

Abstract
Around 350 million people worldwide suffer from rare diseases. These may have a genetic, infectious, or autoimmune basis, and several include an inflammatory component. Launching of effective treatments can be very challenging when there is a low disease prevalence and limited scientific insights into the disease mechanisms. As a key trigger of inflammatory processes, complement has been associated with a variety of diseases and has become an attractive therapeutic target for conditions involving inflammation. In view of the clinical experience acquired with drugs licensed for the treatment of rare diseases such as hereditary angioedema and paroxysmal nocturnal hemoglobinuria, growing evidence supports the safety and efficacy of complement therapeutics in restoring immune balance and preventing aggravation of clinical outcomes. This review provides an overview of the candidates currently in the pharmaceutical pipeline with potential to treat orphan diseases and discusses the molecular mechanisms triggered by complement involved with the disease pathogenesis.
Keywords: autoimmune diseases, C1 inhibitor, complement, compstatin, eculizumab, orphan drugs, rare diseases
1. Introduction
The complement system is invariably considered one of the most ancient defense mechanisms of the body [1]. Since the discovery of the bactericidal properties of serum components more than a century ago, the perception of complement as part of immunity has changed considerably [2]. Initially regarded as a mere co-adjuvant in microbial elimination through opsonization and lysis, it is hard to assume that evolutionary forces would conserve such an intricate system comprising about 50 proteins to act entirely on microbial killing. Today, complement is seen not only as a first line of defense against pathogens but also as a modulator of acquired immunity, being the decisive factor that guides the quality and magnitude of cell activation and also orchestrates several important physiological and pathological processes, such as the clearance of foreign bodies, coagulation, tissue regeneration, and inflammation (Fig. 1) [2–4].
Figure 1.
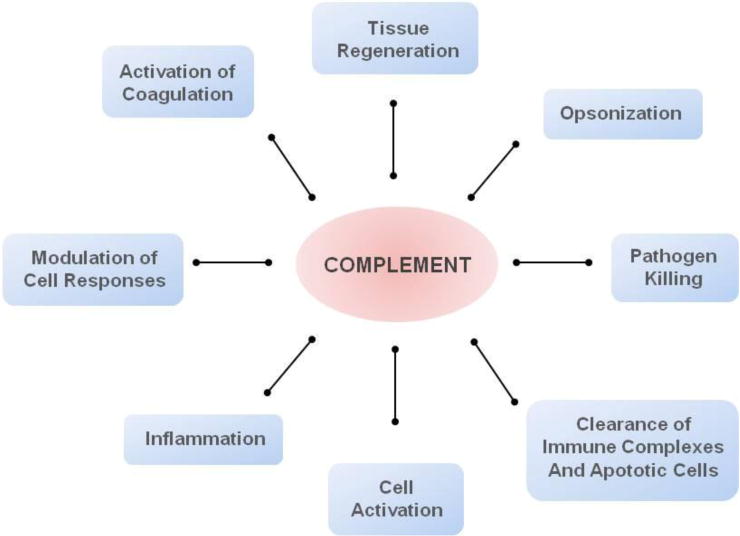
Complement acts as a key mediator of several pathophysiological processes.
The biological functions of complement are elicited as a result of the activation of the classical, alternative, and/or lectin pathways (CP, AP, and LP, respectively). The initial components of each pathway serve as pattern recognition molecules: C1q fixes antigen-antibody complexes, mannose binding lectins (MBL) and ficolins bind to microbial carbohydrates, and molecules of C3b and properdin recognize self-and non-self-structures that are damaged or lack complement-regulatory proteins. The initial trigger for the CP and LP leads to subsequent activation of the components C2 and C4 and the formation of the C3 convertase, C4b2b (Fig. 2). The AP is continuously activated via the spontaneous hydrolysis of C3, resulting in a conformational change that allows the binding of Factor B (FB). Upon cleavage of FB by the serine protease factor D (FD), the C3bBb complex is formed. This complex acts as the C3 convertase of the AP and plays a critical role in the amplification of the AP as a result of the continuous production of C3b molecules (Fig. 2), ensuring an immediate and effective response against danger signals. More recently, properdin has been identified as a pattern recognition molecule that is able to initiate the activation of the AP [5]. In addition, it also acts as a positive regulator of the C3bBb complex, favoring stable and longer activation of the AP. All three pathways converge at the cleavage of C3 by one of the C3 convertases, to generate the fragments C3a and C3b. C5 is similarly activated by C5 convertases (C4b2b3b or C3bBb3b) to produce the fragments C5a, an important inducer of inflammation, and C5b, the initial component of the terminal complement pathway (Fig. 2). The protein fragments produced during this activation cascade can either play a role in further activating the system and/or binding to specific receptors on cell surfaces to induce a functional response. The complement cascade culminates in the formation of the membrane attack complex (MAC: C5bC6C7C8C9n) [2, 6]. In sublytic amounts, particularly on nucleated cells, the MAC significantly affects cell signaling pathways and promotes inflammation [7]. Furthermore, as has been long appreciated, it promotes the osmotic lysis of microbes and of cells lacking proper complement regulation.
Figure 2.
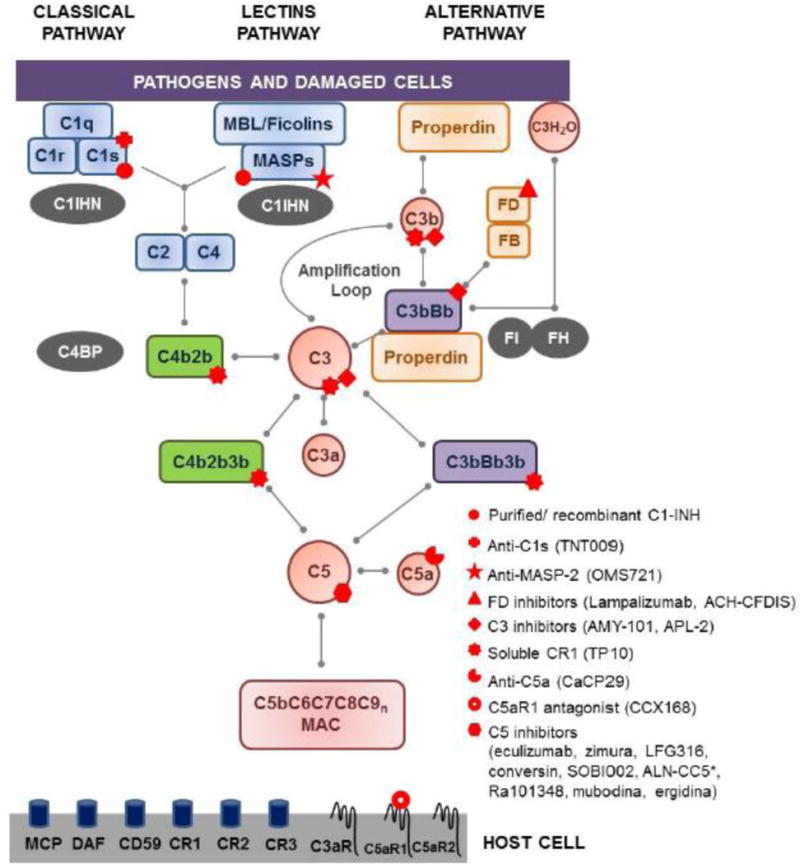
Simplified scheme of the complement cascade. In blue: proteins of the classical and lectins pathways; green: convertases of the classical and lectins pathways; orange: proteins of the alternative pathway; purple: convertases of the alternative pathway; red: proteins common to all the pathways; gray: soluble regulators. Complement cell receptors are depicted at the surface of the host cell. Red symbols: points of therapeutic interventions in rare diseases. Abbreviations: C1INH-C1 inhibitor, C4BP-C4b binding protein, CR-complement receptor, DAF-decay accelerating factor, FB-factor B, FD-factor D, FH-factor H, FI-factor I, MAC-membrane attack complex, MBL-mannose binding lectin, MCP-membrane cofactor protein. *ALN-CC5 inhibits C5 expression.
An essential aspect of complement’s precise operation is the tight regulation of distinct stages of the system. This regulatory task is carried out by several soluble and cell surface-expressed proteins (Fig. 2). The soluble regulators are C1 inhibitor (C1-INH), Factor I (FI), Factor H (FH), and C4b binding protein (C4BP); the regulatory cell receptors are complement receptor type 1 (CR1 or CD35), membrane cofactor protein (MCP or CD46), decay accelerator factor (DAF or CD55), and CD59 [2, 8]. A proper level of regulation prevents uncontrolled activation and amplification of the system and assures the preservation of host cells and tissues while a destructive response is being mounted against the ‘intruder’. In fact, in addition to the presence of a defective complement component or an excessive trigger signal, insufficient regulation is often recognized as a pathogenic driver of complement-related diseases. Indeed, many inflammatory, autoimmune, and infectious diseases have been associated with exacerbated complement activity [9, 10]. More specifically, the discovery of the molecular mechanisms underlying the pathology in dense deposit disease (DDD), paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and age-related macular degeneration (AMD) has revitalized interest in unveiling the mechanisms that drive extreme complement activation in disease. It has also propelled efforts to develop novel strategies for targeting complement activation with the goal of interfering with the disease course.
Influenced by the clinical success of the anti-C5 antibody eculizumab in the treatment of PNH and aHUS, two orphan diseases characterized by an overactive complement system, an array of drugs designed to modulate complement activity is currently in the pharmaceutical pipeline, with the goal of treating rare diseases. In fact, since the implementation of the Orphan Drug Act in 1983, about 400 new drugs have been approved by the Food and Drug Administration (FDA) for rare diseases. In view of the existence of approximately 7,000 different types of rare disease (defined as affecting fewer than 200,000 people in the US) affecting 350 million people worldwide, the introduction of novel therapeutic compounds that could prevent, delay, or even halt the progression of these diseases is highly anticipated by both the medical and patient communities. Here we provide an overview of drug candidates that interfere with complement activation and are currently in clinical development for the treatment of rare diseases.
2. Targeting the Complement System: Where, When, and How Much
Given its upstream position in inflammatory and immunomodulatory processes and the immunological basis of a number of diseases, blockade of complement-mediated functions has long been pursued as a valid therapeutic option [9]. With various potential sites of inhibition to be considered (Fig. 2 and 3), the chosen therapy should reflect a consideration of the pathological mechanisms of the disease, such as the trigger of complement activation, the pathway(s) involved, the location of the complement activation, and the disease course. It is unlikely that the optimal target for a particular pathology will be suited for several distinct classes of disease; furthermore, some conditions may require combined inhibition of two or even all three complement pathways. Also, the delivery route and the point and extent of the inhibition must be carefully defined for each specific indication. Finally, a careful evaluation of adverse effects including drug-related impairment of complement-mediated defense mechanisms should be performed (Fig. 3). Ideally, the optimal therapy should be sufficient to constrain exacerbated complement activation without shutting down necessary biological functions pertinent to immune surveillance or tissue homeostasis. The available data from clinical trials with distinct drug candidates have not yet raised any safety concerns; however, definitive answers to the very relevant questions addressed here will only come after extensive clinical experience has been gained. Some of these issues will be further considered below.
Figure 3.
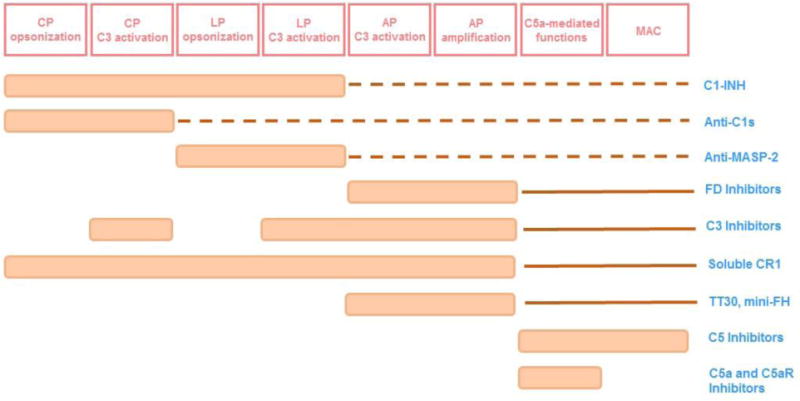
Impact of drug candidates on complement-mediated functions. Abbreviations: AP-alternative pathway, CP-classical pathway, C5aR-C5a receptor 1, C1-INH-C1 inhibitor, FD-factor D, FH-factor H, LP-lectin pathway, MAC-membrane attack complex, MASP-2-mannose binding lectin. Dotted lines indicate impact in the complement functions downstream of C3 due to less generation of C3b. Full lines indicate impact on C5-mediated functions due to inhibited amplification loop.
3. Targeting the Initiators
3.1. C1 Inhibitor
C1-INH is a soluble, highly glycosylated serine protease inhibitor of 110 kDa that circulates with a concentration of ~0.2 mg/ml. Its main function is to control the various proteases involved in the complement, kinin, fibrinolytic, and coagulation systems [11] (Fig. 3). Despite its broader target specificity, preparations of C1-INH are the only complement protease inhibitor currently available in the clinic with a primary indication for the treatment of HAE. However, given the safe and successful outcomes that have been achieved in HAE therapy, C1-INH is now being investigated in trials for several disease states such as sepsis (NCT01766414, NCT00785018, 2011-002222-46), transplant rejection (NCT01134515, NCT01147302), neuromyelitis optica (NMO) (NCT01759602), ischemia-reperfusion injury (IRI) (NCT01886443), and autoimmune hemolytic anemia (2012-003710-13). Four different C1-INH concentrates have been approved by the FDA and European medicine agency (EMA). They are: Berinert (CSL Behring), Cetor (Sanquin), and Cinryze (ViroPharma Biologics) and the recombinant C1-INH concentrate Ruconest (Pharming Group N.V) (Tables 1 and 2).
