents, whereas the overall squaramide is more polar, as evidenced by logD (Fig. 2B–C, Table 1).
In a genetic search for squaramide resistant mutants that was likely non-saturating, amino acid substitutions at eight different positions in the β and β′ subunits were recently identified in the H. influenzae RNAP 27. Of these, four amino acid residues are directly engaged in contacts with squaramides indicating that the squaramide-binding site identified from the crystal structure is biologically significant. The β′A1323V/T mutations (E. coli numbering) would result in a steric clash of a bulky side chain with the benzyl ring of the squaramide (Fig. 2C). The β′L1332F mutation would produce a similar steric clash. The β′K1348 participates not only in a van der Waals contact with a methyl group of the dimethylisoxazole moiety in the squaramide but also neutralizes the negative charge of the βE1272 side chain. Such charge neutralization might be important for the binding of the apolar isoxazole. The β′K1348Q mutation may disturb these functions and thus prevent the squaramide binding. The βL1326 participates in van der Waals contact with a terminal benzyl ring of the squaramide, playing a key role in the squaramide and RNAP interaction (see compounds 5 and 17 in Table 1) and the βL1326T mutation disrupts this contact. An additional four resistance mutations (Q1257H, E1279K, D1296E, P1320S of the β subunit) are located near the switches but since they are within 8 Å of the squaramide binding pocket their effect on squaramide binding may be indirect.
The conformation of the switch 2 itself is altered upon binding of the squaramides. In the apo-form RNAP structure, the middle part of the switch 2 (β′ residues 338–340) forms a short α-helix and occupies the squaramide binding space (Fig 3A). In order to accommodate squaramide binding, these residues are pushed into the DNA binding main channel. This altered switch 2 conformation is predicted to clash with the melted template DNA (positions at +3 and +4), suggesting that squaramides inhibit transcription activity of RNAP by hinge jamming 8, 25 and/or blocking template DNA loading 24, similar to the proposed mechanisms for the Myx-dependent transcription inhibition.
Figure 3.

(A) Comparison of the switch 2 conformations in the apo-form of RNAP (white) and bound with compound 14 (red). The switch 2 conformation when compound 14 is bound clashes with the template DNA of the RNAP-DNA complex. Structures of the apo-form RNAP, the RNAP – compound 14 complex and the RNAP elongation complex (template DNA, green; non-template DNA, cyan) were aligned by superimposing their β′ subunits. The DNA position of the template DNA and the RNAP bridge helix (BH) are labeled. Switch 2 is indicated in red with its disordered region shown as a dashed line. (B) Comparison of binding of compound 8 (magenta) and 14 (yellow) to the switch region of E. coli RNAP. Compounds are shown as stick models and their chemical groups are indicated.
Compound 8 and 14 bind to the same site with an almost identical orientation of the terminal isoxazole and benzyl rings (Fig. 3B). However, the internal squarate and piperidine linkers seem distorted, that of compound 14 having rotated and shifted ~2.5 Å from the switch 2 compared with its position in compound 8. Since the compounds are biochemically equipotent, there seems no net energetic consequence to this change; however, the current resolutions of the RNAP – squaramide complex structures are insufficient to rule out compensatory enthalpic and entropic changes.
Modification of the RHS of the squaramide quickly resulted in compounds with much reduced activity (compounds 1–4, Table 1). The co-crystal structures of RNAP – squaramide provide an explanation, showing that the isoxazole ring of the squaramides fit into a deep and narrow pocket (Fig. 2B–C), with limited tolerance for change. A major conclusion of the SAR of the LHS is the need for the benzyl ring at a fairly precise distance and orientation from the isoxazole. Its absence, compound 5, direct substitution off of the piperidine, compound 6, and attached through a methylene linker off of the 3′ instead of the 4′ carbon, compound 7, each resulted in at least 100-fold reduction in potency (Table 1). This is in good agreement with the aforementioned comparison of the co-crystal structures of compound 8 and 14: in addition to the isoxazole, the benzyl ring is key for binding whereas positioning of the squarate and the piperidine linker is much less critical.
Although the trifluoro modification at the benzyl ring drastically improved biochemical potency by about ten-fold (compounds 13 and 14, Table 1), the co-crystal structure of compound 14 suggests the binding pocket is still not fully occupied (Fig. 2B). Further squaramide modifications, introducing hydrophobic group(s) at the benzyl ring could be considered to enhance the squaramide binding affinity. However, this will need to be balanced with a possibly reduced free fraction that may result as a consequence, as illustrated by the four-fold reduction between compounds 13 and 14 (Table 1). One possible strategy to compensate would be to introduce more polarity in the seemingly more flexible squarate-piperidine linker, as exemplified here by amino substitution of the piperidine or its conversion to piperazine.
The crystal structure of E. coli RNAP holoenzyme in complex with Myx
The crystal structures of bacterial RNAP and Myx complexes including myxopyronin A (MyxA) and desmethyl derivative of myxopyronin B (dMyxB) (Fig. 1A) have been determined using the T. thermophilus RNAP, a model bacterial RNAP for high-resolution crystallographic studies 24, 25. However, a structure of the E. coli RNAP in complex with myxopyronin B (MyxB), determined here, allowed for a direct comparison between the squaramide and Myx binding on the same bacterial RNAP. The absolute stereochemistry of the natural products of MyxA and MyxB has been assigned as (R)-configuration at C7 position 33 (Fig. 1B). However, MyxB used in this paper was synthesized in racemic form (Scheme 5) and the current resolution of the RNAP – MyxB complex structure is insufficient to determine a configuration of MyxB in the RNAP complex. An electron-density map shows a clear density for MyxB located in the RNAP switches (Supplemental Fig. 1C). Both the position of MyxB in its binding pocket as well as its orientation observed in the crystal structure are similar to those described in the T. thermophilus RNAP and Myx complexes 24, 25 (Supplemental Fig. 2).
Scheme 5.

Synthesis of Myxopyronin B (18)
The positions of squaramides and MyxB within E. coli RNAP mostly overlap; however, each compound extends towards different sides of their binding pockets (Fig. 4A–B). The carbamate chain of MyxB forms significant contacts with the three α-helix bundle (the helix-turn-helix flanked by switches 3 and 4 of the β subunit and one helix from the β′ subunit) and the carbamate carbonyl group inserts deep into the three α-helix bundle to reach the C-terminal end of the bridge helix. In contrast, the squaramide contacts only the front edge of the three-helix bundle via its isoxazole ring. The LHS of squaramide overlays with the dienone chain of MyxB, forming interactions with switch 1 and the C-terminal α helix of the β subunit. However, the benzyl moieties of squaramides reach deeper and form a larger interface than does MyxB. Extending either of these scaffolds in a manner consistent with the other may take advantage of additional binding interactions, and thus improve affinity.
Figure 4.

(A) Comparison of the MyxB and squaramide bindings on the E. coli RNAP. Structure of the RNAP – MyxB complex was superposed with the RNAP – compound 14 complex. Only compound 14 is shown in the RNAP – MyxB complex structure. The β (cyan) and β′ (pink) subunit are depicted as a ribbon model, the switches 1 and 2 are indicated as blue and red, respectively. The MyxB (carbon atoms are shown in orange) and compound 14 (carbon atoms are shown in yellow) are shown as stick models. The three α-helix bundle contacting with the carbamate chain of Myx are indicated by red asterisks, the C-terminal α–helix of the β subunit interacting with the dienone chain is indicated by a blue asterisk. The RNAP bridge helix (BH) is labeled. (B) Comparison of the MyxB and compound 14 in the RNAP complexes. Orientation of this panel is the same as in (A). Chemical groups of the MyxB are labeled. (C–D) The switch 2 conformations of the E. coli RNAP – MyxB complex (C) and the E. coli RNAP –compound 14 complex (D).
Like squaramides, binding of MyxB to E. coli RNAP induces a structural rearrangement of the switch 2 (Fig. 4B). However, the conformation of the switch 2 is slightly different from that observed in the RNAP – squaramide complex (Fig. 4C). The relatively narrow dienone chain of MyxB results in a van der Waals contact between the β′K332 side chain and a methyl group of the MyxB dienone chain, thus pulling the switch 2 to the MyxB binding pocket. In case of the squaramide complexes, the bulky piperidine pushes the switch 2 out, positioning it deeper in the DNA binding main channel.
Concluding remarks
An iterative chemistry program of squaramides in conjunction with a crystallographic study of RNAP in complex with representative squaramides has provided a rational way forward for further optimization of these RNAP transcription inhibitors. The terminal isoxazole and benzyl rings of squaramide are crucial for binding of the scaffold to their fairly constrained pockets in the switch region. This interaction leads to dysfunction of the switch region that is predicted to be allosterically (hinge jamming model) or sterically (steric clash model) incompatible with binding of template DNA to the DNA binding main channel, similar to the molecular mechanism of Myx-dependent transcription inhibition. The key role of the squarate plus piperidine linker is to position the isoxazole and benzyl rings at the other side of squaramide binding cleft. There is room to maneuver this bicyclic moiety, suggesting that compensatory modifications to maintain desirable physicochemical properties can be made in this linker.
EXPERIMENTAL SECTION
Synthetic Chemistry
AstraZeneca R&D and Nimbus Discovery performed synthesis of compounds. Compound purity was determined by LC/MS shortly before biological testing and was >95% except for compound 3(92%) and 4 (90%).
Synthesis
Synthesis of squaric amide 1 and analogs followed standard solution phase organic synthesis (Scheme 1) and purification, using dry solvents and reagents from standard vendors without further manipulation.
