

Abstract
Comparisons between structures of the β1-adrenergic receptor (AR) bound to either agonists, partial agonists, or weak partial agonists led to the proposal that rotamer changes of Ser5.46, coupled to a contraction of the binding pocket, are sufficient to increase the probability of receptor activation. (RS)-4-[3-(tert-butylamino)-2-hydroxypropoxy]-1H-indole-2-carbonitrile (cyanopindolol) is a weak partial agonist of β1AR and, based on the hypothesis above, we predicted that the addition of a methyl group to form 4-[(2S)-3-(tert-butylamino)-2-hydroxypropoxy]-7-methyl-1H-indole-2-carbonitrile (7-methylcyanopindolol) would dramatically reduce its efficacy. An eight-step synthesis of 7-methylcyanopindolol was developed and its pharmacology was analyzed. 7-Methylcyanopindolol bound with similar affinity to cyanopindolol to both β1AR and β2AR. As predicted, the efficacy of 7-methylcyanopindolol was reduced significantly compared with cyanopindolol, acting as a very weak partial agonist of turkey β1AR and an inverse agonist of human β2AR. The structure of 7-methylcyanopindolol–bound β1AR was determined to 2.4-Å resolution and found to be virtually identical to the structure of cyanopindolol-bound β1AR. The major differences in the orthosteric binding pocket are that it has expanded by 0.3 Å in 7-methylcyanopindolol–bound β1AR and the hydroxyl group of Ser5.46 is positioned 0.8 Å further from the ligand, with respect to the position of the Ser5.46 side chain in cyanopindolol-bound β1AR. Thus, the molecular basis for the reduction in efficacy of 7-methylcyanopindolol compared with cyanopindolol may be regarded as the opposite of the mechanism proposed for the increase in efficacy of agonists compared with antagonists.
Introduction
The β1 and β2 adrenergic receptors (ARs) are well studied prototypical members of the G protein–coupled receptor superfamily (Venkatakrishnan et al., 2013). In vivo, the receptors are activated by both adrenaline and noradrenaline, with important clinical roles in the modulation of cardiac output (β1AR) and bronchodilatation (β2AR). Recent successes in the structure determination of both β1AR (Warne et al., 2008, 2011, 2012; Moukhametzianov et al., 2011; Christopher et al., 2013; Miller-Gallacher et al., 2014) and β2AR (Cherezov et al., 2007; Wacker et al., 2010; Rosenbaum et al., 2011; Rasmussen et al., 2011a,b) have led to a molecular understanding of receptor activation (Lebon et al., 2012). Binding of a full agonist to the receptors results in a contraction of the ligand-binding pocket by 1.0 Å for β1AR and 1.2 Å for β2AR and a rotamer change of Ser5.46 so that it forms a hydrogen bond with the para-hydroxyl group of the catecholamine moiety of the agonist. However, the agonist-bound receptors remain in an overall conformation that is consistent with an inactive state. The role of the agonist is thus to increase the probability of active state formation, but the agonist is insufficient on its own to stabilize the active state. A crystal structure of β2AR in the active state bound to the heterotrimeric G protein Gs (Rasmussen et al., 2011b) showed that the fully active state is characterized by the formation of a cleft in the intracellular face of the receptor and a 16-Å outward movement of the cytoplasmic end of transmembrane helix 6.
Structures of β1AR bound to full agonists (Warne et al., 2011), partial agonists (Warne et al., 2011), weak partial agonists (Warne et al., 2008; Moukhametzianov et al., 2011; Miller-Gallacher et al., 2014), and biased agonists (Warne et al., 2012) have been determined. All these structures are in an inactive state, so the structures with agonists represent the encounter complex between the ligand and receptor before the receptor becomes activated. As mentioned above, full agonists cause a contraction of the binding pocket and cause a rotamer change of Ser5.46 due to hydrogen bond formation. In addition, there is a rotamer change of Ser5.42. The combination of these changes results in the weakening of helix-helix interactions between H3, H4, and H5, which has been proposed to be the important initial event that increases the probability of the receptor adopting an active conformation (Warne and Tate, 2013). In support of this, partial agonists cause a contraction of the binding pocket and the rotamer change of Ser5.42, but not the rotamer change of Ser5.46. Weak partial agonists cause only the rotamer change of Ser5.42, but not of Ser5.46, and they do not cause the contraction of the ligand-binding pocket due to the additional oxymethylene spacer in the backbone of the ligand (Fig. 1). Thus, there appears to be a direct correlation between specific differences in the structure of the receptor-ligand complex and the efficacy of the ligand.