Table 1.
List of complement-related drugs with orphan status by the FDA and EMEA.
| Compound Name | Manufacturer | Complement Target | Designation Date | Orphan Drug Status | Indication |
|---|---|---|---|---|---|
| Berinert Human C1 esterase inhibitor | CSL Behring | C1r, C1s, MASP-1, MASP-2, other proteases | 10/16/1992-FDA | Approved | Prevention and/or treatment of acute attacks of HAE-Marketed |
| Human C1 esterase inhibitor | Alpha Therapeutic Corporation (now Grifols) | C1r, C1s, MASP-1, MASP – 2, other proteases | 8/21/1996-FDA | Designated | Treatment and prevention of HAE caused by C1-esterase inhibitor deficiency-Undisclosed development stage |
| Ruconest Recombinant C1 esterase inhibitor | Pharming Group N.V. Licenced by Santarus/Salix | C1r, C1s, MASP-1, MASP-2, other proteases | 2/23/1999-FDA | Approved | Treatment of (acute attacks of) HAE caused by hereditary or acquired C1-esterase inhibitor deficiency-Marketed |
| Cinryze Human C1 esterase inhibitor | Viro Pharma Biologics Incorporated (now Shire) | C1r, C1s, MASP-1, MASP-2, other proteases | 7/16/2004-FDA | Approved | Routine prophylaxis against angioedema attacks in patients with HAE-Marketed |
| Ruconest Recombinant human C1 esterase inhibitor | Pharming Group N.V. | C1r, C1s, MASP-1, MASP-2, other proteases | 6/9/2006-FDA | Designated | Prevention and/or treatment of delayed graft function after solid organ transplantation-Phase I |
| Ruconest Recombinant human C1 esterase inhibitor | Pharming Group N.V. | C1r, C1s, MASP-1, MASP-2, other proteases | 6/9/2006-FDA | Designated | Treatment of capillary leakage syndrome-Undisclosed development stage |
| OMS721 Anti human MASP-2 monoclonal antibody | Omeros Corporation | MASP-2 | 12/16/2013-FDA | Designated | Prevention of complement-mediated thrombotic microangiopathy-Phase II |
| APL-2 C3-inhibiting peptide (Pegylated) | Apellis Pharmaceuticals | C3 | 4/20/2014-FDA | Designated | Treatment of PNH-Phase I |
| AMY-101 C3-inhibiting peptide | Amyndas Pharmaceuticals | C3 | 10/9/2014-FDA 10/22/2014-EMEA | Designated | Treatment of PNH-Pre-clinical |
| TP10 (CDX-1135) Soluble complement receptor type 1 | Avant Immunotherapeutics (now Celldex) | C3-convertase, C4b, C3b | 3/6/2000-FDA | Designated | Prevention of post-cardiopulmonary bypass syndrome in children undergoing cardiopulmonary bypass-Phase II |
| Mubodina Recombinant human minibody against component C5 | Adienne S.A. | C5 | 2/4/2009-FDA 10/27/2011-EMEA | Designated | Treatment of primary membranoproliferative glomerulonephritis-Pre-clinical |
| Mubodina Recombinant human minibody against component C5 | Adienne S.A | C5 | 6/7/2011-FDA | Designated | Treatment of aHUS associated with an inherited abnormality of the complement system-Pre-clinical |
| Ergidina Recombinant human minibody against component C5 fused with RGD-motif | Adienne S.A. | C5 | 2/4/2009-FDA | Designated | Prevention of ischemia/reperfusion injury associated with solid organ transplantation-Pre-clinical |
| Eculizumab | Alexion Pharmaceuticals | C5 | 9/21/2000 | Designated | Treatment of dermatomyositis-Phase II |
| Eculizumab | Alexion Pharmaceuticals | C5 | 3/5/2001-FDA | Designated | Treatment of idiopathic membranous glomerular nephropathy-Phase II |
| Eculizumab | Alexion Pharmaceuticals | C5 | 8/20/2003-FDA 10/17/2003-EMEA | Designated/Approved | Treatment of PNH-Marketed |
| Eculizumab | Alexion Pharmaceuticals | C5 | 4/29/2009-FDA 7/24/2009-EMEA | Designated/Approved | Treatment of aHUS-Marketed |
| Eculizumab | Alexion Pharmaceuticals | C5 | 10/18/2011-FDA 4/7/2012-EMEA | Designated/Approved | Treatment of infection-associated hemolytic uraemic syndrome-Phase II/III |
| Eculizumab | Alexion Pharmaceuticals | C5 | 6/24/2013-FDA 5/8/2013-EMEA | Designated | Treatment of neuromyelitis optica-Phase III |
| Eculizumab | Alexion Pharmaceuticals | C5 | 1/10/2014-FDA 2/19/2014-EMEA | Designated | Prevention of delayed graft function after solid organ transplantation-Phase II |
| Eculizumab | Alexion Pharmaceuticals | C5 | 3/26/2014-EMEA | Designated | Prevention of graft rejection following solid organ transplantation-Phase II |
| Eculizumab | Alexion Pharmaceuticals | C5 | 6/13/2014-FDA 7/29/2014-EMEA | Designated | Treatment of myasthenia gravis-Phase III |
| CCX168 C5aR Antagonist | ChemoCentryx | C5aR | 6/2/2014-FDA | Designated | ANCA-associated vasculitis-Phase II |
| CCX168 C5aR Antagonist | ChemoCentryx | C5aR | 11/17/2014-FDA | Designated | Treatment of aHUS associated with an inherited abnormality of the complement system-Phase II |
Table 2.
List of complement-related drugs without orphan designation in pre-clinical and clinical development for rare diseases.
| Compound Name | Manufacturer | Complement Target | Indication |
|---|---|---|---|
| Cetor C1 esterase inhibitor | Sanquin | C1r, C1s, MASP-1, MASP-2, other proteases | Hereditary Angioedema, Acquired C1-inhibitor deficiency |
| TNT009 Anti-C1s Antibody | True North Therapeutics | C1s | Cold Agglutinin Disease Phase l |
| Lampalizumab Anti-Factor D Antibody (FCFD4514S, RG7417) | Genentech | FD | Age-related Macular Degeneration Phase II/III |
| ACH-CFDIS Anti-Factor D inhibitor | Achillion | FD | Pre-clinical |
| CDX-1135 (TP10) soluble CR1 | Celldex | C3-convertase, C4b, C3b | Dense Deposit Disease Phase I |
| TT30 (ALXN1102) | Alexion Pharmaceuticals | C3-convertase, C3b | Paroxysmal Nocturnal Hemoglobinuria Phase I |
| TT32 | Alexion Pharmaceuticals | C3-convertase, C4b, C3b | Pre-clinical |
| Mini-FH | Amyndas | C3-convertase, C3b | Paroxysmal Nocturnal Hemoglobinuria Pre-clinical |
| Zimura (ARC1905) | Ophthotech | C5 | Age-related Macular Degeneration Phase II |
| LFG316 | Novartis | C5 | Dry Age-related Macular Degeneration Phase II |
| Coversin (OmCl) | Volution Immuno-Pharmaceuticals | C5 | Age-related Macular Degeneration Phase I |
| SOBI002 | Swedish Orphan Biovitrum | C5 | Phase I |
| RA101348 | Ra Pharmaceuticals | C5 | Pre-clinical |
| ALN-CC5 (Anti-C5 siRNA) | Alnylam | C5 | Paroxysmal Nocturnal Hemoglobinuria Phase I/II |
| CaCP29 (IFX-1) | InflaRx | C5a | Sepsis Phase II |
Berinert was accorded orphan drug status by the FDA in 1992 (Table 1) and is now marketed in several countries. The active drug consists of purified C1-INH from human plasma. In addition to HAE, Berinert is currently being tested for the prevention of antibody-mediated rejection in human leukocyte antigen (HLA)-sensitized patients. A phase I/II clinical trial completed at the end of 2014 demonstrated that the use of C1-INH is safe in the post-transplant period and may reduce the risk of ischemia-reperfusion injury (IRI) [12].
Like Berinert, C1-INH from human plasma is also the active substance in Cetor [13] and Cinryze. Cinryze, but not Cetor, has held orphan drug status from the FDA for HAE since 2004 (Tables 1 and 2) and has also been tested as an add-on therapy for NMO. A proof-of-concept trial carried out in 10 patients with NMO has demonstrated the safety of C1-INH and promises benefits regarding neurological damage in NMO [14].
In contrast to the compounds described above, Ruconest is a recombinant human protein purified from the milk of transgenic rabbits that has recently been approved by the FDA for acute attacks of HAE. While a lower degree of glycosylation accounts for the shorter half-life of Ruconest (1.6 h vs. >30 h for Berinert, Cetor, and Cinryze) this distinct feature does not have an obvious effect on its clinical efficacy [15, 16]. Along with the FDA approval as an orphan drug for the treatment of HAE (Table 1), it has also been designated for the prevention and treatment of delayed graft function after solid organ transplantation (Table 1); however, the related clinical trial was withdrawn in 2012 (NCT01035593) with the claim that recent improvements in clinical practice have reduced the apparent incidence of antibody-mediated rejection in renal transplantation. Ruconest has also been designated for the treatment of capillary leakage syndrome, a rare and life-threatening condition characterized by episodes of severe hypotension, hypoalbuminemia, and hemoconcentration that develop as a complication of diseases states such as bone marrow/stem cell transplantation, IL-2 therapy, sepsis, and neonatal cardiac surgery; however, no relevant advances or clinical trials have yet been announced by Pharming.
A fifth C1-INH compound, developed by Alpha Therapeutic Corporation, has also received FDA orphan status for the treatment of HAE. No further developments were announced after the company was acquired by Grifols in 2003. While substitution therapy with C1-INH is certainly the most effective treatment for HAE, its elevated costs and laborious production may limit its application for other diseases. However, although the impact is still difficult to evaluate, it is expected that the introduction of Ruconest into the clinic may reduce the therapy costs for HAE.
Hereditary Angioedema
HAE, a potentially life-threatening disease that affects approximately 1:50,000 people worldwide, is associated with an inherited deficiency of C1-INH [17]. HAE can present as low levels of protein (type I) or the production of a dysfunctional protein (type II). HAE type III seems to be independent of C1-INH, and its etiology is still unknown. The disease is characterized by severe episodes of edema in the skin and mucosa that can be extremely debilitating, particularly if the airways are affected. The symptoms are caused by the leakage of fluids from the capillaries as a result of uncontrolled activation of the plasma cascade systems and generation of vasoactive peptides, especially bradykinin [18]. Given its chronic presentation, the disease carries a substantial socio-economic burden, with costs per patient reaching as high as US$230,000 annually [19]. Among the treatment options for HAE, which include androgens, kallikrein inhibitors, bradykinin receptor antagonists, and human C1-INH concentrate, only C1-INH directly targets the fundamental cause of HAE and has been shown to effectively treat and prevent HAE types I and II [18]. Several clinical trials have demonstrated the safety and efficacy of all four C1-INH preparations and have indicated that a threshold baseline level of 0.38 U/ml of the protein is sufficient to protect against HAE attacks [15].
3.2. Anti-C1s Antibody
C1s is an 83-kDa serine protease within the C1 complex, the initiator of the CP. It circulates at a concentration of 0.05 mg/ml as a proenzyme and in complex with C1q, C1r and C1-INH and is critical for the cleavage of the C4 protein, allowing for subsequent activation of the CP (Fig. 2). Deficiency states of this protease are associated with increased susceptibility to infections and autoimmune diseases [20]. Thus far, the clinical experience acquired with the inhibition of complement proteases comes from Nafamostat (FUT-175), currently marketed for pancreatitis in Japan and Korea. However, like C1-INH, Nafamostat inhibits proteases other than C1s, and it is not clear whether its clinical effects are actually derived from complement inhibition [21]. No clinical trials have yet been conducted with a selective strategy for targeting a single initiator of the CP. Although this concept sounds like an interesting approach for diseases resulting from an imbalanced CP, since it leaves the other pathways free to act if required (Fig. 3), it is still not clear whether the disruption of a single pathway is sufficient to alleviate clinical signs. In line with this concept of inhibiting a single pathway, True North Therapeutics has a clinical development program for an humanized anti-C1s antibody (TNT009) for the treatment of cold agglutinin disease (CAD) (Table 2).
Cold Agglutinin Disease
CAD is a type of autoimmune hemolytic anemia in which autoantibodies (cold agglutinins) bind to red blood cells (RBC) at low temperatures to cause RBC agglutination. Anemia occurs as a result of autoantibody-mediated CP activation on the surface of the RBCs, leading to the deposition of complement opsonins that drive extravascular hemolysis in the liver. The annual incidence of CAD is estimated to be between 1/35,000–1/80,000 in North America and Western Europe. The most common symptoms include severe pain in the back and legs, headache, vomiting, diarrhea, dark urine, and hepatosplenomegaly. CAD can be primary (idiopathic) or secondary, caused by an underlying condition, such as infection (Mycoplasma pneumoniae), a lymphoproliferative disorder, systemic autoimmunity, or neoplasm [22]. Mild cases are usually managed non-pharmacologically, i.e., by avoiding cold exposure. In an attempt to treat more severe cases, corticosteroids, alkylating agents, purine nucleoside analogs, and Rituximab have been tested; however, the reported success rate is always below 50% [23].