3-amino-4-ethoxycyclobut-3-ene-1,2-dione (1a)
A solution of 3,4-diethoxycyclobut-3-ene-1,2-dione (2.5 g, 14.69 mmol) in methanol (15 mL) was treated with ammonia (7 M in methanol, 5 mL, 35 mmol) and stirred at room temperature for 16 hours. The resulting suspension was filtered, and the filtrate concentrated to give crude product. This material was suspended in a 1:1 mixture of ethyl acetate and hexanes (70 mL), heated to 60 °C for 2 min, cooled to room temperature, and filtered to give the title compound as an off-white solid (1.54 g, 10.91 mmol, 74.3 %). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.20 – 1.55 (m, 3 H) 4.64 (q, J=6.97 Hz, 2 H) 8.30 (br. s., 2 H).
N-(2-ethoxy-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (1b)
A stirred solution of 3-amino-4-ethoxycyclobut-3-ene-1,2-dione (1a, 2050 mg, 14.53 mmol) in tetrahydrofuran (60 mL) was treated with sodium hydride (1400 mg, 35.00 mmol) to give a bright yellow suspension, this was stirred at room temperature for 20 min followed by treatment with 3,5-dimethylisoxazole-4-sulfonyl chloride (3694 mg, 18.88 mmol) in one portion. This suspension was stirred at room temperature for 24 hours; solvents were removed under reduced pressure. The resulting red paste was suspended in hot ethyl acetate (50 mL), neutralized with 2N HCl in ether (10 mL), filtered hot, the filtrate was concentrated and the residue purified by flash chromatography on silica gel (ethyl acetate, followed by a gradient of 0–10% methanol in dichloromethane), to give the title compound as a yellow oil (2.07 g, 6.89 mmol, 47.5 %). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.22 – 1.35 (m, 3 H) 2.26 (s, 3 H) 2.55 (s, 3 H) 4.56 (q, J=6.97 Hz, 2 H).
3,5-dimethyl-N-(2-(4-(4-methylbenzyl)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)isoxazole-4-sulfonamide (1)
A solution of N-(2-ethoxy-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (1b, 75 mg, 0.25 mmol), 4-(4-methylbenzyl)piperidine hydrochloride (1.25 mmol), diisopropylethylamine (0.75 mL), in 1,4-dioxane (8 mL), was heated to 60 °C for 18 hours. Solvents were removed under reduced pressure and the residue was purified by flash chromatography on silica gel (50–100% acetone:ethyl acetate). Desired product was then precipitated from a hot ethyl acetate/hexane mixture to give the title compound as an off-white powder (58 mg, 53% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.19 (qd, J=12.17, 3.66 Hz, 2 H) 1.62 (d, J=12.88 Hz, 2 H) 1.73 (td, J=6.95, 4.04 Hz, 1 H) 2.27 (s, 4 H) 2.29 (s, 3 H) 2.48 (d, J=6.90 Hz, 1 H) 2.56 (s, 3 H) 2.89 – 3.04 (m, 2 H) 4.48 (d, J=11.87 Hz, 2 H) 5.50 – 5.95 (m, 1 H) 7.01 – 7.13 (m, 4 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.47 (s, 1 C) 12.35 (s, 1 C) 31.68 (s, 1 C) 36.50 (s, 1 C) 41.99 (s, 1 C) 46.29 (s, 1 C) 125.86 (s, 1 C) 128.16(s, 2 C) 129.00 (s, 1 C) 129.04 (s, 2 C) 139.83 (s, 1 C) 157.42 (s, 1 C) 160.54 (s, 1 C) 171.09 (s, 1 C) 171.36 (s, 1 C) 184.13 (s, 1 C) 188.45 (s, 1 C); HRMS: exact mass calculated for C21H24N3O5S [M+H]+, 430.1431; found: 430.1412.
Compounds 5 – 12 were synthesized in an analogous manner to compound 1 using appropriate starting materials as shown in Scheme 1. Purified by reverse phase chromatography on Xbridge C18 OBD 19 mm x 100 mm 5 μm column at 20 mL/min (5–60% acetonitrile in water (0.1% TFA)) to give title compounds as off-white solids.
3,5-dimethyl-N-(2-(4-methylpiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)isoxazole-4-sulfonamide (5)
1H NMR (500 MHz, DMSO-d6) δ ppm 0.91 (d, J=6.31 Hz, 3 H) 1.09 – 1.25 (m, 2 H) 1.53 – 1.64 (m, 1 H) 1.68 (d, J=12.93 Hz, 2 H) 2.31 (s, 3 H) 2.58 (s, 3H) 3.06 (td, J=12.61, 2.21 Hz, 2 H) 4.03 – 4.62 (m, 11 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.47 (s, 1 C) 12.37 (s, 1 C) 21.61 (s, 1 C) 28.95 – 29.92 (m, 2 C) 33.72 (s, 1 C) 46.48 (s, 2 C) 119.34 (s, 1 C) 157.42(s, 1 C) 166.63 (s, 1 C) 171.39 (s, 1 C) 171.44 (s, 1 C) 183.86 (s, 1 C) 188.62 (s, 1 C); HRMS: exact mass calculated for C15H20N3O5S [M+H]+, 354.1118; found: 354.1117.
N-(3,4-dioxo-2-(4-phenylpiperidin-1-yl)cyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (6)
1H NMR (500 MHz, DMSO-d6) δ ppm 1.66 (qd, J=12.51, 4.10 Hz, 2 H) 1.81 (d, J=11.98 Hz, 2 H) 2.28 (s, 3 H) 2.55 (s, 3 H) 2.71 – 2.83 (m, 1 H) 3.09 (t, J=12.14 Hz, 2 H) 4.50 – 4.99 (m, 2 H) 6.82 (br. s., 1 H) 7.16 – 7.22 (m, 1 H) 7.24 – 7.35 (m, 4 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.53 (s, 1 C) 12.19 (s, 1 C) 33.02 (s, 2 C) 41.27 (s, 2 C) 46.03 (br. s., 1 C) 121.28 (s, 1 C) 126.19 (s, 1 C) 126.77 (s,2 C) 128.41 (s, 2 C) 145.62 (s, 1 C) 157.56 (s, 1 C) 169.27 (s, 1 C) 171.36 (s, 1 C) 174.68 (s, 1 C) 185.87 (s, 1 C) 187.60 (s, 1 C); HRMS: exact mass calculated for C20H22N3O5S [M+H]+, 416.1275; found: 416.1265.
N-(2-(3-benzylpiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (7)
1H NMR (500 MHz, DMSO-d6) δ ppm 1.08 – 1.22 (m, 1 H), 1.41 (d, J=9.80 Hz, 1 H) 1.57 – 1.72 (m, 2 H) 1.81 (d, J=3.47 Hz, 1 H) 2.25 (s, 3 H) 2.42 (dd, J=13.56, 8.20 Hz, 1 H) 2.53 (s, 3 H) 2.60 (d, J=5.84 Hz, 1 H) 2.85 – 3.02 (m, 1 H) 3.05 – 3.19 (m, 1 H) 4.14 – 4.57 (m, 2 H) 7.13 – 7.21 (m, 3 H) 7.24 – 7.31 (m, 2 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.49 (s, 1 C) 12.15 (s, 1 C) 24.55 (br. s., 1 C) 29.05 (br. s., 1 C) 37.42 (br. s., 1 C) 45.72 (br. s., 1 C) 46.24 (br. s, 1 C) 51.40 (br. s., 1 C) 121.31 (s, 1 C) 125.86 (s, 1 C) 128.22 (s, 2 C) 128.87 (s, 2 C) 139.45 (s, 1 C) 157.54 (s, 1 C) 169.18 (s, 1 C) 171.45 (s, 1 C) 174.47 (br. s., 1 C) 185.80 (s, 1 C) 187.43 (s, 1 C); HRMS: exact mass calculated for C21H24N3O5S [M+H]+, 430.1431, found: 430.1415.
3,5-dimethyl-N-(2-(4-(4-methylbenzyl)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)isoxazole-4-sulfonamide (8)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.08 – 1.30 (m, 2 H) 1.61 (d, J=12.88 Hz, 2 H) 1.68 – 1.76 (m, 1 H) 2.26 (s, 3 H) 2.28 (s, 3 H) 2.55 (s, 3 H) 2.87 – 3.10 (m, 2 H) 4.48 (d, J=11.87 Hz, 2 H) 5.72 (br. s., 1 H) 7.01 – 7.12 (m, 4 H). HRMS: exact mass calculated for C22H26N3O5S [M+H]+, 444.1588, found: 444.1595.
N-(3,4-dioxo-2-(4-phenethylpiperidin-1-yl)cyclobut-1-en-1-yl)-3,5-dimethylisoxazole-4-sulfonamide (9)
1H NMR (400 MHz, DMSO-d6) δ ppm 1.05 – 1.24 (m, 2 H) 1.39 – 1.61 (m, 3 H) 1.76 (d, J=12.05 Hz, 2 H) 2.27 (s, 1 H) 2.54 (s, 3 H) 2.57 – 2.64 (m, 2 H) 2.94 (t, J=11.67 Hz, 2 H) 4.58 (br. s., 2 H) 7.13 – 7.18 (m, 1 H) 7.19 – 7.22 (m, 2 H) 7.23 – 7.32 (m, 2 H); 13C NMR (101 MHz, DMSO-d6) δ ppm 10.45 (s, 1 C) 12.11 (s, 1 C) 31.97 (s, 2 C) 32.00 (br. s., 1 C) 34.24 (s, 1 C) 37.72 (s, 1 C) 45.68 (br. s., 2 C) 121.29 (s, 1 C) 125.54 (s, 2 C) 128.18 (s, 3 C) 142.21 (s, 1 C) 157.49 (s, 1 C) 169.15 (s, 1 C) 171.31 (s, 1 C) 174.58 (s, 1 C) 185.76 (s, 1 C) 187.57 (s, 1 C); HRMS: exact mass calculated for C22H26N3O5S [M+H]+, 444.1588, found: 444.1587.