Fig. 1.

Structures of β receptor ligands: (A) noradrenaline; (B) isoprenaline; (C) carazolol; (D) ICI118551; (E) cyanopindolol; and (F) 7-methylcyanopindolol. Portions of the structures in blue are regions analogous to those found in noradrenaline. The oxymethylene spacer between the ethanolamine backbone and ligand head group that prevents contraction of the ligand-binding pocket in the antagonists is shown in red. Noradrenaline and isoprenaline are regarded as full agonists (Baker, 2010b), carazolol and cyanopindolol are regarded as weak partial agonists (Baker, 2010b), and ICI18551 is regarded as an inverse agonist (Bond et al., 1995; Azzi et al., 2001).
A prediction of the molecular mechanism for agonist activation of β1AR is that the efficacy of a ligand should be reduced upon the addition of a methyl group in the position analogous to the para-hydroxyl of, for example, isoprenaline (Warne et al., 2011), to prevent the rotamer change of Ser5.46 by steric hindrance (see Fig. 1 for the structures of the ligands discussed in the text). To test this hypothesis, we therefore made the methylated version of (RS)-4-[3-(tert-butylamino)-2-hydroxypropoxy]-1H-indole-2-carbonitrile (cyanopindolol), 4-[(2S)-3-(tert-butylamino)-2-hydroxypropoxy]-7-methyl-1H-indole-2-carbonitrile (7-methylcyanopindolol). Cyanopindolol was used as the starting point for this study because its properties are pharmacologically and structurally well defined. Through the single modification of cyanopindolol to make 7-methylcyanopindolol, we could be certain that the only effect would be on the rotamer conformation of Ser5.46. In this way, we could be certain that any changes in efficacy were due to this modification and not to any other differences in the ligand.
Materials and Methods
Materials
7-Methylcyanopindolol was synthesized as described below. 3H-(–)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one (CGP12177), 3H-adenine, and 14C-cAMP were from Amersham International (Amersham, UK), and Microscint 20 and Ultima Gold XR scintillation fluid were from PerkinElmer (Shelton, CT). Fetal calf serum was from PAA Laboratories (Teddington, UK). 2-Hydroxy-5-(2-[(hydroxy-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]propyl)amino]ethoxy)benzamide was from Tocris Life Sciences (Avonmouth, UK). S-Cyanopindolol, 1-(9H-carbazol-4-yloxy)-3-(propan-2-ylamino)propan-2-ol (carazolol), (–)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)-amino]-2-butanol, isoprenaline, and propranolol were from Sigma Chemicals (Poole, UK). n-Decyl-β-D-maltopyranoside (DM) was purchased from Anatrace (Maumee, OH). Monoolein was from Nu Chek Prep (Elysian, MN). Cholesteryl hemisuccinate (CHS) and cholesterol were purchased from Sigma-Aldrich (Poole, UK).
Cell Culture.
Pharmacological studies were conducted in Chinese hamster ovary (CHO) cells stably expressing either the turkey β1AR [receptor expression level 148 fmol/mg protein, defined as tßtrunc by Baker (2010a)] or the human β2-adrenoceptor (CHO-hβ2) (receptor expression level 466 fmol/mg protein) (Baker et al., 2003). Both of these cell lines also contained the stably transfected reporter gene containing six cAMP response elements upstream of a secreted placental alkaline phosphatase reporter gene (CRE-SPAP). Negative control cells were parental CHO cells (CHO-CRE-SPAP cells), which are CHO cells with the stably transfected reporter but without any transfected receptor (Baker et al., 2003). Cells were grown in Dulbecco’s modified Eagle’s medium nutrient mix F12 containing 10% fetal calf serum and 2 mM of L-glutamine in a 37°C humidified 5% CO2 to 95% air atmosphere.