A recent report has shown that TNT003 (the non-humanized form of TNT009) can prevent cold agglutinin–mediated deposition of C3 fragments onto RBCs and consequent phagocytosis of these opsonized cells in vitro [24]. Furthermore, TNT003 also halted the production of anaphylatoxins, supporting further development of the anti-C1s antibody for use in treating autoimmune hemolytic anemia. TNT009 is the lead drug candidate of True North Therapeutics, and the company is recruiting healthy volunteers and CAD patients to test the safety and tolerability of TNT009. Indeed, it is reasonable to expect that anti-C1s blockage may clinically prevent extravascular and intravascular hemolysis mediated by C3 and the MAC. Because complement activation on RBCs is a typical aspect of antibody-mediated anemias, positive and safe results from this trial could lead to broader application of the anti-C1s antibody.
3.3. Anti-MASP-2 Antibody
The MBL-associated serine protease (MASP)-2, of ~80 kDa, has a concentration of ~0.3 μg/ml in the plasma. Like C1s, it activates C2 and C4 upon binding of MBL and ficolins to carbohydrates [25] (Fig. 2). MASP-2 deficiency has been described in 10 individuals, most of whom are healthy [26]. In contrast, one case report giving more detail has described an individual with increased susceptibility to infections and severe inflammatory conditions [27], indicating that the impact of MASP-2 on pathophysiological mechanisms remains elusive.
Omeros Corporation holds exclusive rights to therapeutic antibodies targeting MASP-2, and the antibody OMS721 (Table 1 and Fig. 3) has received orphan drug designation for the treatment of thrombotic microangiopathy (TMA).
Thrombotic Microangiopathy
The term TMA refers to a group of pathologies that present with endothelial injury and thrombosis in the capillaries and arterioles and may be associated with thrombocytopenia, anemia, purpura, and renal failure. The classic TMAs are HUS and thrombotic thrombocytopenic purpura (TTP). HUS also represents a group of pathologies with similar clinical presentations that are triggered by environmental or genetic factors and have an incidence of 1/100,000. The typical HUS, or STEC-HUS, which accounts for 90% of all cases of HUS, is associated with infection with one of the Escherichia coli strains, which produce Shiga toxin [28]. This type of toxin targets the globotriaosylceramide receptor (Gb3), which is highly expressed by the renal microvascular endothelium and inhibits protein synthesis, causing cell death [29]. In contrast to shiga toxin-producing Escheria coli (STEC)-HUS, atypical HUS (aHUS) results from abnormalities in the control mechanisms of the complement system. More than 100 different mutations have been described in the proteins that regulate complement activation such as FH, MCP, and FI. The proteins C3 and FB have also been implicated in the pathogenesis of aHUS, as well as anti-FH antibodies [28]. TTP, in contrast, is caused by the decreased activity of ADAMTS13, a metalloprotease involved in the cleavage of the von Willebrand factor. In the acquired form of the disease, complement-activating anti-ADAMTS13 antibodies may be responsible for the pathology [30]. The annual incidence of TTP ranges from 1/250,000 to 1/1,000,000. In addition, TMA can be triggered by conditions such as pregnancy, transplantation, and metabolic and autoimmune diseases [31]. Complement dysregulation is considered to be a common factor among TMA diseases and leads to endothelial damage, microvascular thrombosis, and organ damage.
An anti-MASP-2 antibody, OMS721 (Omeros Corporation), is currently being tested for safety and tolerability in phase II trials (NCT02222545 and 2014-001032-11) in TMA patients. OMS721 has received orphan drug designation from the FDA for the prevention of complement-mediated TMAs. Successful trial results are highly anticipated and may validate a role for the lectin pathway in disease pathogenesis that is not yet fully comprehended by the scientific community [32]. In addition, the complement C5 inhibitor eculizumab has been shown to be efficient in the treatment of aHUS, and in some circumstances also in the treatment of HUS and TTP. In fact, eculizumab has been approved by the FDA and EMA for the treatment of aHUS, based on favorable results from separate trials that have demonstrated hematologic normalization, improvement in renal function, and a decrease in thrombotic events [33].
4. Targeting the Amplification Process
4.1. Factor D Inhibitors
Complement FD is a serine protease of 24 kDa with plasma concentrations of ~2 μg/ml, the lowest of all the AP proteins. Given its low concentration, FD is the limiting enzyme for AP activation, being essential for the correct operation of the AP via the activation of FB [34]; therefore, FD blockade is an effective strategy for controlling AP-mediated activation and amplification (Fig. 2 and 3). Cases of FD deficiency have previously been associated with Neisseria infections, and elevated levels of FD have been correlated with numerous disease states [35, 36].
In age-related macular degeneration (AMD), in particular, an increase in the levels of FD has been detected [37], both locally in the eye and systemically, suggesting a potential role for the AP in the pathogenesis of AMD. In support of this concept, Genentech has developed an anti-FD antibody (Lampalizumab) that is currently in phase III trials for the treatment of geographic atrophy (GA), a late-stage form of dry AMD (Table 2). Mechanistically, Lampalizumab binds to a FD site that is essential for the binding of FD with the C3bB proconvertase [38]. Although they are still in the preclinical development phase, a series of small molecules with the ability to inhibit FD have been announced by Achillion Pharmaceuticals (Table 2). The lead compound has shown sub-nanomolar inhibitory potency in in vitro studies, and although used in very high concentrations (100 mg/kg) it was able to abrogate AP activation in non-human primates for more than 24 h after oral administration [39].
Age-related Macular Degeneration
AMD is a late-onset, neurodegenerative disease of the eye that is characterized by progressive degeneration of the macular region of the retina, which results in progressive and irreversible vision loss [40]. AMD is the major cause of blindness in adults over 50 years of age in developed countries; although not a rare disease, it has a tight connection with complement imbalance and the unfolding of the pathology mechanisms has led to increased interest in the development of complement-related therapeutics. It is associated with the progressive deposition of drusen (extracellular debris) in the eye and the consequent death of the retinal pigment epithelial cells (geographic atrophy [GA; dry AMD]) and/or choroidal neovascularization leading to hemorrhage and exudation within the macula (wet AMD); in both cases, vision loss is the ultimate result. In addition to age and smoking, genetic variation in complement genes (FH, C2/BF, C3, and FI) has been implicated as a risk factor [40]. Although wet AMD has been managed with anti-vascular endothelial growth factor (VEGF) treatment, no treatment is available for the dry form of the disease.
Based on compelling evidence that dysregulated complement activation and eye inflammation are a driving force for AMD pathogenesis, blockade of the AP, with consequent inhibition of the AP amplification loop, offers an interesting therapeutic option for treating AMD. A pharmacokinetics study using Lampalizumab in cynomolgus monkeys has shown that therapeutic doses (2 mg/animal) of the antibody injected intravitreally do not affect the AP function systemically [41]. Interestingly, the intravenous injection of high doses (20 mg/animal) of the antibody increased the plasma concentrations of FD by up to 10-fold when compared to pre-injection levels, indicating that the therapeutic dose should be strictly monitored during treatment requiring intravenous administration. A multicenter phase I study to determine the safety, tolerability, and immunogenicity of the antibody has already been performed in patients with GA [42]. Given the promising results, a phase II trial followed (NCT01602120) involving 143 patients with bilateral GA. According to Genentech/Roche reports, the study has shown a 20% reduction in GA lesion progression in patients treated monthly with Lampalizumab, as compared to sham-treated individuals, and a phase III trial is currently evaluating the safety and efficacy of Lampalizumab in delaying the progression of GA. This is the first complement-related therapy for GA to enter phase III.
Apart from Lampalizumab, Apellis is recruiting patients to test the intravitreal application of the C3 inhibitor APL-2-see below (NCT02461771). The C5 inhibitors eculizumab (Soliris; NCT00935883), LFG316 (NCT01255462, NCT01535950, NCT01527500) and Zimura (NCT00709527, NCT00950638) have also been tested for AMD. Eculizumab was used systemically in patients and, although well tolerated, it did not show any improvement in the disease score or drusen volume [43, 44]. Similarly, LFG316 (from Novartis) was well tolerated after intravenous application, but no efficacy was observed in terms of reduction in the need for anti-VEGF treatment (Novartis press release) and the trial was terminated (NCT01624636), pointing toward local AP targeting as a better therapeutic option for AMD. In contrast, based on a phase I trial, Ophthotech reported an improvement in mean visual acuity in patients receiving Zimura intravitreally in combination with anti-VEGF. Although no further trials have been announced for AMD, the company is currently recruiting patients to test the use of Zimura for the treatment of idiopathic polypoidal choroidal vasculopathy (NCT02397954).
5. Targeting the Center
5.1. Peptidic C3 Inhibitors
C3 is the most abundant complement protein in the serum, being present at a concentration of ~1 mg/ml. It is a large protein of 185 kDa that plays a central role in complement activation and is essential for the functioning of all three pathways, as well as for the AP amplification mechanism (Fig. 2). In addition to its role in cascade activation, C3 activation fragments exert diverse biological functions via their cell-surface receptors C3aR, CR1, CR2, CR3 and CR4, complement receptor of the immunoglobulin superfamily (CRIg), DAF, and MCP. C3a is a well-studied pro-inflammatory molecule that acts by signaling via C3aR, a G-protein-coupled receptor (GPCR). It is involved in diverse events that include immune cell activation and promotion of inflammation, mobilization of bone-marrow cells, regulation of lipid metabolism, tissue repair, and angiogenesis [2]. Furthermore, increased levels of C3a are found in several disease states, such as asthma, psoriasis, sepsis, cancer, arthritis, and cardiovascular and renal diseases [45]. Interestingly, recent studies have shown that, contrary to current belief, in some circumstances, C3a offers protection against inflammation by inhibiting C5a-induced neutrophil mobilization and preventing the infection-induced apoptosis of myeloid and lymphoid cells in the spleen [46, 47]. Another essential task conducted by C3 fragments is the opsonization and phagocytosis of microbes and foreign bodies. The C3b fragment and its degradation products constitute a quick and efficient mechanism of body surveillance via tagging of danger-associated molecules and induction of phagocytosis via the cell receptors CR1, CR3, and CRIg in macrophages. Similarly, C3b fragments are responsible for the opsonization of immune complexes, which are subsequently cleared by splenic macrophages. C3b and C3dg fragments have also been shown to regulate the activation and differentiation of T and B cells via the receptors MCP and CR2, respectively [48, 49]. Mutations and polymorphism in the C3 gene have been associated with a risk for diseases such as AMD, aHUS, and DDD as well as the clinical outcome of certain transplantation and vaccination procedures [10].
In light of the multifaceted functions carried out by C3, long-term therapeutic blockage of this component has been regarded with caution, particularly given the susceptibility of C3-deficient patients to certain pyogenic infections. Notably, though, these infections are more frequent during childhood and tend to subside once the patients reach adulthood, perhaps as a result of the acquisition of higher levels of IgG [50]. Furthermore, patients with C3 nephritic factors who present with extremely low levels of C3 do not seem more prone to infections [51]. Undeniably, despite these hypothetical considerations, the therapeutic targeting of C3 is the approach that has the broadest effect because it abrogates all three complement pathways and may therefore benefit diverse disease states that are not completely resolved by the inhibition of a single pathway [52] (Fig.3). Furthermore, the advent of small-sized C3 inhibitors with highly favorable pharmacokinetics profiles (e.g. next-generation compstatin analogs) has made the application of systemic C3 interception possible as it can be swiftly interrupted to allow recovery of C3’s activity in case of acute infections.
The compstatin family of C3 inhibitors comprises a series of cyclic peptides that inhibit the interaction between C3 and the convertases, thereby preventing complement activation along all three pathways [53, 54]. The first C3 inhibitor from the compstatin family tested in the clinic was POT-4 (Potentia Pharmaceuticals). After a successful phase I trial of POT-4 for the treatment of AMD (NCT00473928), phase II studies did not show the expected efficacy, perhaps because of the low dose regimen adopted by the study (NCT01157065, NCT01603043). Also from the compstatin family, both APL-2 from Apellis Pharmaceuticals and the most next-generation molecule AMY-101 from Amyndas Pharmaceuticals have been granted orphan drug status for the treatment of PNH (Table 1). The molecular insights gained regarding compstatin function have led to optimization of the original 13-amino acid peptide and the design of analogs with improved efficacy, stability, and pharmacodynamic properties [54]. Challenging the traditional view that C3 inhibition is unwise and that therapeutic peptides have short half-lives, Cp40 (and the Cp40-based therapeutic AMY-101) have shown a favorable pharmacokinectic profile [55] and promising results in preclinical studies of PNH, periodontal disease, hemodialysis-induced inflammation, and C3 glomerulopathy [52, 54]. One of these analogues (termed Cp40), which showed sub-nanomolar binding affinity for C3, built the base for the development of AMY-101. While the first clinical trial using AMY-101 for the treatment of PNH is expected later in 2015, Apellis is currently recruiting patients to start a phase I study with APL-2 for PNH in patients with a poor response to eculizumab (NCT02264639) and phase II studies for AMD; see above (NCT02461771).
Paroxysmal Nocturnal Hemoglobinuria
PNH is a clonal hematopoietic disorder characterized by hemolytic anemia, bone marrow failure, and thrombotic events [56]. It preferentially affects young adults, with an estimated incidence of 1/1,000,000. PNH occurs as a result of a somatic mutation in the phosphatidyl-inositol glycan class A (PIG-A) gene that affects the biosynthesis of the glycosyl-phosphatidyl-inositol (GPI) anchor in certain hematopoietic stem cells. As a consequence, the expression of GPI-linked cell-surface molecules is impaired [57]. Among these proteins are the complement regulators DAF and CD59. In their absence, the formation of the C3 convertases (DAF) and MAC (CD59) on cell surfaces is not properly regulated, making the erythrocytes susceptible to complement-mediated intravascular lysis [58]. PNH is a life-threatening disease, with death usually occurring as a result of thrombosis, hemorrhage, or infections secondary to bone marrow failure.