N-(2-(4-(4-cyanobenzyl)piperazin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (10)
1H NMR (500 MHz, DMSO-d6) δ ppm 2.28 (s, 3 H) 2.56 (s, 3 H) 3.15 – 3.51 (m, 4 H) 3.73 (br. s., 4 H) 4.41 (br. s., 2 H) 7.69 (d, J=8.20 Hz, 2 H) 7.98 (d, J=8.20 Hz, 2 H) 10.00 (br. s., 1 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.55 (s, 1 C) 12.20 (s, 1 C) 42.51 (br. s., 2 C) 50.59 (br. s., 2 C) 58.52 (br. s., 1 C) 112.38 (br. s., 1 C) 118.37 (s, 1 C) 120.95 (s, 2 C) 132.31 (br. s., 1 C) 132.71 (s, 2 C) 157.54 (s, 1 C) 158.30 (s, 1 C) 169.55 (s, 1 C) 170.85 (s, 1 C) 175.48 (s, 1 C) 186.94 (s, 1 C) 187.06 (s, 1 C); HRMS: exact mass calculated for C21H22N5O5S [M+H]+, 456.1336, found: 456.1334.
N-(2-(4-(4-methoxybenzyl)piperazin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (11)
1H NMR (500 MHz, DMSO-d6) δ ppm 2.28 (s, 3 H) 2.56 (s, 3 H) 3.18 (d, J=8.83 Hz, 2 H) 3.29 – 3.43 (m, 4 H) 3.46 – 3.73 (m, 8 H) 3.78 (s, 3 H) 4.26 (br. s., 2 H) 4.63 (br. s., 2 H) 7.03 (d, J=8.83 Hz, 2 H) 7.41 (d, J=8.51 Hz, 2 H) 9.71 (br. s., 1 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.56 (s, 1 C) 12.21 (s, 1 C) 42.48 (br. s., 1 C) 50.06 (s, 2 C) 55.26 (s, 2 C) 58.86 (s, 1 C) 114.24 (s, 2 C) 120.94 (br.s., 1 C) 120.97 (s, 1 C) 132.88 (s, 2 C) 157.55 (s, 1 C) 160.21 (s, 1 C) 169.54 (s, 1 C) 170.88 (s, 1 C) 175.55 (s, 1 C) 186.98 (s, 1 C) 187.06 (s, 1 C); HRMS: exact mass calculated for C21H25N4O6S [M+H]+, 461.1489, found: 461.1486.
N-(3,4-dioxo-2-(4-phenethylpiperazin-1-yl)cyclobut-1-en-1-yl)-3,5-dimethylisoxazole-4-sulfonamide (12)
1H NMR (400 MHz, DMSO-d6) δ ppm 2.28 (s, 3 H) 2.56 (s, 3 H) 2.93 – 3.05 (m, 2 H) 3.24 (d, J=9.29 Hz, 2 H) 3.35 – 3.39 (m, 2 H) 3.40 – 3.53 (m, 2 H) 3.64 (d, J=11.29 Hz, 2 H) 4.65 (d, J=10.29 Hz, 2 H) 7.29 (d, J=7.28 Hz, 3 H) 7.32 – 7.40 (m, 2 H) 9.72 (br. s., 1 H); 13C NMR (101 MHz, DMSO-d6) δ ppm 10.51 (s, 1 C) 12.15 (s, 1 C) 29.28 (br. s., 1 C) 42.43 (br. s., 2 C) 50.59 (br. s., 2 C) 56.46 (br. s., 1 C) 120.96 (s, 1 C) 126.85 (s, 1 C) 128.63 (s, 2 C) 128.67 (s, 2 C) 136.61 (br. s., 1 C) 157.50 (s, 1 C) 169.47 (s, 1 C) 170.81 (s, 1 C) 175.59 (s, 1 C) 186.95 (s, 1 C) 187.06 (s, 1C); HRMS: exact mass calculated for C21H25N4O5S [M+H]+, 445.154, found: 445.1523.
Compound 13 was synthesized as in Scheme 2.
Scheme 2.

Synthesis of squaramide (13)
tert-butyl-1-(2-(3,5-dimethylisoxazole-4-sulfonamido)-3,4-dioxocyclobut-1-enyl)piperidin-4-ylcarbamate (13a)
A solution of N-(2-ethoxy-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (1b, 650 mg, 2.16 mmol) and tert-butyl piperidin-4-ylcarbamate (490 mg, 2.45 mmol) in tetrahydrofuran (20 mL) was heated to 50 °C for 16 hours, solvents were removed under reduced pressure and the residue was purified by flash chromatography on silica gel (20–100% acetone in ethyl acetate), to give the title compound as a pale yellow solid (670 mg, 1.474 mmol, 68.1 %).
N-(2-(4-aminopiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (13b)
A solution of tert-butyl 1-(2-(3,5-dimethylisoxazole-4-sulfonamido)-3,4-dioxocyclobut-1-enyl)piperidin-4-ylcarbamate (13a, 670 mg, 1.47 mmol) in dichloromethane (20 mL) was treated with trifluoroacetic acid (2 mL, 25.96 mmol), and stirred at room temperature for 17 hours, volatiles were removed and the residue was used without further manipulation. 1H NMR (300 MHz, DMSO-d6) δppm 1.41 – 1.57 (m, 2 H) 1.89 – 2.00 (m, 2 H) 2.25 – 2.31 (m, 3 H) 2.55 (s, 3 H) 3.07 (t, J=11.59 Hz, 2 H) 3.27 (d, J=5.09 Hz, 1 H) 4.59 (br. s., 2 H) 7.83 (br. s., 3 H).
N-(2-(4-(4-fluorobenzylamino)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (13)
A solution of N-(2-(4-aminopiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide, trifluoro acetate salt (13b, 0.35 mmol) in absolute ethanol (10 mL) was treated with triethyl amine (0.25 mL), 4-fluorobenzaldehyde (200 mg), and molecular sieves (4A, powder, activated), was heated to 70 °C for one hour, cooled to room temperature and treated with sodium cyanoborohydride (71 mg), stirred at room temperature for 17 hours, and the resulting suspension was concentrated under reduced pressure, the reside suspended in methanol, filtered, and the filtrate was purified by reverse phase chromatography on Xbridge C18 OBD 19 mm x100mm 5 μm column at 20 mL/min (5–60% acetonitrile in water (0.1% TFA)) to give the title compound trifluoroacetate salt as a white powder. 1H NMR (500 MHz, DMSO-d6) δ ppm 1.56 (qd, J=12.19, 4.41 Hz, 1 H) 2.16 (d, J=10.72 Hz, 2 H) 2.28 (s, 3 H) 2.55 (s, 3 H) 3.05 (t, J=12.45 Hz, 2 H) 3.34 (d, J=4.41 Hz, 1 H) 4.15 – 4.27 (m, 2 H) 4.66 (br. s., 2 H) 7.25 – 7.38 (m, 1 H) 7.54 – 7.63 (m, 1 H) 8.79 (br. s., 1 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.54 (s, 1 C) 12.20 (s, 1 C) 28.34 (s, 1 C) 43.89 (br. s., 2 C) 46.31 (s, 1 C) 53.51 (s, 1 C) 115.56 (s, 1 C) 115.73 (s, 2 C) 121.11 (s, 1 C) 128.32 (d, J=2.75 Hz, 1 C) 128.90 (s, 1 C) 132.37 (d, J=8.25 Hz, 2 C) 157.51 (s, 1 C) 162.41 (d, J=245.61 Hz, 1 C) 169.45 (s, 1 C) 171.40 (s, 1 C) 175.25 (s, 1 C) 186.36 (s, 1 C) 187.49 (s, 1 C); 19F NMR (282 MHz, DMSO-d6) δ ppm −112.64 (s, 1 F) −74.37 (s, 3 F); HRMS: exact mass calculated for C21H23FN4O5S [M+H]+, = 463.1446, found: 463.1439.
N-(3,4-dioxo-2-(4-(4-(trifluoromethyl)benzylamino)piperidin-1-yl)cyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (14)
Synthesized in an analogous manner to 13 using 4-trifluoromethylbenzaldehyde as shown in Scheme 2, isolated as trifluoroacetate salt. 1H NMR (500 MHz, DMSO-d6) δ ppm 1.57 (qd, J=12.30, 4.41 Hz, 2 H) 2.17 (d, J=10.72 Hz, 2 H) 2.28 (s, 3 H) 2.55 (s, 3 H) 3.06 (t, J=12.30 Hz, 2 H) 3.31 – 3.45 (m, 1 H) 4.28 – 4.40 (m, 2 H) 4.62 (d, J=16.71 Hz, 2 H) 7.74 (d, J=8.20 Hz, 2 H) 7.86 (d, J=8.20 Hz, 2 H) 8.89 (br. s., 2H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.53 (s, 1 C) 12.20 (s, 1 C) 28.33 (s, 2 C) 43.90 (br. s., 2 C) 51.33 (s, 1 C) 53.79 (s, 1 C) 121.07 (s, 1 C) 124.05 (q, J=272.20 Hz, 1 C) 125.62 (q, J=3.70 Hz, 2 C) 130.77 (s, 2 C) 136.75 (s, 1 C) 157.50 (s, 1 C) 169.49 (s, 1 C) 171.41 (s, 1 C) 175.11 (s, 1 C) 186.33 (s, 1 C) 187.51 (s, 1 C); 19F NMR (282 MHz, DMSO-d6) δ ppm −61.18 (s, 3F), −74.88 (s, 3F); HRMS: exact mass calculated for C22H24F3N4O5S [M+H]+, 513.1414, found: 513.1407.
tert-butyl-1-(2-(3,5-dimethylisoxazole-4-sulfonamido)-3,4-dioxocyclobut-1-enyl)piperidin-4-yl(methyl) carbamate (15a)
Prepared in a manner analogous to 13a using tert-butyl methyl(piperidin-4-yl)carbamate in Scheme 2 to give the title compound as an orange foam (1.52 g, 3.24 mmol, 69.6 %) 1H NMR (300 MHz, CHLOROFORM-d) δppm 1.47 (s, 10 H) 1.73 (br. s., 4 H) 2.36 (s, 3 H) 2.57 (s, 3 H) 2.61 – 2.81 (m, 3 H) 3.09 (br. s., 2 H)4.68 (br. s., 2 H).