3H-CGP12177 Whole Cell Binding.
Ligand affinity was measured using 3H-CGP12177 whole cell binding as previously described (Baker, 2010b). Briefly, cells were grown to confluence in white-sided, clear-bottomed 96-well view plates. The following day, the media was removed and replaced with 100 μl of serum-free media containing the competing ligand at twice the final required concentration, followed immediately by 100 μl of 3H-CGP12177. After 2 hours of incubation (37°C, 5% CO2), the cells were washed twice by the addition and removal of 200 μl of phosphate-buffered saline (4°C). A white base was applied to the plate, 100 μl of Microscint 20 was added to each well, a sealant top was applied to the top of the plate, and after at least 8 hours at room temperature, the plates were counted on a TopCount (PerkinElmer) at 21°C for 2 minutes per well. Propranolol (10 μM final concentration) was used to determine nonspecific binding in all cases. All data points were performed in triplicate, and each 96-well plate also contained six determinations of total and nonspecific binding.
3H-cAMP Accumulation.
Cells were grown to confluence in 24-well plates. The following day, the cells were prelabeled with 3H-adenine (2 hours of incubation with 0.5 ml of media containing 1 μCi 3H-adenine). The 3H-adenine was removed, the cells were washed by the addition and removal of 1 ml of serum-free media, and then 1 ml of serum-free media containing 1 mM of 1-methyl-3-(2-methylpropyl)-7H-purine-2,6-dione was added to each well. After 15 minutes, 10 μl of ligand was added to each well and incubated for 5 hours (37°C, 5% CO2). The reaction was terminated by the addition of 50 μl of concentrated HCl per well, the plates were frozen and thawed, and 3H-cAMP separated from other 3H-nucleotides by sequential Dowex and alumina column chromatography, as previously described (Baker, 2010a). Basal activity and a positive control (response to 10 μM of isoprenaline) were included in all plates for every experiment. Responses are therefore expressed as a percentage of this maximum or, in the case of inverse agonists, as a percentage of the basal response.
Data Analysis.
For whole cell binding, a sigmoidal binding curve (eq. 1) was fitted to the concentration response curves using Prism 2.01 (GraphPad Software, La Jolla, CA) and the IC50 was then determined as the concentration required to inhibit 50% of the specific binding.
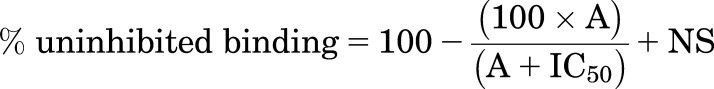 |
(1) |
where A is the concentration of the competing ligand, IC50 is the concentration at which half of the specific binding of 3H-CGP12177 has been inhibited, and NS is the nonspecific binding.
From this IC50 value and the known concentration of 3H-CGP12177, a KD value (concentration at which half the receptors are bound by the competing ligand) was calculated using eq. 2. The KD values for 3H-CGP12177 were 0.42 nM at the turkey β1AR (Baker, 2010a) and 0.17 nM at the human β2AR (Baker, 2010b).
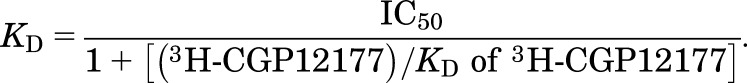 |
(2) |
For functional responses (3H-cAMP accumulation and CRE-SPAP gene transcription), most agonist responses were described by a one-site sigmoidal concentration response curve (eq. 3)
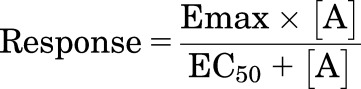 |
(3) |
where Emax is the maximum agonist response, [A] is the agonist concentration, and EC50 is the concentration of agonist that produces 50% of the maximal response.