Until the beginning of the 2000s, the treatment for PNH was only symptomatic and included transfusions, erythropoietin, glucocorticoids, and anticoagulants. The introduction of the anti-C5 antibody eculizumab into the clinic constituted a breakthrough in the treatment of PNH, since the inhibition of MAC formation prevented intravascular hemolysis. Consequently, eculizumab therapy became the standard of care for PNH patients because it achieved a reduction in the number of transfusions needed (or even eliminated them entirely), as well as a reduction in the number of fatal thromboembolic events [59, 60]. Despite the unquestionable benefits of anti-C5 for PNH therapy, the clinical response toward eculizumab is not uniform. The presence of aplastic anemia and polymorphic variants of the C5 and CR1 genes that render the individual resistant to the drug [61, 62] may account for the variability. Furthermore, patients on eculizumab develop C3-mediated extravascular hemolysis as a consequence of the uncontrolled complement activation upstream of C5 (because of the absence of DAF). Therefore, there is an accumulation of PNH erythrocytes opsonized with C3 fragments, and these erythrocytes are eventually phagocytosed by liver and spleen macrophages [63, 64]. Given this scenario, therapeutic intervention at the level of C3 could offer a more effective approach for PNH. Additional C5 inhibitors (see below, Table 2) are being tested as alternative therapeutics for PNH. A phase I trial (NCT02083666) with the C5-targetting affibody SOBI002 has been suspended because of adverse events, and the company Alnylam is recruiting healthy individuals and PNH patients to evaluate the safety, tolerability, and pharmacokinetics of the anti-C5 RNAi ALN-CC5 in a phase I/II study (NCT02352493).
Alongside the C5 inhibitors, the AP inhibitors TT30 and mini-FH have been shown to completely inhibit the hemolysis of PNH erythrocytes and to prevent the deposition of C3 fragments on the surviving erythrocytes in an in vitro PNH assay [65, 66]. While both of these strategies may be able to prevent both intravascular and extravascular hemolysis in PNH patients, further evidence in human trials is still lacking, since mini-H is still in the pre-clinical development phase, and a clinical trial with TT30 has been prematurely terminated because of enrollment issues (NCT01335165). In addition, as mentioned above, the Cp40-based C3 inhibitor AMY-101 and APL-2 have received orphan drug designation for the treatment of PNH. Cp40 and a PEGylated derivative thereof have been shown to effectively protect PNH erythrocytes from hemolysis and prevent the deposition of C3 fragments in vitro [67]; Apellis Pharmaceuticals is currently recruiting PNH patients with a poor response to eculizumab for a phase I trial (NCT02264639).
5.2. Profiting from the Natural Regulators
An alternative and elegant way to block C3 would be to take advantage of the natural C3 inhibitors found in vivo, such as CR1 and FH. Indeed, the use of complement regulators would offer a more physiologic option, especially in the treatment of chronic diseases. Since CR1 can inhibit all three pathways by acting as a cofactor for FI and promoting the decay of the C3 and C5 convertases (Fig. 3), a recombinant form of soluble CR1 (also known as TP-10 and CDX-1135) (Table 2) has been expressed in Chinese hamster ovary cells and has shown efficacy in reducing myocardial IRI in a rat model [68]. CDX-1135 has been evaluated in clinical studies by Celldex, who performed a phase I trial for DDD (NCT01791686). Although this trial was terminated because of slow enrollment, an earlier phase II trial with TP10 for the treatment of cardiopulmonary bypass syndrome has been completed by Avant (NCT00082121). Although it has been shown to be safe, well tolerated, able to protect vascular function in children undergoing cardiopulmonary bypass and to decrease the incidence of mortality and myocardial infarction in males [69], CDX-1135 is no longer in the pipeline at Celldex. Interestingly, though, the University of Iowa is sponsoring a phase I trial for TP10 in the treatment of DDD (NCT02302755). In a case of compassionate use of TP10, an 8-year-old patient with severe DDD with a rapid progression received seven doses of the inhibitor, which was able to improve the child’s levels of serum C3 and normalize the activity of the terminal pathway, therefore encouraging further studies of its potential use for C3 glomerulopathy [70]. While this compound bears promise with regard to efficiently and selectively inhibiting complement, such a large-sized protein may encounter problems with production and elevated costs.
In an attempt to use the inhibitory potential of FH, the iC3b/C3dg-binding domain of human CR2 was fused with the AP inhibitory domain of human FH to create TT30, of 65 kDa (Alexion Pharmaceuticals) (Table 2, Fig. 3). This elegant design yielded a chimeric protein that is not only able to selectively modulate the AP but also to provide local inhibition of complement only in the cells and tissues affected by C3 deposition [71]. As mentioned above, TT30 has shown promise in models of PHN [65]; however, no further plans for clinical development have been announced by Alexion. In the case of mini-FH, protein engineering was used to express a recombinant molecule containing the FH complement control protein domains (CCP) 1–4 linked to CCP19-20 via a polyglycine linker, with CCP1-4 corresponding to the regulatory domains and CCP19-20 to the C3 binding site. This resulting mini FH molecule, (43 kDa; compared to 150 kDa of the parent protein FH) (Table 2, Fig. 3) retained the regulatory activity of FH and proved to be very efficient in inhibiting the in vitro deposition of C3b on erythrocytes with a PNH-induced phenotype [66]. Despite the promise of such targeted inhibitors, none of the FH-based therapies seems to be in active preclinical or clinical development.
C3 glomerulopathy
According with the latest consensus report by Pickering et al., C3 glomerulopathy refers to a group of rare renal disorders characterized by the presence of C3 and the absence or limited deposition of immunoglobulins in the renal tissue, where the pathology is triggered by the local imbalance in complement activation and leads to proteinuria, hematuria, and renal failure [72]. This condition has an annual incidence of 1/1,000,000. C3 glomerulopathy is subdivided into DDD and C3 glomerulonephritis (C3GN). Whereas DDD presents with mesangial and intramembranous highly electron-dense deposits, C3GN shows isolated and less-dense deposits in the mesangial, subepithelial, subendothelial and intramenbranous areas of the glomeruli [73]. The local complement imbalance is triggered by the presence of nephritic factors: autoantibodies against the C3 convertase that stabilize and prolong the convertase’s half-life, generating a hyperactive AP, autoantibodies against other components of the AP such as FB and FH, or mutations in the AP regulators FH or FI [73, 74]. Half of the patients suffering from this condition progress to kidney failure within 10 years of diagnosis. Further, kidney transplantation is not recommended, because allograft loss and disease recurrence are observed in more than 50% of all cases. Current management of the disease includes drugs for blood pressure control, such as angiotensin-converting enzyme inhibitors, and immunosuppressive therapy [73].
Since C3 glomerulonephritis is a complement-mediated disease, one would expect that anti-complement therapy would improve patients’ quality of life or even resolve all of the symptoms if applied soon after diagnosis. Unfortunately, the phase I clinical trial (NCT01791686) to evaluate the safety and efficacy of CDX-1135 in the treatment of patients with DDD was terminated in March 2014 as a result of declared portfolio prioritization because of slow enrollment and the variable spectrum of potential complement abnormalities in DDD patients.
The antibody eculizumab and the recombinant minibody Mubodina, both targeting C5, have received orphan drug designation for C3 glomerulopathy (Table 1). Although no further progress has been reported for Mubodina, eculizumab has been used in eight patients with C3 glomerulopathy [75] and also tested in six patients enrolled in an open-label study [76, 77]. The anti-C5 therapy resulted in decreased proteinuria and decreased serum levels of albumin and creatinine in seven out of the eight cases, but one patient presented with a progression of the disease. As for the six patients in the open-label study, four showed varied degrees of improvement, but two patients experienced declining renal function [76, 77]. These results indicate that while eculizumab may be beneficial in some cases, other blockade approaches need to be considered.
In line with the notion of alternative approaches, TP10 is currently being tested for DDD (NCT02302755). Also, preclinical evidence has suggested the value of using the C3 inhibitor AMY-101 of the compstatin family to treat C3 glomerulopathy [78]. Because the pathology is indicative of a dysfunctional AP, C3 blockade could constitute a useful treatment strategy for preventing the formation of C3 convertases and the amplification loop. Indeed, the compstatin analog Cp40 prevents complement-mediated lysis of sheep erythrocytes in sera from patients, complement dysregulation in the presence of patient-derived autoantibodies to the C3 and C5convertases, and complement dysregulation associated with disease-causing genetic mutations in in vitro studies [78].
6. Targeting the Effectors
6.1. C5 Inhibitors
Like complement C3, C5 also plays a pivotal role in complement activation, since all three pathways culminate in the cleavage of C5. C5 is ~185 kDa, has a structure similar to that of C3, and is present at ~75 μg/ml in the plasma. Once C5 is cleaved and the fragments C5a and C5b are formed, the C5b fragments attach to the cell membrane, where they participate in the further assembly of the MAC (Fig. 2). Conversely, C5a is released and is able to bind to the C5aR1 receptors that are highly expressed on myeloid cells or to the C5aR2 (also known as C5L2) receptors, which are mainly expressed on neutrophils. Whereas the physiological role of C5aR2 is still debatable, C5aR1 is one of the most-studied molecules in the complement field, reflecting the fact that C5a-induced C5aR1 signaling is a potent trigger of cell activation and inflammation: It stimulates the induction of cytokines, the polarization of T-cell responses, and the exacerbation of many pathological conditions such as cancer, neurologic diseases, sepsis, arthritis, periodontitis, and liver disease, among others [45, 79]. C5 deficiency has been described in approximately 50 individuals worldwide and is associated with recurrent infections caused by Gram-negative bacteria, particularly those of the genus Neisseria. Whereas the MAC, and consequently C5, is important for the immune response against Neisseria because phagocytosis and intracellular killing do not appear to be sufficient to eradicate Neisseria, the impact of the MAC on the control of other bacterial infections is currently under debate [50].
In view of the critical role of C5 in inflammation and MAC formation, targeting of C5 is currently the most-explored complement-related therapeutic strategy (Fig. 3). As of this date, six different molecules that intercept complement at the level of C5/C5a are undergoing clinical trials for the treatment of various diseases, and other inhibitors are close to the clinical phase. Whereas such interest likely reflects the assumption that intervention downstream of C3 would have less impact on host defense function, it may limit the applicability in certain diseases (see above).
Eculizumab (Soliris) by Alexion Pharmaceuticals (Table 1), a monoclonal antibody directed against C5, simultaneously blocks the generation of C5a and MAC assembly. It is the first product licensed specifically to inhibit the complement system and has been the prevailing treatment for PHN and aHUS since 2007 and 2011, respectively. Clinical trials have established the safety and efficacy of this inhibitor for PNH and aHUS [60, 80]; however, as a cautionary measure, individuals under eculizumab treatment are vaccinated against meningitis prior to the treatment. Apart from PNH and aHUS, eculizumab has received orphan drug status for dermatomyositis, idiopathic membranous glomerular nephropathy, NMO, myasthenia gravis (MG), and the prevention of graft rejection following solid organ transplantation (Table 1). To date, this C5 inhibitor has been tested in 28 clinical trials and is currently under evaluation in an additional 25 trials for numerous pathologies. While this drug has undoubtedly revolutionized the treatment of PNH and brought the attention of pharmaceutical companies to complement therapeutics, it has a major drawback: Its elevated cost of ~US$500,000 per patient per year has led to Forbes magazine awarding eculizumab the title of “most expensive drug in the world” [81].
Eculizumab’s success has clearly awakened pharmaceutical companies to complement therapeutics. As such, a number of startup and even some large companies have been investing in the development of anti-C5 molecules. A second C5 antibody, LFG316, developed by Novartis, is in phase II trials for AMD (NCT01527500). Regardless of its initial disappointing results (see above), a new trial is currently evaluating the impact of different dosages of the drug. This compound has also entered phase II trials for panuveitis (NCT01526889). Adienne Pharma and Biotech is developing recombinant minibodies against C5, in which the heavy and light chain variable fragments are fused and linked as a single chain with the IgG hinge region. The resulting compound, Mubodina, has been designated an orphan drug for the treatment of membranoproliferative glomerulonephritis and aHUS (Table 1). In addition, a Mubodina derivative called Ergidina, which bears an arginine-glycine-aspartic acid (RGD) tail to target endothelial cells, has received orphan drug status for the prevention of posttransplantation IRI (Table 1).
Efforts to circumvent the high production cost of antibodies has impelled the development of other classes of molecules with therapeutic capability, such as aptamers, small proteins, affibodies, and RNAi. Zimura (ARC1905), a PEGylated aptamer developed by Ophthotech, has completed phase I trials for AMD (NCT00709527, NCT00950638) and shown positive safety results. Currently, the company is recruiting patients for a phase II trial for the treatment of idiopathic polypoidal chroroidal vasculopathy (NCT02397954). Also, the 16-kDa small protein Conversin (also termed OmCI, from Volution Immuno-Pharmaceuticals) (Table 2), originally isolated from the tick Ornithodoros moubata, is able to prevent MAC-and leukotriene B4-derived effects and has shown promising results in preclinical disease models [82]. One caveat, though, is potential immunogenicity because the molecule is derived from a pathogen; this concern should be carefully considered when advancing to clinical protocols. Also, the small molecule SOBI002 (12 kDa) based on the Affibody platform, by Swedish Orphan Biovitrum, is fused to an albumin-binding protein aimed at increasing the half-life of the molecule (2 weeks) [83]. Unfortunately, the phase I trial to test the safety, tolerability, and pharmacokinetics of the compound in healthy volunteers bas been recently suspended because of adverse events (NCT02083666), and no further development has been announced. Finally, the RNAi ALN-CC5, by Alnylam Pharmaceuticals, is being tested in healthy volunteers and PNH patients in a phase I/II trial (NCT02352493), and preliminary results from 12 healthy volunteers have shown acceptable safety and efficacy [84]. It is still unclear, though, whether this therapy reaches the level of effectiveness achieved by eculizumab.