3,5-dimethyl-N-(2-(4-(methylamino)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)isoxazole-4-sulfonamide (15b) was prepared analogously to 13b
N-(2-(4-((4-(difluoromethoxy)benzyl)(methyl)amino)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (15) was prepared in a manner analogous to 13, using amine 15b and 4-difluoromethoxybenzaldehyde. 1H NMR (500 MHz, DMSO-d6) δppm 1.72 – 1.85 (m, 2 H) 2.05 – 2.19 (m, 2 H) 2.29 (s, 3 H) 2.55 (s, 3 H) 2.61 (br. s., 3 H) 3.02 (t, J=12.14 Hz, 2 H) 3.51(br. s., 1 H) 4.14 – 4.26 (m, 1 H) 4.48 (d, J=11.35 Hz, 1 H) 4.63 – 4.95 (m, 2 H) 7.28 (d, J=8.20 Hz, 2 H) 7.31 (t, J=73.80 Hz, 1 H) 7.60 (d, J=8.20 Hz, 2 H) 9.45 (br. s., 1 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.57 (s, 1 C) 12.21 (s, 1 C) 25.23 (br. s., 1 C) 26.64 (br. s., 1 C) 35.31 (s, 1 C) 44.19 (br. s., 2 C) 55.05 (s, 1 C) 61.38 (s, 1 C) 116.11 (t, J=257.98 Hz, 1 C) 118.72 (s, 2 C) 121.17 (s, 2 C) 126.96 (s, 1 C) 133.21 (s, 1 C) 151.76 (br. s., 1 C) 157.57 (s, 1 C) 169.42 (s, 1 C) 171.17 (s, 1 C) 175.07 (s, 1 C) 186.27 (s, 1 C) 187.41 (s, 1 C); 19F NMR (282 MHz, DMSO-d6) δ ppm −74.07 (s, 3F), −74.23 (s, 2F); HRMS: exact mass calculated for C23H27F2N4O6S [M+H]+, 525.1614, found: 525.1618.
N-(2-(4-((4-chlorobenzyl)(methyl)amino)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (16) prepared analogously to 15 using 4-chlorobenzaldehyde. 1H NMR (500 MHz, DMSO-d6) δ ppm 1.68 – 1.89 (m, 2 H) 2.03 – 2.18 (m, 2 H) 2.29 (s, 3 H) 2.55 (s, 3 H) 2.61 (d, J=4.73 Hz, 3 H) 2.96 – 3.11 (m, 2 H) 3.51(t, J=10.56 Hz, 1 H) 4.20 (dd, J=12.93, 7.57 Hz, 1 H) 4.49 (dd, J=12.93, 2.84 Hz, 1 H) 4.74 (br. s., 2 H) 7.57 (s, 4 H) 9.47 (br. s., 1 H); 13C NMR (126 MHz, DMSO-d6) δppm 10.57 (s, 1 C) 12.21 (s, 1 C) 25.18 (s, 1 C) 26.62 (s, 1 C) 35.36 (s, 1 C) 44.12 (br. s., 2 C) 54.99 (s, 1 C) 61.41 (s, 1 C) 121.13 (s, 1 C) 128.92 (s, 2 C) 129.21 (s, 1 C) 133.16 (s, 2 C) 134.45 (s, 1 C) 157.55 (s, 1 C) 169.44 (s, 1 C) 171.14 (s, 1 C) 174.92 (s, 1 C) 186.22 (s, 1 C) 187.41 (s, 1 C); HRMS: exact mass calculated for C22H26ClN4O5S [M+H]+, 493.1307, found: 493.1302.
N-(2-(4-((cyclohexylmethyl)(methyl)amino)piperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-sulfonamide (17) was prepared analogously to 15 using cyclohexane carboxaldehyde. 1H NMR (500 MHz, DMSO-d6) δ ppm 0.84 – 1.04 (m, 2 H) 1.07 – 1.20 (m, 1 H) 1.20 – 1.35 (m, 2 H) 1.57 – 1.85 (m, 6 H) 1.92 – 2.11 (m, 2 H) 2.28 (s, 3 H) 2.55 (s, 3 H) 2.74 (d, J=4.41 Hz, 4 H) 2.80 (dt, J=12.61, 6.31 Hz, 1 H) 2.95 – 3.16 (m, 4 H) 3.41 – 3.52 (m, 1 H) 4.54 – 4.93 (m, 2 H) 8.73 (br. s., 1 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 10.56 (s, 1 C) 12.21 (s, 1 C) 24.73 (s, 1 C) 25.01 (s, 1 C) 25.35 (s, 1 C) 25.49 (s, 1 C) 26.04 (s, 1 C) 29.86 (s, 1 C)30.24 (s, 1 C) 32.66 (s, 1 C) 37.41 (s, 1 C) 44.09 (br. s., 2 C) 58.40 (s, 1 C) 61.63 (s, 1 C) 121.17 (s, 1 C) 157.55 (s, 1 C) 169.39 (s, 1 C) 171.19 (s, 1 C)175.05 (s, 1 C) 186.26 (s, 1 C) 187.41 (s, 1 C); HRMS: exact mass calculated for C22H33N4O5S [M+H]+, 465.2166, found: 465.2162.
Compound 2 was synthesized as in Scheme 3.
Scheme 3.

Synthesis of squaramide (2)
N-(2-butoxy-3,4-dioxocyclobut-1-enyl)benzenesulfonamide (2a)
A solution of 3,4-dimethoxycyclobut-3-ene-1,2-dione (200 mg, 1.41 mmol) and benzenesulfonamide (245 mg, 1.56 mmol) in butan-1-ol (3 mL, 1.41 mmol) was heated to 110 °C for 15 hours. Volatiles were removed and the residue was suspended in dichloromethane, filtered, and the filtrate purified by flash chromatography on silica gel (methanol in dichloromethane) to give the title compound as a pale yellow oil.
N-(2-(4-benzylpiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)benzenesulfonamide (2)
A solution of N-(2-butoxy-3,4-dioxocyclobut-1-enyl)benzenesulfonamide (2a, 70 mg, 0.23 mmol), and 4-benzylpiperidine (0.2 mL, 0.23 mmol) in isopropanol (5 mL) was heated to 65 °C for 3 hours, the reaction was cooled to room temperature, filtered, concentrated, and the residue was purified by flash chromatography on silica gel (methanol in dichloromethane) to give a colorless film, this was triturated with ethyl acetate/hexanes, to give the title compound as a white solid (20 mg, 0.049 mmol, 21.53 %). 1H NMR (500 MHz, DMSO-d6) δ ppm 1.16 (qd, J=12.24, 4.26 Hz, 2 H) 1.58 (d, J=12.30 Hz, 2 H) 1.66 – 1.76 (m, 1 H) 2.51 – 2.53 (m, 2 H) 2.89 (t, J=11.82 Hz, 2 H) 3.31 (s, 2 H) 4.58 (d, J=13.87 Hz, 1 H) 7.13 – 7.20 (m, 3 H) 7.24 – 7.30 (m, 2 H) 7.35 – 7.43 (m, 3 H) 7.83 – 7.95 (m, 2 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 31.85 (s, 2 C) 36.76 (s, 1 C) 42.16 (s, 1 C) 45.51 (br. s., 1 C) 125.81 (s, 1 C) 126.77 (s, 2 C) 127.96 (s, 2 C) 128.13 (s, 2 C) 129.03 (s, 2 C) 129.92 (s, 1 C) 139.91 (s, 1 C) 146.07 (s, 1 C) 171.45 (s, 1 C) 175.18 (s, 1 C) 185.89 (s, 1 C) 187.34 (s, 1C); HRMS: exact mass calculated for C22H23N2O4S [M+H]+, 411.1373, found: 411.1366.
1-(difluoromethyl)-N-(2-ethoxy-3,4-dioxocyclobut-1-enyl)-3-methyl-1H-pyrazole-4-sulfonamide (3a) was synthesized in a manner analogous to 1b following Scheme 1 using 1-(difluoromethyl)-3-methyl-1H-pyrazole-4-sulfonyl chloride
N-(2-(4-benzylpiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-1-(difluoromethyl)-3-methyl-1H-pyrazole-4-sulfonamide (3)
Synthesized in a manner analogous to 1 using 3a in 92% purity. 1H NMR (500 MHz, DMSO-d6) δ ppm 1.09 – 1.28 (m, 2 H) 1.60 (d, J=11.66 Hz, 2 H) 1.74 (ddd, J=10.88, 7.25, 3.63 Hz, 1 H) 2.29 (s, 2 H) 2.61 – 2.69 (m, 1 H) 2.72 – 2.80 (m, 1 H) 2.92 (t, J=11.82 Hz, 2 H) 4.54 (br. s., 1 H) 7.17 (d, J=7.88 Hz, 3 H) 7.25 – 7.31 (m, 2 H) 7.69 (t, J=59.30 Hz, 1 H) 8.36 (s, 1 H); 13C NMR (126 MHz, DMSO-d6) δ ppm 12.38 (s, 1 C) 31.84 (s, 2 C) 36.73 (s, 1 C) 42.14 (s, 1 C) 42.67 (s, 2 C) 109.64 (t, J=248.36 Hz, 1 C) 125.83 (s, 1 C) 128.14 (s, 2 C) 128.25 (br. s., 1 C) 129.05 (s, 2 C) 132.31 (s, 1 C) 139.91 (s, 1 C) 148.96 (s, 1 C) 171.27 (s, 1 C) 174.51 (s, 1 C) 185.85 (s, 1 C) 187.49 (s, 1C); HRMS: exact mass calculated for C21H23F2N4O4S [M+H]+, 465.1403, found: 465.1379.