However, several of the responses were best described by a two-site concentration response (eq. 4)
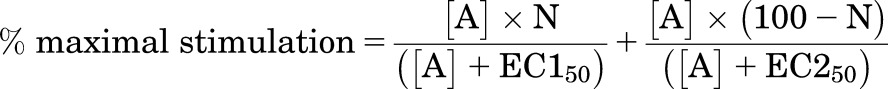 |
(4) |
where N is the percentage of site 1, [A] is the concentration of the agonist, and EC150 and EC250 are the respective EC50 values for the two agonist sites.
All data are presented as the mean ± S.E.M. of n separate experiments.
Synthesis of 7-Methylcyanopindolol
The synthesis of 7-methylcyanopindolol was performed in an eight-step procedure (Fig. 2) that is described in detail below (Steps 1–8). Where no preparative routes are included, the relevant intermediate is commercially available. Commercial reagents were used without further purification. Room temperature refers to approximately 20–27°C. 1H NMR spectra were recorded at 400 MHz on either a Bruker (Bruker, Berlin, Germany) or Jeol (JEOL, Peabody, MA) instrument. Chemical shift values are expressed in ppm, i.e., (δ)-values. The following abbreviations are used for the multiplicity of the NMR signals: s = singlet, br = broad, d = doublet, t = triplet, and m = multiplet. Coupling constants are listed as J values measured in Hertz. NMR and mass spectroscopy results were corrected to account for background peaks. Chromatography refers to column chromatography performed using 60–120 mesh silica gel and executed under nitrogen pressure (flash chromatography) conditions. Thin layer chromatography for monitoring reactions refers to thin layer chromatography run using the specified mobile phase and the Silica gel F254 as a stationary phase from Merck (Kenilworth, NJ).
Fig. 2.

Synthesis of 7-methylcyanopindolol. Reagents and conditions: (a) benzyl bromide, K2CO3, DMF, 65°C, 6 hours; (b) (i) methyl azidoacetate, sodium metal, MeOH, −8°C, 3 hours, (ii) xylene, 130°C, 18 hours; (c) (i) 2 N NaOH, EtOH, 90°C, 4 hours, (ii) 3 N HCl (aqueous) room temperature; (d) (i) SOCl2, Et2O, 0°C, 5 hours, (ii) NH3, Et2O, −20°C, 5 minutes; (e) H2, 10% Pd/C, MeOH, room temperature, 8 hours; (f) POCl3, 1,4-dioxane, 75°C, 15 minutes; (g) (S)-epichlorohydrine, NaOH, 5:2 H2O-1,4-dioxane, room temperature, 16 hours; (h) tert-butylamine, 70°C, 8 hours.
Liquid chromatography–mass spectometry (LCMS) experiments were typically carried out using electrospray conditions as specified for each compound under the following conditions.
Method A
Instrument.
A Waters Acquity H-class ultra performance liquid chromatography with SQ detector using ethylene bridged hybrid C18 (Milford, MA) (50*2.1 mm, i.d. 1.7 μm) and water (0.1% ammonium hydroxide) and MeCN (0.1% ammonium hydroxide) as the mobile phase was used. The eluent gradient program was MeCN (0.1% ammonium hydroxide) from 10 to 100% for 2.5 minutes, 100% MeCN (0.1% ammonium hydroxide) for 2 minutes, and MeCN (0.1% ammonium hydroxide) from 100 to 10% for 0.5 minutes. The flow rate was 0.3 ml/min.
Method B
Instruments.
HP1100 hplc system (Agilent Technologies, Santa Clara, CA) with the HP DAD G1315A detector and Micromass ZQ (Waters, Milford, CT) using a Phenomenex Gemini-NX C-18 (Torrance, CA), 3 μm, 2.0 × 30 mm column and water (0.1% ammonium hydroxide) and MeCN (0.1% ammonium hydroxide) as the mobile phase were used. The eluent gradient program was MeCN (0.1% ammonium hydroxide) from 0 to 95% for 8.4 minutes, 95% MeCN (0.1% ammonium hydroxide) for 1 minute, and MeCN (0.1% ammonium hydroxide) from 100 to 10% for 0.5 minutes. The flow rate was 1.5 ml/min, injection volume was 3 μl, column temperature was 45°C, and UV detection was from 230 to 400 nM.