C5 inhibitors are also being tested for the treatment of several other orphan diseases, as discussed below.
Dermatomyositis
Dermatomyositis is an idiopathic inflammatory myopathy characterized by skeletal muscle inflammation that causes skin lesions and muscle weakness. It presents at an annual incidence of 9/1,000,000 [85] and is associated with high morbidity, given that the development of muscle weakness adversely affects physical capacities over time, with potential involvement of the pulmonary, gastrointestinal, and cardiac systems. Furthermore, patients diagnosed at an older age or with rapid onset of the disease, the presence of skin necrosis, or a low baseline level of complement C4 have an increased risk of developing malignancy [86].
The exact pathogenesis of dermatomyositis has not yet been elucidated; however, patients usually present with autoantibodies directed in particular against synthetases and nuclear factors; environmental triggers such as infections may also play a role in the disease. Complement is implicated in the disease, since hyperactivation and MAC formation lead to lysis of endothelial cells and damage to the capillaries. As a result, the number of capillaries is reduced throughout the muscle, which therefore suffers from ischemia. Complement dysregulation also causes a local inflammatory environment by triggering the release of pro-inflammatory cytokines and the recruitment of inflammatory cells [87]. The therapeutic goal is to improve muscle strength and enable the individual to perform daily activities. As with the majority of autoimmune diseases, current treatment aims at controlling the exacerbated inflammation with corticosteroids and/or immunosuppressants [88]. The use of high-dose intravenous immunoglobulin (IVIg) has also proved effective [89].
In 2000, eculizumab was granted orphan drug status from the FDA to treat dermatomyositis (Table 1). A pilot study (NCT00005571) was then conducted to evaluate the safety and efficacy of the drug on 13 patients with the disease undergoing concomitant treatment with moderate doses of methotrexate or steroids. Though the trial results were positive regarding safety and tolerability, no further information was released by the company. Nevertheless, eculizumab has been used to treat a 16-year-old girl with severe symptoms of dermatomyositis and TMA who failed to respond to a variety of corticosteroids, immunosuppressants, and IVIG [90]. Treatment with eculizumab was effective in improving her clinical condition and biological parameters, indicating the importance of further studies of complement-related therapy for dermatomyositis.
Neuromyelitis Optica
NMO is an inflammatory disease of the central nervous system characterized by demyelination of the spinal cord and optical nerve; it has an incidence of 0.05–0.4/100,000. Patients present with severe, acute attacks of paralysis, blindness, and sensory impairment. The mean age of onset ranges from 32.6 and 45.7 years, and the disease is more frequent in females than males [91]. The disease is characterized by the presence of autoantibodies against the astrocyte water channel protein aquaporin-4 [92]. These autoantibodies activate the CP on the astrocytes, which are devoid of complement regulators such as DAF, MCP, and CD59 [93]; therefore, they cause a local inflammatory response with infiltration of granulocytes and macrophages, leading to oligodendrocyte damage, demyelination, and neuronal death [94]. It is noteworthy that in animal models, IgG from NMO patients is unable to cause inflammation, demyelination, or neuronal death in the absence of complement [95]. Furthermore, the concentrations of the activation product C3a are directly correlated with the severity of the neurological disability and the levels of anti-aquaporin antibodies [96, 97].
Current NMO therapy is focused on the control of acute disease, improvement of neurological disability, and prevention of future attacks. The standard procedure follows the use of corticosteroids and plasma exchange for acute and chronic cases, with the addition of immunosuppressants if required [98]. Eculizumab has recently been tested in a pilot study in 14 female NMO patients (average age: 41.1 years) who were positive for aquaporin antibodies. Overall, the anti-C5 therapy was well tolerated, reduced the attack frequency, and stabilized or reduced the neurological disabilities in the patients. During the treatment, 12 of the 14 patients had no attacks, and no patient presented with any worsened disability [99], indicating that complement has a pivotal role in the pathogenesis of NMO. Notably, the patients were off the usual therapy with corticosteroids and immunosuppressants during the trial. A phase III trial is currently being conducted to evaluate the safety and efficacy of the drug in patients with relapsing disease (NCT01892345).
The safety of C1 INH has also been evaluated in 10 NMO patients. Here, C1-INH was added to the current standard therapy, i.e. high doses of corticosteroids. The combined therapy was safe, and although the trial was not designed to measure efficacy, the scores for disease severity decreased from a median of 4.5 on admission to 4.0 on discharge, and then down to 2.5 at the 30-day follow-up, when they did not differ from the median baseline score [14]. Whereas no comparison can yet be drawn regarding the efficacy of anti-C5 versus anti-C1 inhibition strategies, these trials clearly indicate the potential for targeting complement in NMO disease.
Myasthenis gravis
MG is a heterogeneous autoimmune disorder characterized by a defective postsynaptic neuromuscular transmission that results in fatigable weakness of the voluntary muscles. It has an incidence of 1/250,000 and affects mainly females before the age of 40 and both males and females after the age of 50. It is caused by the presence of autoantibodies against components of the neuromuscular junction, specifically, the anti-acetylcholine receptor (AChR), anti-muscle-specific receptor tyrosine kinase (MuSK) and/or anti-low density lipoprotein receptor-related protein 4 (LRP4). The muscles that control the eyes and eyelid movement are particularly susceptible to weakness, but other muscles for chewing, talking, and swallowing are frequently involved. In addition, the respiratory muscles can be affected, leading to acute respiratory failure [100].
Approximately 90% of the patients with generalized MG have antibodies against the AChRs, which are essential for muscle contraction. These antibodies are mainly of the IgG1 and IgG3 isotypes and trigger the pathology by direct blockade of the ACh binding site, cross-linking of the AChRs, and endocytosis and degradation of the receptors and activation of the complement system [101]. In contrast, the MuSK antibodies are of the IgG4 isotype and are thus unable to activate complement. Unfortunately, anti-MuSK MG is associated with an increased risk of myasthenic crises, and these patients do not respond well to acetylcholinesterase inhibitors and thymectomy (although Rituximab appears to be more effective in these patients than in patients with AChR MG) [102]. The third type of antibody, the anti-LRP-4 MG, accounts for 1% of all generalized MG [103] and, like the AChR type, the antibodies are of the IgG1 isotype, and complement is involved in the damage to the postsynaptic membranes [101].
Approximately 10% of all MG patients have a thymoma, and 60% present with thymic hyperplasia, which may play a role in the initiation of the disease through the expression of self-antigens and the impaired negative selection of auto-reactive T cells [101]. Whereas it is well appreciated that thymic cells produce AChR antibodies [104], thymectomy does not eliminate the antibody production, indicating the presence of other sources of autoantibodies in MG. These patients usually respond well to acetylcholinesterase inhibitors and immunosuppressive drugs, and thymectomy may be recommended in some cases [105].
The involvement of complement in MG pathophysiology is well appreciated. Apart from the presence of the MAC at the neuromuscular junction during the disease, the blockade of complement components such as C1q, C3, C5, and C6 has been shown to protect against experimental MG [106]. Furthermore, the absence of the complement regulators DAF and CD59 is directly correlated with a more severe course of the disease during experimental MG [107]. Whereas complement targeting is not expected to work as therapy for patients positive for MuSK autoantibodies, complement intervention may be beneficial for the other cases. A phase II trial has been conducted to test the safety and efficacy of eculizumab in patients with severe, generalized, and refractory MG who are positive for AChR autoantibodies. Fourteen patients were enrolled and received treatment for two periods of 16 weeks in a cross-over fashion with eculizumab or placebo. The results were promising, showing significant improvement in the clinical manifestations [108] and, as a result, a phase III trial is currently recruiting patients (NCT01997229).
Guillain-Barré Syndrome (GBS)
The term GBS refers to a variety of post-infectious neuropathies that are clinically heterogeneous and predominantly seen in males, with an incidence of 1/100,000. GBS typically occurs after a bout of infectious disease in which antibodies that crossreact with gangliosides at nerve membranes are generated, causing nerve damage or impairment of nerve conduction. The type and location of the infection determine the type and clinical course of GBS. Serological studies have shown that Campylobacter jejuni is the most frequent antecedent infection, but other associated infections include cytomegalovirus, Epstein-Barr virus, hepatitis E virus, Mycoplasma pneumonia, and Haemophilus influenza [109]. The most characteristic clinical feature is limb weakness. Lumbar and muscle pain, autonomic dysfunction, and respiratory problems are also observed. Currently, IVIg and plasma exchange are the mainstays of treatment, and the prognosis is variable, depending on the form of GBS, and the outcomes can range from complete recovery to death [109, 110].
It has been shown that C. jejuni, Haemophilus, and cytomegalovirus have ganglioside-like structures that may be responsible for the production of antibodies that crossreact with gangliosides on the nerve cells. Such antibodies are of the IgG1 and IgG3 subclasses, which activate complement leading to MAC formation and take part in the myelin destruction and local inflammatory response that includes infiltration of macrophages. Whereas the proinflammatory effects of C3a and C5a are not excluded in this disease, animal studies have shown that the major acute injury only occurs in the presence of the MAC, and an injury model has been formulated in which an uncontrolled influx of calcium through the MAC results in mitochrondrial and cytoskeletal dysfunction [111].
Because studies have shown that eculizumab prevents the electrophysiological sequelae and lesions when anti-ganglioside antibodies are incubated with neuromuscular junctions in vitro, with normal human serum as a source of complement [111], a phase II trial has been planned and is currently recruiting patients to evaluate the safety and efficacy of eculizumab in the treatment of GBS (NCT02029378).
Multifocal Motor Neuropathy (MMN)
MMN is a motor neuropathy characterized by progressive distal asymmetric limb weakness that usually starts and predominates in the upper limbs, with minimal or no sensory impairment, and follows a chronic evolution. The onset usually occurs before the age of 50, and males are predominantly affected, with a prevalence of 0.6/100,000. The morbidity linked to the disease is variable and can lead to a moderate-to-major impairment of daily activities, including walking ability. Although the pathophysiological mechanisms are not fully understood, the disease is often associated with serum IgM antibodies against the ganglioside GM1. MNN does not respond well to corticosteroids and, plasmapheresis and IVIg, though the first-line therapy offer only transient results, often requiring repeated infusions [112, 113].
An animal model of MMN is not available, but serum from patients with anti-GM1 IgM antibodies is known to trigger complement activation in vitro. The extent of the complement deposition is correlated with the antibody titer and is reduced after IVIg treatment in a dose-dependent manner [114]. It is likely that complement is involved in the pathological mechanisms of MMN as it is in GBS disease. An open-label trial in 13 MMN patients has been conducted to evaluate whether co-administration of eculizumab with high-dose IVIg is safe and efficient. Whereas complement targeting abrogated MAC formation in the plasma regardless of the concomitant use of IVIg, and it resulted in increased muscle strength, it did not affect the requirement for IVIg, indicating that anti-C5 therapy alone is not sufficient to prevent the disease, at least with such a short treatment period Fitzpatrick [115]. No further studies have been announced in this area.
Transplantation
Complement has long been implicated in the outcome of solid organ transplantation, and the activation product C4d is currently being used as a biomarker of chronic antibody-mediated rejection (AMR) [116]. Both the tissue injury induced by IRI and the rejection episodes are strongly correlated with excessive complement activation and the associated consequent trigger of diverse inflammatory pathways. Both systemic complement and components locally produced by the transplant organ and infiltrating immune cells appear to play critical roles. Indeed, C3 synthesis in the transplanted kidney was shown to be increased and dependent on the duration of cold ischemia [117]. C3 production by the donor kidney tipically reaches a peak after reperfusion, indicating the potential for complement inhibition during organ collection as means of decreasing tissue damage and improving the transplant outcome. IRI is particularly critical in the organs collected after circulatory arrest of the donor, since local production of complement proteins and increased levels of pro-inflammatory markers have been observed in the kidneys of deceased donors [118]. Several inflammatory pathways participate in the genesis of IRI, and complement is a key modulator of most, if not all, of these processes [119]. The resulting cellular injury is mainly attributed to the terminal pathway products C5a and C5b–9, with consequent release of pro-inflammatory factors, neutrophil degranulation, and endothelial cell activation and death [120, 121]. While current evidence strongly favors a role for the AP and LP in triggering complement-mediated IRI, the role of the CP is still controversial, and key information, including which complement components are involved in the injury and where they are produced and activated, still remains unknown [121].
In transplantation settings, complement activation is observed not only in response to IR but also during episodes of cellular and antibody-mediated rejection. Increased levels of complement activation products have been detected in the plasma, urine, and transplanted organ in patients showing rejection of cardiac or renal transplants. During episodes of cell-mediated rejection, the transplanted kidney contributes with 10% (5% in physiologic conditions) of the circulating pool of C3 [122]. Imbalanced complement activation leads to direct deposition of MAC and indirect damage to the renal parenchyma, by enhancing T cell responses against the donor organ [117, 123]. Furthermore, in episodes of AMR, pre-formed or induced antibodies present in the host recognize antigens in the graft and activate complement and inflammation, ultimately leading to graft rejection [124]. Notably, C3 fragments have been shown to modulate B-cell activation and antibody production upon signaling via CR2 in B cells [125]. Episodes of antibody-mediated rejection, as observed in ABO-incompatible and HLA-sensitized transplant patients, are particularly of concern, since they result in catastrophic hyperacute AMR and an extremely high rate of graft loss [124].