Compound 4 was synthesized as in Scheme 4.
Scheme 4.

Synthesis of squaramide (4)
3-amino-4-(4-benzylpiperidin-1-yl)cyclobut-3-ene-1,2-dione (4a)
A solution of 3-amino-4-ethoxycyclobut-3-ene-1,2-dione (1a, 1.75 g, 12.40 mmol), DIEA (6.50 mL, 37.20 mmol) and 4-benzylpiperidine (2.83 mL, 16.12 mmol) in DMF (73.3 mL) was heated to 80 °C for 12 hours, after cooling to room temperature, majority of the volatiles were removed under reduced pressure, and the residue was dissolved in hot ethyl acetate, washed with saturated aqueous sodium bicarbonate, dried over magnesium sulfate, and purified by flash chromatography on silica gel (methanol in dichloromethane) to give the title compound as a poorly soluble white solid (730 mg, 21%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.16 – 1.30 (m, 2 H) 1.62 (d, J=11.62 Hz, 2 H) 1.78 (td, J=7.39, 3.66 Hz, 1 H) 2.54 (d, J=7.07 Hz, 2 H) 3.05 (td, J=12.76, 2.53 Hz, 2 H) 3.32 (s, 2 H) 7.13 – 7.23 (m, 3 H) 7.25 – 7.34 (m, 2 H) 7.62 (s, 2 H).
N-(2-(4-benzylpiperidin-1-yl)-3,4-dioxocyclobut-1-enyl)-3,5-dimethylisoxazole-4-carboxamide (4)
A solution of 3-amino-4-(4-benzylpiperidin-1-yl)cyclobut-3-ene-1,2-dione (4a, 66 mg, 0.24 mmol) and sodium hydride (9.77 mg, 0.24 mmol) in tetrahydrofuran (10 mL), was treated with 3,4-dimethylisoxazole carbonyl chloride (100 uL) and stirred at room temperature for 16 hours, quenched with acetic acid (0.5 mL), volatiles removed, and the residue purified by flash chromatography on silica gel (ethyl acetate/hexanes) to give a pale yellow oil, this was triturated with ethyl acetate/hexanes to give the title compound as a pale yellow solid (15 mg, 15.62 %) in 90% purity. 1H NMR (300 MHz, DMSO-d6) δ ppm 1.21 – 1.42 (m, 2 H) 1.56 – 1.76 (m, 2 H) 1.83 (br. s., 1 H) 2.31 (s, 3 H) 2.54 (s, 3 H) 3.05 – 3.25 (m, 2 H)3.63 (d, J=12.24 Hz, 1 H) 4.56 (d, J=12.43 Hz, 1 H) 7.12 – 7.23 (m, 3 H) 7.23 – 7.35 (m, 2 H) 10.88 (s, 1 H)); HRMS: exact mass calculated for C22H24N3O4 [M+H]+, 394.1761, found: 394.1757.
Chemical synthesis of racemic myxopyronin
Myxopyronin was synthesized as reflected in Scheme 5 and procedures detailed below 33, 34.
tert-Butyl(3-iodopropoxy)dimethylsilane (20)
Iodine (174 g, 683.2 mmol, 1.1 equiv) was added at 0 °C to a solution of triphenylphosphine (180 g, 683.2 mmol, 1.1 equiv) and imidazole (127 g, 1863.3 mmol, 3 equiv) in dichloromethane (2.2 L). The reaction was kept in the dark and stirred at 0°C for 10 minutes. A solution of 3-((tert-Butyldimethylsilyl)oxy)propan-1-ol (118 g, 621.1 mmol, 1 equiv) in dichloromethane (1.5 L) was added slowly via addition funnel at 0 °C. The reaction was allowed to slowly warm to room temperature and stirred for 16 hours in the dark. The reaction was quenched with saturated sodium hydrosulfite (24 mL), diluted with water (2 L), extracted with dichloromethane (2 x 0.5 L), dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was triturated with heptanes (2 L) and filtered. The filtrate was concentrated under reduced pressure to give the desired product as orange oil (173.2 g, 93% yield). LCMS (ES, m/z): 299 [M-H]−, 1H NMR (300 MHz, CDCl3): δ 3.59 (t, 2H), 3.20 (t, 2H), 1.99-1.81 (m, 2H), 0.81 (s, 9H), 0.01 (s, 6H).
(E)-Methyl 3-methylhept-2-enoate (21)
To a suspension of copper iodide (24.5 g, 128.4 mmol, 0.63 equiv) in anhydrous THF (300 mL) was slowly added 2.5 M n-butyllithium solution in hexane (103 mL, 256.9 mmol, 1.26 equiv) via an addition funnel at −45 °C. The reaction was allowed to stir at −45 °C for 30 minutes, then was cooled to −78°C. A solution of methyl 2-butynoate (20.0 g, 203.9 mmol, 1 equiv) in anhydrous THF (20 mL) was added drop-wise at −78 °C. The reaction was allowed to stir at −78 °C for 30 minutes, at which point TLC indicated the reaction was completed. The reaction was quenched with by drop-wise addition of methanol (25 mL), followed by saturated ammonium chloride (120 mL). The mixture was extracted with methyl tert-butyl ether (MTBE) (3 x 200 mL), dried over sodium sulfate, filtered, concentrated under reduced pressure. The residue was purified over silica gel using 100% heptanes to give the desired product as an orange oil (23.1 g, 73% yield). LCMS (ES, m/z): 156.1 M+, 1H NMR (300 MHz, CDCl3): δ 5.63 (s, 1H), 3.62 (s, 3H), 2.17 (s, 3H), 1.50-1.20 (m, 6H), 0.99-0.89 (m, 3H).
(E)-3-Methylhept-2-en-1-ol (22)
A solution of 1.0 M lithium aluminum hydride in THF (602 mL, 602 mmol, 2 equiv) was added drop-wise over 1 hour at −40°C to a solution of compound 21 (47 g, 301 mmol, 1 equiv) in anhydrous THF (460 mL). The reaction was allowed to slowly warm to room temperature and stirred for 16 hours. The reaction was cooled to 0 °C and quenched with sodium sulfate decahydrate until bubbling ceased. Celite (10 spatula full) was added to the reaction, which was stirred for 15 minutes. The reaction was filtered, concentrated under reduced pressure at 20 °C and purified on an AnaLogix system eluting with a gradient of 0 to 10% ethyl acetate in pentanes over 40 minutes to give the desired product as clear oil (19 g, 50% yield). LCMS (ES, m/z): 127.8 [M-H]−; 1H NMR (300 MHz, CDCl3): δ 5.43-5.38 (m, 1H), 4.17- 4.13 (m, 1H), 3.71-3.54 (m, 2H), 2.04-1.99 (dd, 1H), 1.74-1.42 (m, 3H), 1.39-1.10 (m, 5H), 0.89 (d, 3H).
(E)-3-Methylhept-2-enal (23)
Tetrapropylammonium perruthenate (TPAP) (1.8 g, 5.16 mmol, 0.06 equiv) was added at 0 °C in one portion to a suspension of compound 22 (11.0 g, 85.9 mmol, 1 equiv), activated 4 molecular sieves (44 g) and N-methylmorpholine-N-oxide (20.1 g, 171.8 mmol, 2 equiv) in dichloromethane (200 mL). The reaction was allowed to stir at 0 °C for 30 minutes, filtered, concentrated under reduced pressure at 20°C and purified on an AnaLogix system eluting with a gradient of 0 to 15% ether in pentanes to give the desired product as clear oil (9 g, 82% yield). The product was used subsequently. LCMS (ES, m/z): 127.2 [M+H]+; 1H NMR (300 MHz, CDCl3): δ 9,99 (d, 1H), 5.88 (d, 1H), 2.21 (t, 2H), 2.16 (s, 3H), 1.52-1.47 (m, 2H), 1.38-1.30 (m, 2H), 0.93 (t, 3H).
3-Oxopentanoic acid (24)
Ethyl propionylacetate (100 g, 694 mmol, 1 equiv) was stirred in a 1N sodium hydroxide solution (694 mL, 694 mmol, 1 equiv) under nitrogen for 24 hours. After being washed with ether (250 mL) the mixture was cooled with an ice bath and acidified to pH 2 with concentrated hydrochloric acid and the mixture was extracted with ethyl acetate (3 x 500 mL). The combined organics layers were dried over sodium sulfate, concentrated under reduced pressure and dried at room temperature in a vacuum oven for 4 hours to give the desired product as a white solid which was used crude into the next step (50 g, 62% yield).