In line with these findings, complement inhibition during transplantation has been shown to be effective in diminishing IRI and increasing graft survival in several animal models, and the efficacy of complement-targeted therapeutics in clinical settings of transplantation is currently being evaluated [116]. The use of Eculizumab also has showed promising results, for preventing AMR in patients with HLA-specific antibodies and reversed established rejection [126, 127]. Furthermore, in a presensitized non-human primate (NHP) model of kidney transplantation, complement depletion with cobra venom factor (CVF) resulted in organ accommodation; i.e., endothelial cell resistance to antibody-mediated destruction, and long-term engraftment [128]. Despite these promising results, further work is still needed to better understand the mechanisms by which complement influences the balance between graft tolerance and rejection and to characterize the most efficient targeting approach for improving kidney transplant outcomes. Eculizumab, ergidina and ruconest have been granted with orphan drug status for the prevention of injury, delayed graft function or rejection after transplantation (Table 1). Ten distinct clinical trials are underway to test eculizumab for AMR and delayed graft function mainly in kidney transplantation. In addition, the C1-INH Berinert has been shown to be safe and well tolerated when used after kidney transplantation in HLA sensitized patients [12] and a second trial is currently recruiting patients to evaluate the efficacy of Berinert in preventing delayed graft function and IRI (NCT02134314).
6.2. C5a and C5aR Inhibitors
As an alternative to the prevention of C5 cleavage, a distinct strategy to interfere with the effector functions of complement is the abrogation of C5a-mediated effects. This approach may offer clinical benefits for diverse conditions in which inflammation is a major driving force for the disease. It is particularly interesting because the targeting of C5a can hamper inflammation without affecting either opsonization or MAC-mediated functions (Fig. 3). Blocking the C5a-C5aR interaction with small molecules has long been pursued as a therapeutic strategy [129, 130]. However, thus far none of these molecules has produced the expected clinical results, and most have not survived the process of clinical development. The small molecule CCX168 (from ChemoCentryx) (Table 1) is the only C5aR antagonist that is currently being tested in clinical trials. It holds an orphan drug designation for anti-neutrophil cytoplasmic antibody (ANCA) vasculitis and aHUS and is in phase II trials for ANCA vasculitis (NCT01363388, NCT02222155) as well as IgA nephropathy (NCT02384317) and aHUS (NCT02464891). CaCP29 (IFX-1, from InflaRx) (Table 2) is a monoclonal antibody against C5a that has completed a phase I trial (NCT01319903) for safety evaluation and is currently in phase II testing for the treatment of sepsis (NCT02246595). Of note, C5a has an extremely rapid turnover, which may undermine the therapeutic capacity of the C5a inhibitors.
ANCA-associated Vasculitis
ANCA-associated vasculitis comprises a group of clinical heterogeneous disorders characterized by inflammation of the small blood vessels and anti-neutrophil antibodies directed against either proteinase 3 or myeloperoxidase [131]. Although the clinical features are heterogeneous and reflect the location of the vessels affected, some common symptoms include fever, anemia, weight loss, rash, synovitis, and involvement of the respiratory tract and kidneys [132]. As is true for other heterogeneous and immune-related diseases, treatment is based on corticosteroids and/or immunosuppressants and plasma exchange in more severe cases [131].
Both autoantigens, proteinase 3 and myeloperoxidase, are typically retained within neutrophilic granules. When an inflammatory stimulus such as a pro-inflammatory cytokine, complement activation, or ANCA-IgG production occurs, the extracellular expression of the two antigens is increased, leading to the recruitment of inflammatory cells and further activation of neutrophils, with subsequent organ damage and dysfunction [133, 134]. Because IgGs are absent or only scarcely found in the affected tissues [132], ANCA-induced neutrophil activation is considered the main factor in the disease pathogenesis. In line with this concept, C4-deficient mice are not protected against the disease, whereas FB or C5 deficiency confers protection [135]. Furthermore, FB and properdin have been detected in the glomeruli of patients, indicating a role for the AP in renal damage; also, systemic activation of AP has been demonstrated in the plasma of patients with active disease [136, 137]. In addition, C5aR is essential for the development of the disease [134], since C5a has been defined as a key factor in the priming of neutrophils, with consequent expression of the autoantigens proteinase 3 and myeloperoxidase [138–140].
Based on our understanding of the pathophysiological mechanisms of the disease, therapeutic intervention targeting either the AP or the C5a-C5aR interaction seems a logical approach. Indeed, a phase II trial is recruiting patients to test the safety and efficacy of the C5aR antagonist CCX168 in the treatment of ANCA-associated renal vasculitis (NCT0136388). A second trial with eculizumab was withdrawn because of difficulties in enrolling patients (NCT01275287).
7. Conclusions
The perception of the complement system as a critical factor that tips the balance between homeostasis and pathological states has brought new enthusiasm to the field of complement. Aided by animal models and genomic and structural studies, the unfolding of complement-related mechanisms that drive health and disease has advanced considerably in the past decade. The acquired knowledge has prompted the design of distinct and, in some cases, very creative strategies to interfere therapeutically at various stages of the cascade. Some of these approaches, such as the use of C1-INH, anti-C1s and anti-MASP antibodies, are very specific, aiming at the disruption of a single pathway. While it is likely that the immune response will be less compromised by this approach, it remains to be determined whether such specific targeting will be sufficient to induce relevant clinical benefit, and in which disease states. More comprehensive strategies that envisage the blockade at the level of C3 may fulfill the requirement for more robust inhibition in cases in which more than one pathway is involved in the pathology. In addition, strategies based on complement regulators that are directed to sites of complement activation may offer a type of intervention closely resembling the physiological control of complement. In addition, the abrogation of C5-mediated effects is a good option for diseases in which the MAC and/or C5a is the driving force.
The clinical experience acquired with the drugs eculizumab and C1-INH has undeniably shown that complement-targeted therapeutics can be safe, effective, and profitable. Notably, the stock price of Alexion has increased by more than 600% in the past 5 years. The high costs for eculizumab and C1-INH, and the therapeutic focus on the orphan disease market, raise a red flag as to whether the interest of the pharmaceutical companies in complement-target therapy is predominantly financial. Whereas the clinical trials currently evaluating eculizumab for the rare diseases mentioned above are of extreme importance because they may offer more effective therapies and lead to improved quality of life for the patients, the licensing of eculizumab for numerous diseases may also put enormous pressure on the public health system. As discussed in this review, small molecules and peptidic compounds could offer more cost-effective treatment options. Hence, it is important that more effort is placed on the development of additional complement-related therapies with lower cost and even higher effectiveness.
At this date, over 40 clinical trials are ongoing to evaluate different classes of complement inhibitors in numerous diseases. The next few years will hopefully bring valuable information concerning the safety and efficacy of distinct strategies for complement targeting. For orphan diseases in particular, this may represent a great improvement in the management of clinical signs and quality of life. Traditionally, rare pathological conditions and diseases with an immune component have often been tackled with corticosteroids and immunosuppressants that are known to induce an array of side effects. Whereas complement may not be the main trigger in several of these disorders, it may still play a pivotal role in the maintenance of an inflammatory environment that fuels the pathology. Certainly, further research will be required to select the proper strategy for complement inhibition and to validate the involvement of complement in specific diseases, since virtually any disease with an inflammatory component may benefit from the disruption of complement activation.
Highlights.
It is estimated that about 350 million people worldwide suffer from rare diseases.
The launch of effective treatments for rare diseases is challenging.
Complement is an attractive therapeutic target for inflammatory diseases.
Clinical evidence shows the safety and efficacy of complement therapeutics.
Further effort should be put on the development of complement therapeutics.
Acknowledgments
We thank Deborah McClellan for her excellent editorial assistance. This work was supported by National Institutes of Health grants AI068730, AI030040, and AI097805, the National Science Foundation grant 1423304, and by funding from the European Community’s Seventh Framework Programme under grant agreement number 602699 (DIREKT).
List of Abbreviations
- AChR
acetylcholine receptor
- aHUS
atypical hemolytic uremic syndrome
- AMD
age-related macular degeneration
- AMR
antibody-mediated rejection
- ANCA
anti-neutrophil cytoplasmic antibody
- AP
alternative pathway
- C1-INH
C1 esterase Inhibitor
- C3GN
C3 glomerulonephritis
- C5aR
C5a receptor
- C4BP
C4b binding protein
- CAD
cold agglutinin disease
- CCP
complement control protein domains
- CP
classical pathway
- CRIg
complement receptor of the immunoglobulin superfamily
- DAF
decay accelerating factor
- DDD
dense deposit disease
- EMA
European medicine agency
- FB
Factor B
- FD
Factor D
- FDA
Food and Drug Administration
- FH
Factor H
- FI
Factor I
- GA
geographic atrophy
- GBS
Guillain-Barré syndrome
- GPCR
G-protein-coupled receptor
- GPI
glycosyl-phosphatidyl-inositol
- HAE
hereditary angioedema
- HLA
human leukocyte antigen
- HUS
hemolytic uremic syndrome
- IRI
ischemia-reperfusion injury
- IVIG
intravenous immunoglobulin
- LRP
low density lipoprotein receptor-related protein
- LP
lectin pathway
- MAC
membrane attack complex
- MASP
MBL-associated serine protease
- MBL
mannose-binding lectin
- MCP
membrane cofactor protein
- MG
myasthenia gravis
- MMN
multifocal motor neuropathy
- MuSK
muscle-specific receptor tyrosine kinase
- NMO
neuromyelitis optica
- PIG-A
phosphatidyl-inositol glycan class A
- PNH
paroxysmal nocturnal hemoglobinuria
- RBC
red blood cells
- RGD
arginine-glycine-aspartic acid
- SERPIN
serine protease inhibitor
- STEC
shiga toxin-producing Escherichia coli
- TMA
thrombotic microangiopathy
- TTP
thrombotic thrombocytopenic purpura
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
JD Lambris and D Ricklin are inventors of patent applications that describe the use of complement inhibitors for therapeutic purposes. JD Lambris is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors. AM Risitano received research funding from Alexion Pharmaceuticals, Amyndas Pharmaceuticals, Alnylam Pharmaceuticals, Rapharma and Novartis and is a consultant for Alnylam and RA Pharma. The other authors declare no financial interest or conflict.
References
- 1.Zarkadis IK, Mastellos D, Lambris JD. Phylogenetic aspects of the complement system. Dev Comp Immunol. 2001;25:745–762. doi: 10.1016/s0145-305x(01)00034-9. [DOI] [PubMed] [Google Scholar]
- 2.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kemper C, Kohl J. Novel roles for complement receptors in T cell regulation and beyond. Mol Immunol. 2013;56:181–190. doi: 10.1016/j.molimm.2013.05.223. [DOI] [PubMed] [Google Scholar]
- 4.Kolev M, Le FG, Kemper C. Complement–tapping into new sites and effector systems. Nat Rev Immunol. 2014;14:811–820. doi: 10.1038/nri3761. [DOI] [PubMed] [Google Scholar]
- 5.Spitzer D, Mitchell LM, Atkinson JP, Hourcade DE. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol. 2007;179:2600–2608. doi: 10.4049/jimmunol.179.4.2600. [DOI] [PubMed] [Google Scholar]
- 6.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I – Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liszewski K, Atkinson JP. Complement regulators in human disease: lessons from modern genetics. J Intern Med. 2015;277:294–305. doi: 10.1111/joim.12338. [DOI] [PubMed] [Google Scholar]
- 11.Feussner A, Kalina U, Hofmann P, Machnig T, Henkel G. Biochemical comparison of four commercially available C1 esterase inhibitor concentrates for treatment of hereditary angioedema. Transfusion. 2014;54:2566–2573. doi: 10.1111/trf.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vo AA, Zeevi A, Choi J, Cisneros K, Toyoda M, Kahwaji J, Peng A, Villicana R, Puliyanda D, Reinsmoen N, Haas M, Jordan SC. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation. 2015;99:299–308. doi: 10.1097/TP.0000000000000592. [DOI] [PubMed] [Google Scholar]
- 13.Hofstra JJ, Kleine Budde I, van Twuyver E, Choi G, Levi M, Leebeek FW, de Monchy JG, Ypma PF, Keizer RJ, Huitema AD, Strengers PF. Treatment of hereditary angioedema with nanofiltered C1-esterase inhibitor concentrate (Cetor(R)): multi-center phase II and III studies to assess pharmacokinetics, clinical efficacy and safety. Clin Immunol. 2012;142:280–290. doi: 10.1016/j.clim.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Levy M, Mealy MA. Purified human C1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm. 2014;1:e5. doi: 10.1212/NXI.0000000000000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy. 2012;67:123–130. doi: 10.1111/j.1398-9995.2011.02716.x. [DOI] [PubMed] [Google Scholar]
- 16.van Doorn MB, Burggraaf J, van D T, Eerenberg A, Levi M, Hack CE, Schoemaker RC, Cohen AF, Nuijens J. A phase I study of recombinant human C1 inhibitor in asymptomatic patients with hereditary angioedema. J Allergy Clin Immunol. 2005;116:876–883. doi: 10.1016/j.jaci.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 17.Kelemen Z, Moldovan D, Mihaly E, Visy B, Szeplaki G, Csuka D, Fust G, Farkas H, Varga L. Baseline level of functional C1-inhibitor correlates with disease severity scores in hereditary angioedema. Clin Immunol. 2010;134:354–358. doi: 10.1016/j.clim.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Caccia S, Suffritti C, Cicardi M. Pathophysiology of Hereditary Angioedema. Pediatr Allergy Immunol Pulmonol. 2014;27:159–163. doi: 10.1089/ped.2014.0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernstein JA. HAE update: epidemiology and burden of disease. Allergy Asthma Proc. 2013;34:3–6. doi: 10.2500/aap.2013.34.3623. [DOI] [PubMed] [Google Scholar]
- 20.Truedsson L. Classical pathway deficiencies – A short analytical review. Mol Immunol. 2015 doi: 10.1016/j.molimm.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Keck T. Site-specific therapeutic effects of protease inhibitors: effect of route of administration in experimental pancreatitis. Pancreatology. 2001;1:656–661. doi: 10.1159/000055877. [DOI] [PubMed] [Google Scholar]
- 22.Berentsen S, Sundic T. Red blood cell destruction in autoimmune hemolytic anemia: role of complement and potential new targets for therapy. Biomed Res Int. 2015;2015:363278. doi: 10.1155/2015/363278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swiecicki PL, Hegerova LT, Gertz MA. Cold agglutinin disease. Blood. 2013;122:1114–1121. doi: 10.1182/blood-2013-02-474437. [DOI] [PubMed] [Google Scholar]
- 24.Shi J, Rose EL, Singh A, Hussain S, Stagliano NE, Parry GC, Panicker S. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood. 2014;123:4015–4022. doi: 10.1182/blood-2014-02-556027. [DOI] [PubMed] [Google Scholar]
- 25.Matsushita M, Endo Y, Fujita T. Structural and functional overview of the lectin complement pathway: its molecular basis and physiological implication. Arch Immunol Ther Exp (Warsz) 2013;61:273–283. doi: 10.1007/s00005-013-0229-y. [DOI] [PubMed] [Google Scholar]
- 26.Olszowski T, Poziomkowska-Gesicka I, Jensenius JC, Adler G. Lectin pathway of complement activation in a Polish woman with MASP-2 deficiency. Immunobiology. 2014;219:261–262. doi: 10.1016/j.imbio.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Stengaard-Pedersen K, Thiel S, Gadjeva M, Moller-Kristensen M, Sorensen R, Jensen LT, Sjoholm AG, Fugger L, Jensenius JC. Inherited deficiency of mannan-binding lectin-associated serine protease 2. N Engl J Med. 2003;349:554–560. doi: 10.1056/NEJMoa022836. [DOI] [PubMed] [Google Scholar]
- 28.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–633. doi: 10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 29.Johannes L, Romer W. Shiga toxins–from cell biology to biomedical applications. Nat Rev Microbiol. 2010;8:105–116. doi: 10.1038/nrmicro2279. [DOI] [PubMed] [Google Scholar]
- 30.Ren G, Hack BK, Minto AW, Cunningham PN, Alexander JJ, Haas M, Quigg RJ. A complement-dependent model of thrombotic thrombocytopenic purpura induced by antibodies reactive with endothelial cells. Clin Immunol. 2002;103:43–53. doi: 10.1006/clim.2002.5168. [DOI] [PubMed] [Google Scholar]
- 31.Riedl M, Fakhouri F, Le QM, Noone DG, Jungraithmayr TC, Fremeaux-Bacchi V, Licht C. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost. 2014;40:444–464. doi: 10.1055/s-0034-1376153. [DOI] [PubMed] [Google Scholar]
- 32.Heitzeneder S, Seidel M, Forster-Waldl E, Heitger A. Mannan-binding lectin deficiency – Good news, bad news, doesn’t matter? Clin Immunol. 2012;143:22–38. doi: 10.1016/j.clim.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, Delmas Y, Douglas K, Furman RR, Gaber OA, Goodship T, Herthelius M, Hourmant M, Legendre CM, Remuzzi G, Sheerin N, Trivelli A, Loirat C. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87:1061–1073. doi: 10.1038/ki.2014.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volanakis JE, Narayana SV. Complement factor D, a novel serine protease. Protein Sci. 1996;5:553–564. doi: 10.1002/pro.5560050401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biesma DH, Hannema AJ, van Velzen-Blad H, Mulder L, van Z R, Kluijt I, Roos D. A family with complement factor D deficiency. J Clin Invest. 2001;108:233–240. doi: 10.1172/JCI12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sprong T, Roos D, Weemaes C, Neeleman C, Geesing CL, Mollnes TE, van D M. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. 2006;107:4865–4870. doi: 10.1182/blood-2005-07-2820. [DOI] [PubMed] [Google Scholar]
- 37.Stanton CM, Yates JR, den Hollander AI, Seddon JM, Swaroop A, Stambolian D, Fauser S, Hoyng C, Yu Y, Atsuhiro K, Branham K, Othman M, Chen W, Kortvely E, Chalmers K, Hayward C, Moore AT, Dhillon B, Ueffing M, Wright AF. Complement factor D in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:8828–8834. doi: 10.1167/iovs.11-7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katschke KJ, Jr, Wu P, Ganesan R, Kelley RF, Mathieu MA, Hass PE, Murray J, Kirchhofer D, Wiesmann C, van Lookeren CM. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J Biol Chem. 2012;287:12886–12892. doi: 10.1074/jbc.M112.345082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan BP, Thanassi J, Podos S, Yang G, Phadke A, Gadhachanda V, Pais G, Hashimoto A, Wang Q, Chen D, Wang X, Agarwal A, Deshpande M, Huang Y, Wiles J, Huang M. Novel Small Molecule Inhibitors Targeting Complement Factor D for Therapy of Paroxysmal Nocturnal Hemoglobinuria. Abstract presented at the 56thAnnual Meeting of the American Society of Hematology; San Francisco, California, USA. December 6–9, 2014; 2014. [Google Scholar]
- 40.Bora NS, Matta B, Lyzogubov VV, Bora PS. Relationship between the complement system, risk factors and prediction models in age-related macular degeneration. Mol Immunol. 2015;63:176–183. doi: 10.1016/j.molimm.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Loyet KM, Good J, Davancaze T, Sturgeon L, Wang X, Yang J, Le KN, Wong M, Hass PE, van Lookeren CM, Haughney PC, Morimoto A, mico-Beyer LA, DeForge LE. Complement inhibition in cynomolgus monkeys by anti-factor d antigen-binding fragment for the treatment of an advanced form of dry age-related macular degeneration. J Pharmacol Exp Ther. 2014;351:527–537. doi: 10.1124/jpet.114.215921. [DOI] [PubMed] [Google Scholar]
- 42.Do DV, Pieramici DJ, van Lookeren CM, Beres T, Friesenhahn M, Zhang Y, Strauss EC. A phase ia dose-escalation study of the anti-factor D monoclonal antibody fragment FCFD4514S in patients with geographic atrophy. Retina. 2014;34:313–320. doi: 10.1097/IAE.0b013e3182979ddd. [DOI] [PubMed] [Google Scholar]
- 43.Yehoshua Z, CA DAGF, Nunes RP, Gregori G, Penha FM, Moshfeghi AA, Zhang K, Sadda S, Feuer W, Rosenfeld PJ. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: the COMPLETE study. Ophthalmology. 2014;121:693–701. doi: 10.1016/j.ophtha.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia Filho CA, Yehoshua Z, Gregori G, Nunes RP, Penha FM, Moshfeghi AA, Zhang K, Feuer W, Rosenfeld PJ. Change in drusen volume as a novel clinical trial endpoint for the study of complement inhibition in age-related macular degeneration. Ophthalmic Surg Lasers Imaging Retina. 2014;45:18–31. doi: 10.3928/23258160-20131217-01. [DOI] [PubMed] [Google Scholar]
- 45.Klos A, Wende E, Wareham KJ, Monk PN. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev. 2013;65:500–543. doi: 10.1124/pr.111.005223. [DOI] [PubMed] [Google Scholar]
- 46.Wu MC, Brennan FH, Lynch JP, Mantovani S, Phipps S, Wetsel RA, Ruitenberg MJ, Taylor SM, Woodruff TM. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci U S A. 2013;110:9439–9444. doi: 10.1073/pnas.1218815110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mueller-Ortiz SL, Morales JE, Wetsel RA. The receptor for the complement C3a anaphylatoxin (C3aR) provides host protection against Listeria monocytogenes-induced apoptosis. J Immunol. 2014;193:1278–1289. doi: 10.4049/jimmunol.1302787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardone J, Le FG, Vantourout P, Roberts A, Fuchs A, Jackson I, Suddason T, Lord G, Atkinson JP, Cope A, Hayday A, Kemper C. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol. 2010;11:862–871. doi: 10.1038/ni.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37:199–207. doi: 10.1016/j.immuni.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lachmann PJ, Smith RA. Taking complement to the clinic–has the time finally come? Scand J Immunol. 2009;69:471–478. doi: 10.1111/j.1365-3083.2009.02258.x. [DOI] [PubMed] [Google Scholar]
- 51.Skattum L, Martensson U, Sjoholm AG. Hypocomplementaemia caused by C3 nephritic factors (C3 NeF): clinical findings and the coincidence of C3 NeF type II with anti-C1q autoantibodies. J Intern Med. 1997;242:455–464. doi: 10.1111/j.1365-2796.1997.tb00018.x. [DOI] [PubMed] [Google Scholar]
- 52.Ricklin D, Lambris JD. Therapeutic control of complement activation at the level of the central component C3. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol. 1996;157:884–891. [PubMed] [Google Scholar]
- 54.Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, Lupu F, Nilsson B, Risitano AM, Ricklin D, Lambris JD. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45:423–440. doi: 10.1111/eci.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, Tzekou A, DeAngelis RA, Resuello RR, Lupu F, Barlow PN, Lambris JD. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Risitano AM. Paroxysmal nocturnal hemoglobinuria and the complement system: recent insights and novel anticomplement strategies. Adv Exp Med Biol. 2013;735:155–172. doi: 10.1007/978-1-4614-4118-2_10. [DOI] [PubMed] [Google Scholar]
- 57.Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 58.Mastellos DC, Ricklin D, Yancopoulou D, Risitano A, Lambris JD. Complement in paroxysmal nocturnal hemoglobinuria: exploiting our current knowledge to improve the treatment landscape. Expert Rev Hematol. 2014;7:583–598. doi: 10.1586/17474086.2014.953926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, Gaya A, Coyle L, de C C, Fu CL, Maciejewski JP, Bessler M, Kroon HA, Rother RP, Hillmen P. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111:1840–1847. doi: 10.1182/blood-2007-06-094136. [DOI] [PubMed] [Google Scholar]
- 60.Hillmen P, Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L, Schrezenmeier H, Szer J, Brodsky RA, Hill A, Socie G, Bessler M, Rollins SA, Bell L, Rother RP, Young NS. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110:4123–4128. doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]
- 61.Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski A, Tamburini P, Inazawa J, Kinoshita T, Kanakura Y. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370:632–639. doi: 10.1056/NEJMoa1311084. [DOI] [PubMed] [Google Scholar]
- 62.Rondelli T, Risitano AM, Peffault de LR, Sica M, Peruzzi B, Ricci P, Barcellini W, Iori AP, Boschetti C, Valle V, Fremeaux-Bacchi V, De AM, Socie G, Luzzatto L, Notaro R. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2014;99:262–266. doi: 10.3324/haematol.2013.090001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, Ricci P, Alfinito F, Camera A, Gianfaldoni G, Amendola A, Boschetti C, Di BE, Fratellanza G, Barbano F, Rodeghiero F, Zanella A, Iori AP, Selleri C, Luzzatto L, Rotoli B. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113:4094–4100. doi: 10.1182/blood-2008-11-189944. [DOI] [PubMed] [Google Scholar]