6-Ethyl-4-hydroxy-3-propionyl-2H-pyran-2-one (25)
A solution of compound 24 (144 g, 1.24 mol, 1 equiv) in tetrahydrofuran (1.2 L) was treated portion-wise with carbonyldiimidazole (267 g, 1.65 mol, 1.33 equiv) to control off-gassing. The reaction was stirred under nitrogen for 16 hours at room temperature, when TLC (1:1 ethyl acetate/heptanes) indicated that all the starting material had been consumed. The reaction was acidified with 0.5 M hydrochloric acid to pH 1 and the mixture was extracted with ethyl acetate (3 x 1 L). The combined organics layers were washed with saturated brine (1 L), dried over sodium sulfate and concentrated under reduced pressure. The residue was triturated with a minimal amount of heptanes to remove insoluble solids and the filtrates were concentrated under reduced pressure to give the desired product as clear oil (110 g, 45% yield). LCMS (ES, m/z): 197.1 [M+H]+; 1H NMR (300 MHz, DMSO-d6): δ 6.27 (s, 1H), 3.01 (q, 2H), 2.56 (q, 2H), 1.15 (t, 3H), 1.05 (t, 3H).
(rac)-6-(5-((tert-Butyldimethylsilyl)oxy)pentan-2-yl)-4-hydroxy-3-propionyl-2H-pyran-2-one (26)
To a suspension of diisopropylamine (235 mL, 1679.4 mmol, 3.2 equiv) in anhydrous THF (2.2 L) at −40 °C was added 2.5 M n-butyllithium solution in hexane (672 mL, 1679.4 mmol, 3.2 equiv) via addition funnel. The reaction was allowed to warm to 0°C and stirred for 30 minutes. The solution was cooled to −78 °C, then compound 25 (102.9 g, 524.8 mmol, 1 equiv) in anhydrous THF (700 mL) was slowly added to the reaction via addition funnel at −78°C and allowed to stir for 1 hour. A solution of compound 20 (173.2 g, 577.3 mmol, 1.1 equiv) in anhydrous THF (700 mL) was slowly added via addition funnel at −78 °C, followed by hexamethylphosphoramide (HMPA) (292 mL, 1679.4 mmol, 3.2 equiv). The reaction was allowed to stir at −78 °C for 30 minutes, quenched with saturated NH4Cl (4 L), extracted with ethyl acetate (3 x 1.5 L), dried over sodium sulfate, filtered, concentrated under reduced pressure. The residue was purified over silica gel eluting with a gradient of 0 to 50% ethyl acetate in heptanes to give the desired product as orange oil (59.8 g, 31% yield). LCMS (ES, m/z): 369.3 [M+H]+; δ 5.92 (s, 1H), 3.60 (m, 2H), 3.11 (q, 2H), 2.58 (m, 1H), 1.7 (m, 1H), 1.59 – 1.17 (m, 3H), 1.26 (d, 3H), 1.16 (t, 3H), 0.88 (s, 9H), 0.04 (s, 6H).
(rac)-6-(5-((tert-Butyldimethylsilyl)oxy)pentan-2-yl)-4-hydroxy-1-methyl-3-propionyl-pyridin-2(1H)-one (27)
Compound 26 (59.5 g, 161.8 mmol, 1 equiv) was stirred at room temperature in a 2M methylamine solution in methanol (566 mL, 1132.4 mmol, 7 equiv) for 16 hours. The reaction was concentrated under reduced pressure, diluted with ethyl acetate (1 L), washed with saturated NH4Cl (1 L), saturated brine (1 L), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified over silica gel eluting with a gradient of 0 to 30% ethyl acetate in heptanes to give the desired product as light orange oil (47.9 g, 78% yield).). LCMS (ES, m/z): 382.2 [M+H]+; δ 5.68 (s, 1H), 3.59 (t, 2H), 3.20 (s, 3H), 3.10 (m, 2H), 2.45 (m, 1H), 1.71-1.32 (m, 4H), 1.26 (t, 3H), 1.20 (d 3H), 0.88 (s, 9H), 0.04 (s, 6H).
(rac)-6-(5-((tert-Butyldimethylsilyl)oxy)pentan-2-yl)-3-((2E,4E)-2,5-dimethylnona-2,4-dienoyl)-4-hydroxy-1-methylpyridin-2(1H)-one (28)
A 2.5 M n-BuLi solution in hexane (17.1 mL, 42.7 mmol, 2.5 equiv) was added drop-wise to a solution of compound 27 (6.5 g, 17.1 mmol, 1 equiv) in anhydrous THF (100 mL) at −78 °C. The reaction was allowed to stir at −78 °C for 45 minutes. A solution of compound 23 (7.1 g, 56.3 mmol, 3.3 equiv) in anhydrous THF (50 mL) was added to the above reaction slowly at −78 °C. The reaction was allowed to stir at −78 °C for 30 minutes, warmed to 0 °C and stirred an additional 3 hours. The reaction was slowly poured into saturated NH4Cl (1.3 L), extracted with dichloromethane (3 x 400 mL), dried over sodium sulfate, filtered, concentrated under reduced pressure. The residue was allowed to sit at room temperature for 16 hours, at which point TLC indicated the formation of the product was complete (elimination). The residue was purified on an AnaLogix system eluting a gradient of 0 to 40% ethyl acetate in heptanes to give the desired product as orange oil (1.15 g, 14% yield). LCMS (ES, m/z): 490.2 [M+H]+; δ 6.09 (s, 2H), 5.68 (s, 1H), 3.59 (m, 2H), 3.10 (s, 3H), 2.43 (m, 1H), 2.12 (m, 2H), 2.00 (s, 3H), 1.73 (s, 3H), 1.67-1.26 (m, 8H), 1.19 (d 3H), 0.88 (s, 9H), 0.85 (m, 3H), 0.03 (s, 3H), 0.0 (s, 3H).
(rac)-3-((2E,4E)-2,5-Dimethylnona-2,4-dienoyl)-4-hydroxy-6-(5-hydroxypentan-2-yl)-1-methyl-pyridin-2(1H)-one (29)
Compound 28 (3.7 g, 7.6 mmol, 1 equiv) was allowed to stir in a 3:1:1 mixture of acetic acid (81 mL), THF (27 mL) and water (27 mL) at room temperature for 16 hours. The reaction was diluted with water (700 mL), extracted with ether (3 x 200 mL), washed with saturated brine, dried over sodium sulfate, filtered, concentrated under reduced pressure. The resulting residue was dried in vacuum oven to give the desired product as orange oil (2.87 g, 100% yield). LCMS (ES, m/z): 376.2 [M+H]+; δ 8.63 (s, br, 1H), 6.09 (s, 2H), 5.76 (s, 1H), 3.64 (m, 2H), 3.10 (s, 3H), 2.40 (m, 1H), 2.14 (m, 2H), 1.99 (s, 3H), 1.73 (s, 3H), 1.61-1.24 (m, 8H), 1.20 (d 3H), 0.91 (m, 9H).
(rac)-4-(5-((2E,4E)-2,5-Dimethylnona-2,4-dienoyl)-4-hydroxy-1-methyl-6-oxo-1,6-dihydropyridin-2-yl)pentanal (30)
Pyridine (2.7 mL, 33.44 mmol, 5.5 equiv), followed by Dess-Martin periodinane (9.02 g, 21.28 mmol, 3.5 equiv) was added to a solution of compound 29 (2.28 g, 6.08 mmol, 1 equiv) in dichloromethane (60 mL). The reaction was allowed to stir at room temperature for 3 hours, quenched with saturated NaHCO3 200 mL), extracted with dichloromethane (6 x 100 mL), dried over sodium sulfate, filtered, concentrated under reduced pressure. The residue was purified on an AnaLogix system eluting with a gradient of 0 to 45% ethyl acetate in heptanes to give the desired product as yellow oil (1.18 g, 52% yield). LCMS (ES, m/z): 374.1 [M+H]+; δ 13.6 (s, br, 1H), 9.75 (s, 1H), 6.10 (s, 2H), 5.70 (s, 1H), 3.12 (s, 3H), 2.47 (m, 3H), 2.17 (m, 2H), 2.04 (s, 3H), 1.73 (s, 3H), 1.65-1.21 (m, 6H), 0.87 (d 3H), 0.85 (m, 3H).
(rac)-(E)-Methyl-6-(5-((2E,4E)-2,5-dimethylnona-2,4-dienoyl)-4-hydroxy-1-methyl-6-oxo-1,6-dihydropyridin-2-yl)hept-2-enoate (31)
Methyl 2-(triphenylphosphoranylidene)acetate (1.16 g, 3.48 mmol, 1.1 equiv) was added to a solution of compound 30 (1.18 g, 3.16 mmol, 1 equiv) in dichloromethane (40 mL). The reaction was allowed to stir at room temperature for 16 hours. The reaction was concentrated under reduced pressure and purified on an AnaLogix system eluting with a gradient of 0 to 30% ethyl acetate in heptanes over 30 minutes to give the desired product as yellow oil (1.15 g, 85% yield). LCMS (ES, m/z): 430.2 [M+H]+; 1H NMR (300 MHz, CDCl3): δ 13.6 (brs, 1H), 6.91 (dt, 1H), 6.09 (s, 2H), 5.8 (d, 1H), 5.69 (s, 1H), 3.72 (s, 3H), 3.10 (d, 3H), 2.49-2.40 (m, 1H), 2.25-2.04 (m, 4H), 2.00 (s, 3H), 1.96-1.76 (m, 1H), 1,73 (s, 3H), 1.69-1.50 (m, 1H), 1.49 -1.27 (m, 4H), 1.20 (d, 3H), 0.92 (t, 3H).
(rac)-(E)-6-(3-((2E,4E)-2,5-Dimethylnona-2,4-dienoyl)-4-hydroxy-2-oxo-2H-pyran-6-yl)hept-2-enoic acid (32)
A 1M lithium hydroxide solution (40.2 mL, 40.2 mmol, 15 equiv) was added to a solution of compound 31 (1.15 g, 2.68 mmol, 1 equiv) in THF (160 mL). After stirring at room temperature for 2 days, the reaction was quenched with saturated NH4Cl (200 mL), acidified with 1N HCl (aq.) until pH 2 and extracted with ethyl acetate (3 x 100 mL). The combined organic layers were washed with saturated brine, dried over sodium sulfate, filtered, concentrated under reduced pressure. The residue was dried in a vacuum oven to give the desired product as an orange oil (0.91 g, 84% yield). LCMS (ES, m/z): 403.2 [M+H]+; 1H NMR (300 MHz, CDCl3): δ 8.83 (s, br, 1H), 6.38 (d, 1H), 6.26 (d, 1H), 6.08 (s, 1H), 5.20 (m, 1H), 2.57 (m, 2H), 2.24-2.12 (m, 2H), 2.03-1.98 (m, 2H), 1.94 (s, 3H), 1.82 (s, 3H), 1.76-1.20 (m, 7H), 1.25 (d, 3H), 0.94 (t, 3H).
(rac)-Methyl-((E)-5-(3-((2E,4E)-2,5-dimethylnona-2,4-dienoyl)-4-hydroxy-2-oxo-2H-pyran-6-yl)-hex-1-en-1-yl)carbamate (racemic-Myxopyronin B) (18)
N,N-Diisopropylethylamine (0.46 mL, 2.63 mmol, 2.4 equiv) and ethyl chloroformate (0.23 mL, 2.40 mmol, 2.2 equiv) were added sequentially at 0 °C to a solution of compound 32 (0.44 g, 1.09 mmol, 1 equiv) in anhydrous acetone (22 mL). The reaction was allowed to stir at 0 °C for 1.5 hour. A solution of sodium azide (0.34 g, 5.23 mmol, 4.8 equiv) in water (6 mL) was added and the reaction was allowed to stir at 0°C for an additional 70 minutes. The reaction was quenched with ice-water (50 mL), extracted with toluene (3 x 50 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was allowed to reflux in a mixture of anhydrous toluene (130 mL) for 2 hours and concentrated under reduced pressure. Anhydrous methanol (65 mL) was added and the reaction was allowed to reflux for an additional 1.5 hours. The solvent was removed under reduced pressure and the residue was purified on a preparatory HPLC eluting with a 70:30 mixture of methanol and water containing acetic acid to give the desired product as an orange oil (0.19 g, 40% yield). LCMS (ES, m/z): 432.3 [M+H]+; 1H NMR (300 MHz, CDCl3): δ 7.15 (d, 1H), 6.39 (d, 1H), 6.25 (d, 1H), 6.1 (s, 1H), 5.06-4.99 (m, 1H), 3.66 (s, 3H), 2.71-2.64 (m, 1H), 2,24-2.19 (m, 2H), 2.03-1.98 (m, 2H), 1.93 (s, 3H), 1.82 (s, 3H), 1.76-1.64 (m, 2H), 1.62-1.59 (m, 1H), 1.57-1.28 (m, 4H), 1.25 (d, 3H), 0.93 (t, 3H); HRMS: exact mass calculated for C24H34NO6 [M+H]+, 432.2386; found: 432.2383.
Protein expression and purification
The E. coli RNAP σ70 holoenzyme for crystallization and in vitro transcription assays was prepared as described 30.
X-ray crystal structure determinations of the E. coli RNAP σ70 holoenzyme in complex with squaramides and Myx
Crystals of E. coli RNAP holoenzyme were prepared as described previously 30. The holoenzyme crystals were soaked in a crystallization solution containing 30% PEG400 and 1 mM compounds for 12 hours at 22 °C followed by flash frozen by liquid nitrogen.
The X-ray crystallographic datasets were collected at the Macromolecular Diffraction at the Cornell High Energy Synchrotron Source (MacCHESS) A1 and F1 beamlines (Cornell University, Ithaca, NY), and the data were processed by HKL2000 35. The E. coli RNAP holoenzyme structure 30 was used as the initial model for molecular replacement followed by rigid body and positional refinements with non-crystallographic symmetry by using the program Pheix 36. The resulting maps contained additional electron densities, which allowed squaramides and Myx not present in the initial search model to be built in using Coot 37. Final coordinates and structure factors were submitted to the PDB with ID codes listed in Table 2.
Refinement of the E. coli RNAP holoenzyme structure
The structure of E. coli RNAP holoenzyme published in Murakami 30 (PDB 4IGC) showed discontinued electron density map of switch 2 and therefore did not contain the switch 2 model. This structure was re-refined using TLS refinement, resulted in connectivity improvement of the electron density map of switch 2, allowing building the entire switch 2. The new coordinate has been deposited as PDB 4YG2.
Biological Assays
The transcription-coupled translation assay was performed according to Buurman et al. 27. Antibacterial activity against H. influenzae acrB 27 of biochemically potent compounds was determined according to the CLSI guidelines (Clinical and Laboratory Standards Institute, 2009).
Physicochemical Properties
Plasma protein binding and logD7.4 determinations were performed as described by Reck et al 38. Equilibrium solubility was measured as described 27.
Supplementary Material
Supplemental Figure 1. Gray meshes showing the Fo – Fc electronic density map (4 σ) of the compound 8 (A), compound 14 (B) and MyxB 18 (C) overlays on their chemical models. β (cyan) and β′ (pink) subunit are depicted as a ribbon model. Orientation of these panels are the same as in the Fig. 2A. The trifluoro modification at the benzyl ring of the compound 14 is indicated by a red arrow.
Supplemental Figure 2. Comparison of Myx binding on the T. thermophilus and E. coli RNAP. The structure of the T. thermophilus RNAP – dMyxB complex (PDB: 3EQL) was superposed with the E. coli RNAP – MyxB complex. From the E. coli RNAP – MyxB complex structure, the β (cyan) and β′ (pink) subunit are depicted as a ribbon model and the switch 2 is indicated as red. The Myx are shown as stick models (orange, in the E. coli RNAP complex; green, in the T. thermophilus RNAP complex). The three α-helix bundle contacting with the carbamate chain of Myx are indicated by red asterisks and the RNAP bridge helix (BH) is labeled. (B) Schematic summary of contacts between E. coli RNAP and MyxB. Amino acids belonging to the switches are labeled with those identified by resistance mutations highlighted in blue. Chemical moieties of MyxB are labeled.
Supplemental File: Molecular formula strings discussed in the manuscript
Acknowledgments
We thank the staff at the MacCHESS for supporting crystallographic data collection. This work was supported by NIH grant GM087350 (K.S.M.). Figures were prepared using PyMOL, Molsoft ICM and Maestro element.
Abbreviations used
- RNAP
RNA polymerase
- Myx
myxopyronin
- TT
transcription-coupled translation
- LHS
left hand side
- RHS
right hand side
- SAR
Structure Activity Relationships
- MIC
minimum inhibitory concentrations
- IC50
half maximal inhibitory concentration
Footnotes
PDB ID Codes: PDB IDs are indicated in Table 2.
Supporting Information Available: The Fo – Fc electronic density maps of the compound 8, compound 14 and MyxB in the RNAP complex (Supplemental Fig. 1). Comparison of the Myx bindings on the T. thermophilus and E. coli RNAPs (Supplemental Fig. 2A). Schematic summary of contacts between E. coli RNAP and MyxB. Amino acids belonging to the switches are labeled with those identified by resistance mutations highlighted in blue. Chemical groups of the MyxB are labeled.
Author Contributions: V.M. and K.S.M. crystallized E. coli RNAP complex with inhibitors and determined their X-ray crystal structures. C.J.E. and A.D.F. supported the refinements of the RNAP structures. P.R.F., M.A.F., D.C.M., and C.E.M. designed and synthesized compounds. V.M., P.R.F., C.E.M., E.T.B, and K.S.M. designed experiments. V.M., P.R.F., D.C.M., E.T.B., and K.S.M. wrote the manuscript, and all authors discussed the results and commented on the manuscript.