- 64.Lin Z, Schmidt CQ, Koutsogiannaki S, Ricci P, Risitano AM, Lambris JD, Ricklin D. Complement C3dg-mediated erythrophagocytosis: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2015 doi: 10.1182/blood-2015-02-625871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Risitano AM, Notaro R, Pascariello C, Sica M, Del VL, Horvath CJ, Fridkis-Hareli M, Selleri C, Lindorfer MA, Taylor RP, Luzzatto L, Holers VM. The complement receptor 2/factor H fusion protein TT30 protects paroxysmal nocturnal hemoglobinuria erythrocytes from complement-mediated hemolysis and C3 fragment. Blood. 2012;119:6307–6316. doi: 10.1182/blood-2011-12-398792. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt CQ, Bai H, Lin Z, Risitano AM, Barlow PN, Ricklin D, Lambris JD. Rational engineering of a minimized immune inhibitor with unique triple-targeting properties. J Immunol. 2013;190:5712–5721. doi: 10.4049/jimmunol.1203548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, Lin Z, Pascariello C, Raia M, Sica M, Del VL, Pane F, Lupu F, Notaro R, Resuello RR, DeAngelis RA, Lambris JD. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weisman HF, Bartow T, Leppo MK, Marsh HC, Jr, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 69.Li JS, Jaggers J, Anderson PA. The use of TP10, soluble complement receptor 1, in cardiopulmonary bypass. Expert Rev Cardiovasc Ther. 2006;4:649–654. doi: 10.1586/14779072.4.5.649. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, Meyer NC, Hunsicker LG, Sethi S, Smith RJ. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–1829. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fridkis-Hareli M, Storek M, Mazsaroff I, Risitano AM, Lundberg AS, Horvath CJ, Holers VM. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011;118:4705–4713. doi: 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, Alpers CE, Bajema IM, Bedrosian C, Braun M, Doyle M, Fakhouri F, Fervenza FC, Fogo AB, Fremeaux-Bacchi V, Gale DP, Goicoechea de JE, Griffin G, Harris CL, Holers VM, Johnson S, Lavin PJ, Medjeral-Thomas N, Paul MB, Nast CC, Noel LH, Peters DK, Rodriguez de CS, Servais A, Sethi S, Song WC, Tamburini P, Thurman JM, Zavros M, Cook HT. C3 glomerulopathy: consensus report. Kidney Int. 2013;84:1079–1089. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, Theis JD, Dogan A, Smith RJ. C3 glomerulonephritis: clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465–473. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Servais A, Noel LH, Roumenina LT, Le QM, Ngo S, Dragon-Durey MA, Macher MA, Zuber J, Karras A, Provot F, Moulin B, Grunfeld JP, Niaudet P, Lesavre P, Fremeaux-Bacchi V. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 75.Bomback AS. Eculizumab in the treatment of membranoproliferative glomerulonephritis. Nephron Clin Pract. 2014;128:270–276. doi: 10.1159/000368592. [DOI] [PubMed] [Google Scholar]
- 76.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, Stokes MB, Markowitz GS, D’Agati VD, Canetta PA, Radhakrishnan J, Appel GB. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7:748–756. doi: 10.2215/CJN.12901211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, Appel GB, D’Agati VD. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol. 2012;23:1229–1237. doi: 10.1681/ASN.2011121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, Smith RJ. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220:993–998. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46:2753–2766. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Legendre CM, Licht C, Loirat C. Eculizumab in atypical hemolyticuremic syndrome. N Engl J Med. 2013;369:1379–1380. doi: 10.1056/NEJMc1308826. [DOI] [PubMed] [Google Scholar]
- 81.Herper M. The most expensive drugs in the world. Forbes Magazine. 2010 [Google Scholar]
- 82.Barratt-Due A, Thorgersen EB, Lindstad JK, Pharo A, Lissina O, Lambris JD, Nunn MA, Mollnes TE. Ornithodoros moubata complement inhibitor is an equally effective C5 inhibitor in pigs and humans. J Immunol. 2011;187:4913–4919. doi: 10.4049/jimmunol.1101000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stromberg P. Introducing SOBI002, a samll Affibody-ABD fusion protein targeting complement component C5., Abstract presented at the 7th International Conference on Complement Therapeutics. Aegean Conferences. 2014;82 [Google Scholar]
- 84.Borodovsky A, Y K, Sprague A, Banda NK, Holers VM, Vaishnaw A, Maier M, Kallanthottathil R, Charisse K, Kuchimanchi S, Manoharan M, Salant DJ, Fitzgerald K, Meyers R, Sorensen B. Aln-CC5, an Investigational RNAi Therapeutic Targeting C5 for Complement Inhibition. Blood. 2014;214 [Google Scholar]
- 85.Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26–30. doi: 10.1001/archdermatol.2009.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fardet L, Dupuy A, Gain M, Kettaneh A, Cherin P, Bachelez H, Dubertret L, Lebbe C, Morel P, Rybojad M. Factors associated with underlying malignancy in a retrospective cohort of 121 patients with dermatomyositis. Medicine (Baltimore) 2009;88:91–97. doi: 10.1097/MD.0b013e31819da352. [DOI] [PubMed] [Google Scholar]
- 87.Dalakas MC. Immunotherapy of myositis: issues, concerns and future prospects. Nat Rev Rheumatol. 2010;6:129–137. doi: 10.1038/nrrheum.2010.2. [DOI] [PubMed] [Google Scholar]
- 88.Dalakas MC. Therapeutic advances and future prospects in immune-mediated inflammatory myopathies. Ther Adv Neurol Disord. 2008;1:157–166. doi: 10.1177/1756285608097463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dalakas MC. Mechanistic effects of IVIg in neuroinflammatory diseases: conclusions based on clinicopathologic correlations. J Clin Immunol. 2014;34(Suppl 1):S120–126. doi: 10.1007/s10875-014-0024-5. [DOI] [PubMed] [Google Scholar]
- 90.Vanoni1 F, J C, Parvex P, Chizzolini C, Hofer M. A difficult case of juvenile dermatomyositis complicated by thrombotic microangiopathy and purtscher-like retinopathy. Pediatric Rheumatology. 2014;12:275. [Google Scholar]
- 91.Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, Kleiter I, Chitnis T, G.I.C. Consortium, O. Biorepository for Neuromyelitis Demographic and clinical features of neuromyelitis optica: A review. Mult Scler. 2015;21:845–853. doi: 10.1177/1352458515572406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11:535–544. doi: 10.1016/S1474-4422(12)70133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saadoun S, Papadopoulos MC. Role of membrane complement regulators in neuromyelitis optica. Mult Scler. 2015 doi: 10.1177/1352458515571446. [DOI] [PubMed] [Google Scholar]
- 94.Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6:383–392. doi: 10.1038/nrneurol.2010.72. [DOI] [PubMed] [Google Scholar]
- 95.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nytrova P, Potlukova E, Kemlink D, Woodhall M, Horakova D, Waters P, Havrdova E, Zivorova D, Vincent A, Trendelenburg M. Complement activation in patients with neuromyelitis optica. J Neuroimmunol. 2014;274:185–191. doi: 10.1016/j.jneuroim.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 97.Jones MV, Fox-Talbot K, Levy M. Evidence for classic complement activity in neuromyelitis optica. Clin Neuropathol. 2014;33:251–252. doi: 10.5414/NP300697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Papadopoulos MC, Bennett JL, Verkman AS. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol. 2014;10:493–506. doi: 10.1038/nrneurol.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, O’Toole O, Wingerchuk DM. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12:554–562. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]
- 100.Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37:141–149. doi: 10.1002/mus.20950. [DOI] [PubMed] [Google Scholar]
- 101.Ha JC, Richman DP. Myasthenia gravis and related disorders: Pathology and molecular pathogenesis. Biochim Biophys Acta. 2015;1852:651–657. doi: 10.1016/j.bbadis.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 102.az-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, Munoz-Blanco JL, Mazia C, Straasheijm KR, Gallardo E, Juarez C, Verschuuren JJ, Illa I. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189–193. doi: 10.1212/WNL.0b013e3182407982. [DOI] [PubMed] [Google Scholar]
- 103.Zhang B, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, Lewis RA, Xiong WC, Lisak RP, Tzartos SJ, Mei L. bAutoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. 2012;69:445–451. doi: 10.1001/archneurol.2011.2393. [DOI] [PubMed] [Google Scholar]
- 104.Lisak RP, Levinson AI, Zweiman B, Kornstein MJ. Antibodies to acetylcholine receptor and tetanus toxoid: in vitro synthesis by thymic lymphocytes. J Immunol. 1986;137:1221–1225. [PubMed] [Google Scholar]
- 105.Sieb JP. Myasthenia gravis: an update for the clinician. Clin Exp Immunol. 2014;175:408–418. doi: 10.1111/cei.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kusner LL, Kaminski HJ, Soltys J. Effect of complement and its regulation on myasthenia gravis pathogenesis. Expert Rev Clin Immunol. 2008;4:43–52. doi: 10.1586/1744666X.4.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kusner LL, Halperin JA, Kaminski HJ. Cell surface complement regulators moderate experimental myasthenia gravis pathology. Muscle Nerve. 2013;47:33–40. doi: 10.1002/mus.23448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Howard JF, Jr, Barohn RJ, Cutter GR, Freimer M, Juel VC, Mozaffar T, Mellion ML, Benatar MG, Farrugia ME, Wang JJ, Malhotra SS, Kissel JT. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48:76–84. doi: 10.1002/mus.23839. [DOI] [PubMed] [Google Scholar]
- 109.B van den B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10:469–482. doi: 10.1038/nrneurol.2014.121. [DOI] [PubMed] [Google Scholar]
- 110.Winer JB. An update in guillain-barre syndrome. Autoimmune Dis. 2014;2014:793024. doi: 10.1155/2014/793024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Willison HJ, Halstead SK, Beveridge E, Zitman FM, Greenshields KN, Morgan BP, Plomp JJ. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain-Barre syndrome. J Neuroimmunol. 2008;201–202:172–182. doi: 10.1016/j.jneuroim.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 112.Lawson VH, Arnold WD. Multifocal motor neuropathy: a review of pathogenesis, diagnosis, and treatment. Neuropsychiatr Dis Treat. 2014;10:567–576. doi: 10.2147/NDT.S39592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Leger JM, Guimaraes-Costa R, Iancu FR. The pathogenesis of multifocal motor neuropathy and an update on current management options. Ther Adv Neurol Disord. 2015;8:109–122. doi: 10.1177/1756285615575269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yuki N, Watanabe H, Nakajima T, Spath PJ. IVIG blocks complement deposition mediated by anti-GM1 antibodies in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2011;82:87–91. doi: 10.1136/jnnp.2010.205856. [DOI] [PubMed] [Google Scholar]
- 115.Fitzpatrick AM, Mann CA, Barry S, Brennan K, Overell JR, Willison HJ. An open label clinical trial of complement inhibition in multifocal motor neuropathy. J Peripher Nerv Syst. 2011;16:84–91. doi: 10.1111/j.1529-8027.2011.00328.x. [DOI] [PubMed] [Google Scholar]
- 116.Sacks SH, Zhou W. The role of complement in the early immune response to transplantation. Nat Rev Immunol. 2012;12:431–442. doi: 10.1038/nri3225. [DOI] [PubMed] [Google Scholar]
- 117.Farrar CA, Zhou W, Lin T, Sacks SH. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J. 2006;20:217–226. doi: 10.1096/fj.05-4747com. [DOI] [PubMed] [Google Scholar]
- 118.Damman J, Seelen MA, Moers C, Daha MR, Rahmel A, Leuvenink HG, Paul A, Pirenne J, Ploeg RJ. Systemic complement activation in deceased donors is associated with acute rejection after renal transplantation in the recipient. Transplantation. 2011;92:163–169. doi: 10.1097/TP.0b013e318222c9a0. [DOI] [PubMed] [Google Scholar]
- 119.Farrar CA, Asgari E, Schwaeble WJ, Sacks SH. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front Immunol. 2012;3:341. doi: 10.3389/fimmu.2012.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL, Sacks SH. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest. 2000;105:1363–1371. doi: 10.1172/JCI8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.de Vries B, Kohl J, Leclercq WK, Wolfs TG, van Bijnen AA, Heeringa P, Buurman WA. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J Immunol. 2003;170:3883–3889. doi: 10.4049/jimmunol.170.7.3883. [DOI] [PubMed] [Google Scholar]
- 122.Tang S, Zhou W, Sheerin NS, Vaughan RW, Sacks SH. Contribution of renal secreted complement C3 to the circulating pool in humans. J Immunol. 1999;162:4336–4341. [PubMed] [Google Scholar]
- 123.Kwan WH, Hashimoto D, Paz-Artal E, Ostrow K, Greter M, Raedler H, Medof ME, Merad M, Heeger PS. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J Clin Invest. 2012;122:2234–2238. doi: 10.1172/JCI61019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol. 2012;8:670–678. doi: 10.1038/nrneph.2012.212. [DOI] [PubMed] [Google Scholar]
- 125.Carroll MC. The complement system in B cell regulation. Mol Immunol. 2004;41:141–146. doi: 10.1016/j.molimm.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 126.Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, Cosio FG, Gandhi MJ, Kremers W, Gloor JM. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11:2405–2413. doi: 10.1111/j.1600-6143.2011.03757.x. [DOI] [PubMed] [Google Scholar]
- 127.Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, King KE, Kraus E, Lees LM, Melancon JK, Stewart ZA, Warren DS, Zachary AA, Montgomery RA. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant. 2009;9:231–235. doi: 10.1111/j.1600-6143.2008.02451.x. [DOI] [PubMed] [Google Scholar]
- 128.Chen Song S, Zhong S, Xiang Y, Li JH, Guo H, Wang WY, Xiong YL, Li XC, Shi S Chen, Chen XP, Chen G. Complement inhibition enables renal allograft accommodation and long-term engraftment in presensitized nonhuman primates. Am J Transplant. 2011;11:2057–2066. doi: 10.1111/j.1600-6143.2011.03646.x. [DOI] [PubMed] [Google Scholar]
- 129.Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. 2007;152:429–448. doi: 10.1038/sj.bjp.0707332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–1642. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 131.Kallenberg CG. Key advances in the clinical approach to ANCA-associated vasculitis. Nat Rev Rheumatol. 2014;10:484–493. doi: 10.1038/nrrheum.2014.104. [DOI] [PubMed] [Google Scholar]
- 132.McKinney EF, Willcocks LC, Broecker V, Smith KG. The immunopathology of ANCA-associated vasculitis. Semin Immunopathol. 2014;36:461–478. doi: 10.1007/s00281-014-0436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A. 1990;87:4115–4119. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xing GQ, Chen M, Liu G, Heeringa P, Zhang JJ, Zheng X, J E, Kallenberg CG, Zhao MH. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–291. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 137.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83:129–137. doi: 10.1038/ki.2012.313. [DOI] [PubMed] [Google Scholar]
- 138.Hao J, Meng LQ, Xu PC, Chen M, Zhao MH. p38MAPK, ERK and PI3K signaling pathways are involved in C5a-primed neutrophils for ANCA-mediated activation. PLoS One. 2012;7:e38317. doi: 10.1371/journal.pone.0038317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux-Bacchi V, Lesavre P, Halbwachs-Mecarelli L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 140.Wang C, Wang H, Hao J, Chang DY, Zhao MH, Chen M. Involvement of high mobility group box 1 in the activation of C5a-primed neutrophils induced by ANCA. Clin Immunol. 2015;159:47–57. doi: 10.1016/j.clim.2015.04.008. [DOI] [PubMed] [Google Scholar]