References
- 1.Fair RJ, Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect Medicin Chem. 2014;6:25–64. doi: 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong W, Bai XC, Brown A, Fernandez IS, Hanssen E, Condron M, Tan YH, Baum J, Scheres SH. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. Elife. 2014;3:e03080. doi: 10.7554/eLife.03080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinsberger S, Husecken K, Groh M, Negri M, Haupenthal J, Hartmann RW. Discovery of novel bacterial RNA polymerase inhibitors: pharmacophore-based virtual screening and hit optimization. J Med Chem. 2013;56:8332–8338. doi: 10.1021/jm400485e. [DOI] [PubMed] [Google Scholar]
- 4.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 5.Villain-Guillot P, Bastide L, Gualtieri M, Leonetti JP. Progress in targeting bacterial transcription. Drug Discov Today. 2007;12:200–208. doi: 10.1016/j.drudis.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 6.Jun SH, Warner BA, Murakami KS. RNA polymerase reaction in bacteria. In: Lane WJLD, editor. Encyclopedia of Biological Chemistry. Academic Press; Waltham: 2013. pp. 167–172. [Google Scholar]
- 7.Murakami KS, Darst SA. Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol. 2003;13:31–39. doi: 10.1016/s0959-440x(02)00005-2. [DOI] [PubMed] [Google Scholar]
- 8.Chakraborty A, Wang D, Ebright YW, Korlann Y, Kortkhonjia E, Kim T, Chowdhury S, Wigneshweraraj S, Irschik H, Jansen R, Nixon BT, Knight J, Weiss S, Ebright RH. Opening and closing of the bacterial RNA polymerase clamp. Science. 2012;337:591–595. doi: 10.1126/science.1218716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho MX, Hudson BP, Das K, Arnold E, Ebright RH. Structures of RNA polymerase-antibiotic complexes. Curr Opin Struct Biol. 2009;19:715–723. doi: 10.1016/j.sbi.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Degen D, Feng Y, Zhang Y, Ebright KY, Ebright YW, Gigliotti M, Vahedian-Movahed H, Mandal S, Talaue M, Connell N, Arnold E, Fenical W, Ebright RH. Transcription inhibition by the depsipeptide antibiotic salinamide A. Elife. 2014;3:e02451. doi: 10.7554/eLife.02451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Degen D, Ho MX, Sineva E, Ebright KY, Ebright YW, Mekler V, Vahedian-Movahed H, Feng Y, Yin R, Tuske S, Irschik H, Jansen R, Maffioli S, Donadio S, Arnold E, Ebright RH. GE23077 binds to the RNA polymerase ‘i’ and ‘i+1’ sites and prevents the binding of initiating nucleotides. Elife. 2014;3:e02450. doi: 10.7554/eLife.02450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aristoff PA, Garcia GA, Kirchhoff PD, Showalter HD. Rifamycins – obstacles and opportunities. Tuberculosis. 2010;90:94–118. doi: 10.1016/j.tube.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Venugopal AA, Johnson S. Fidaxomicin: a novel macrocyclic antibiotic approved for treatment of Clostridium difficile infection. Clin Infect Dis. 2012;54:568–574. doi: 10.1093/cid/cir830. [DOI] [PubMed] [Google Scholar]
- 14.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 15.Molodtsov V, Nawarathne IN, Scharf NT, Kirchhoff PD, Showalter HD, Garcia GA, Murakami KS. X-ray crystal structures of the Escherichia coli RNA polymerase in complex with benzoxazinorifamycins. J Med Chem. 2013;56:4758–4763. doi: 10.1021/jm4004889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon HS, Choi H, Lee M, Glynne RJ, Barnes SW, Walker JR, Batalov S, Yusim K, Feng S, Tung CS, Theiler J, Via LE, Boshoff HI, Murakami KS, Korber B, Barry CE, 3rd, Cho SN. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol Microbiol. 2014;91:1106–1119. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zumla A, George A, Sharma V, Herbert N Baroness Masham of I. WHO’s 2013 global report on tuberculosis: successes, threats, and opportunities. The Lancet. 2013;382:1765–1767. doi: 10.1016/S0140-6736(13)62078-4. [DOI] [PubMed] [Google Scholar]
- 18.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 19.Tupin A, Gualtieri M, Leonetti JP, Brodolin K. The transcription inhibitor lipiarmycin blocks DNA fitting into the RNA polymerase catalytic site. EMBO J. 2010;29:2527–2537. doi: 10.1038/emboj.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neill A, Oliva B, Storey C, Hoyle A, Fishwick C, Chopra I. RNA polymerase inhibitors with activity against rifampin-resistant mutants of Staphylococcus aureus. Antimicrob Agents Chemother. 2000;44:3163–3166. doi: 10.1128/aac.44.11.3163-3166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fruth M, Plaza A, Hinsberger S, Sahner JH, Haupenthal J, Bischoff M, Jansen R, Muller R, Hartmann RW. Binding mode characterization of novel RNA polymerase inhibitors using a combined biochemical and NMR approach. ACS Chem Biol. 2014;9:2656–2663. doi: 10.1021/cb5005433. [DOI] [PubMed] [Google Scholar]
- 22.Kurabachew M, Lu SH, Krastel P, Schmitt EK, Suresh BL, Goh A, Knox JE, Ma NL, Jiricek J, Beer D, Cynamon M, Petersen F, Dartois V, Keller T, Dick T, Sambandamurthy VK. Lipiarmycin targets RNA polymerase and has good activity against multidrug-resistant strains of Mycobacterium tuberculosis. J Antimicrob Chemother. 2008;62:713–719. doi: 10.1093/jac/dkn269. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava A, Talaue M, Liu S, Degen D, Ebright RY, Sineva E, Chakraborty A, Druzhinin SY, Chatterjee S, Mukhopadhyay J, Ebright YW, Zozula A, Shen J, Sengupta S, Niedfeldt RR, Xin C, Kaneko T, Irschik H, Jansen R, Donadio S, Connell N, Ebright RH. New target for inhibition of bacterial RNA polymerase: ‘switch region’. Curr Opin Microbiol. 2011;14:532–543. doi: 10.1016/j.mib.2011.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belogurov GA, Vassylyeva MN, Sevostyanova A, Appleman JR, Xiang AX, Lira R, Webber SE, Klyuyev S, Nudler E, Artsimovitch I, Vassylyev DG. Transcription inactivation through local refolding of the RNA polymerase structure. Nature. 2009;457:332–335. doi: 10.1038/nature07510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay J, Das K, Ismail S, Koppstein D, Jang M, Hudson B, Sarafianos S, Tuske S, Patel J, Jansen R, Irschik H, Arnold E, Ebright RH. The RNA polymerase “switch region” is a target for inhibitors. Cell. 2008;135:295–307. doi: 10.1016/j.cell.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sahner JH, Groh M, Negri M, Haupenthal J, Hartmann RW. Novel small molecule inhibitors targeting the “switch region” of bacterial RNAP: structure-based optimization of a virtual screening hit. Eur J Med Chem. 2013;65:223–231. doi: 10.1016/j.ejmech.2013.04.060. [DOI] [PubMed] [Google Scholar]
- 27.Buurman ET, Foulk MA, Gao N, Laganas VA, McKinney DC, Moustakas DT, Rose JA, Shapiro AB, Fleming PR. Novel rapidly diversifiable antimicrobial RNA polymerase switch region inhibitors with confirmed mode of action in Haemophilus influenzae. J Bacteriol. 2012;194:5504–5512. doi: 10.1128/JB.01103-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeitlinger MA, Derendorf H, Mouton JW, Cars O, Craig WA, Andes D, Theuretzbacher U. Protein binding: do we ever learn? Antimicrob Agents Chemother. 2011;55:3067–3074. doi: 10.1128/AAC.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moy TI, Daniel A, Hardy C, Jackson A, Rehrauer O, Hwang YS, Zou D, Nguyen K, Silverman JA, Li Q, Murphy C. Evaluating the activity of the RNA polymerase inhibitor myxopyronin B against Staphylococcus aureus. FEMS Microbiol Lett. 2011;319:176–179. doi: 10.1111/j.1574-6968.2011.02282.x. [DOI] [PubMed] [Google Scholar]
- 30.Murakami KS. The X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J Biol Chem. 2013;288:9126–9134. doi: 10.1074/jbc.M112.430900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pupov D, Kuzin I, Bass I, Kulbachinskiy A. Distinct functions of the RNA polymerase sigma subunit region 3.2 in RNA priming and promoter escape. Nucleic Acids Res. 2014;42:4494–4504. doi: 10.1093/nar/gkt1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an RNA polymerase II elongation complex at 3. 3 Å resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 33.Hu T, Schaus JV, Lam K, Palfreyman MG, Wuonola M, Gustafson G, Panek JS. Total synthesis and preliminary antibacterial evaluation of the RNA polymerase inhibitors (±)-myxopyronin A and B. J Org Chem. 1998;63:2401–2406. [Google Scholar]
- 34.Lira R, Agrios KA, Doundoulakis T, Simonsen KB, Webber SE, Xiang AX. An efficient synthesis of (±)-myxopyronin B via versatile pyridone intermediates. Heterocycles. 2006;68:1099–1103. [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Afonine PV, Mustyakimov M, Grosse-Kunstleve RW, Moriarty NW, Langan P, Adams PD. Joint X-ray and neutron refinement with phenix.refine. Acta Crystallogr D, Biol Crystallogr. 2010;66:1153–1163. doi: 10.1107/S0907444910026582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D, Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Reck F, Alm R, Brassil P, Newman J, Dejonge B, Eyermann CJ, Breault G, Breen J, Comita-Prevoir J, Cronin M, Davis H, Ehmann D, Galullo V, Geng B, Grebe T, Morningstar M, Walker P, Hayter B, Fisher S. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II: broad-spectrum antibacterial agents with reduced hERG activity. J Med Chem. 2011;54:7834–7847. doi: 10.1021/jm2008826. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Gray meshes showing the Fo – Fc electronic density map (4 σ) of the compound 8 (A), compound 14 (B) and MyxB 18 (C) overlays on their chemical models. β (cyan) and β′ (pink) subunit are depicted as a ribbon model. Orientation of these panels are the same as in the Fig. 2A. The trifluoro modification at the benzyl ring of the compound 14 is indicated by a red arrow.
Supplemental Figure 2. Comparison of Myx binding on the T. thermophilus and E. coli RNAP. The structure of the T. thermophilus RNAP – dMyxB complex (PDB: 3EQL) was superposed with the E. coli RNAP – MyxB complex. From the E. coli RNAP – MyxB complex structure, the β (cyan) and β′ (pink) subunit are depicted as a ribbon model and the switch 2 is indicated as red. The Myx are shown as stick models (orange, in the E. coli RNAP complex; green, in the T. thermophilus RNAP complex). The three α-helix bundle contacting with the carbamate chain of Myx are indicated by red asterisks and the RNAP bridge helix (BH) is labeled. (B) Schematic summary of contacts between E. coli RNAP and MyxB. Amino acids belonging to the switches are labeled with those identified by resistance mutations highlighted in blue. Chemical moieties of MyxB are labeled.
Supplemental File: Molecular formula strings discussed in the manuscript