Step 1: Synthesis of 2-(benzyloxy)-5-methylbenzaldehyde.
2-Hydroxy-5-methylbenzaldehyde (5.00 g, 36 mmol) was dissolved in dimethylformamide (30 ml); K2CO3 (6.07 g, 44 mmol) and benzyl bromide (4.36 ml, 36 mmol) were added; and the reaction mixture was warmed to 65°C and stirred for 6 hours. The reaction mixture was cooled to room temperature and poured into water (500 ml). The precipitate was filtered, washed with water (100 ml), and dried to give 2-(benzyloxy)-5-methylbenzaldehyde (7.0 g, 84% yield) as a white solid. LCMS (Method A): m/z 227 [M+H]+ (ES+) at 3.14 minutes, 99.6%. 1H NMR: (400 MHz, DMSO-d6) δ: 2.28 (s, 3H), 5.27 (s, 2H), 7.23 (d, J = 8.5, 1H), 7.33–7.36 (m, 1H), 7.39–7.42 (m, 2H), 7.43–7.47 (m, 1H), 7.48–7.51 (m, 3H), and 10.41 (br second, 1H).
Step 2: Synthesis of methyl 4-(benzyloxy)-7-methyl-1H-indole-2-carboxylate.
A solution of 2-(benzyloxy)-5-methylbenzaldehyde (1.87 g, 8.26 mmol) and methyl azidoacetate (4 ml, 40.8 mmol) in methanol (20 ml) was added dropwise to a solution of sodium metal (0.75 g, 33.04 mmol) in methanol (5 ml) at −20°C. The mixture was stirred at −8°C for 3 hours and then poured onto ice and filtered, and the precipitate was dissolved in xylene (20 ml). The reaction mixture was heated at 130°C for 18 hours. The reaction mixture was then cooled to 0°C, and the resulting precipitate was filtered and dried to give methyl 4-(benzyloxy)-7-methyl-1H-indole-2-carboxylate (0.9 g, 36% yield) as a white solid. LCMS (Method A): m/z 296 [M+H]+ (ES+) at 3.47 minutes, 100%. 1H NMR: (400 MHz, DMSO-d6) δ: 2.43 (s, 3H), 3.87 (s, 3H), 5.21 (s, 2H), 6.55 (d, J = 7.5, 1H), 6.94 (d, J = 7.5, 1H), 7.17–7.18 (m, 1H), 7.32–7.35 (m,1H), 7.41 (t, J = 7.5, 2H), 7.50–7.52 (m, 2H), and 11.81 (br second, 1H).
Step 3: Synthesis of 4-(benzyloxy)-7-methyl-1H-indole-2-carboxylic acid.
Methyl 4-(benzyloxy)-7-methyl-1H-indole-2-carboxylate (0.9 g, 3.0 mmol) was dissolved in ethanol (30 ml) and 2 N NaOH was added (41.4 ml, 105 mmol). The reaction mixture was stirred for 4 hours at 90°C. The reaction mixture was cooled to room temperature and acidified to pH 4 with aqueous 3 N HCl. The precipitate was filtered, washed with water (100 ml), and dried to give 4-(benzyloxy)-7-methyl-1H-indole-2-carboxylic acid (0.7 g, 82% yield) as a white solid. LCMS (Method A): m/z 280 [M−H]+ (ES−) at 3.16 minutes, 100%. 1H NMR: (400 MHz, DMSO-d6) δ: 2.41 (s, 3H), 5.19 (s, 2H), 6.49 (d, J = 7.5, 1H), 6.84 (d, J = 7.5, 1H), 6.97 (s, 1H), 7.31–7.34 (m, 1H), 7.40 (t, J = 7.5, 2H), 7.49–7.51 (m, 2H), and 11.27 (br second, 1H). One exchangeable proton was not observed.
Step 4: Synthesis of 4-(benzyloxy)-7-methyl-1H-indole-2-carboxamide.
4-(Benzyloxy)-7-methyl-1H-indole-2-carboxylic acid (0.9 g, 3.1 mmol) was dissolved in diethyl ether (18 ml). Thionyl chloride (0.78 g, 6.5 mmol) was added at 0°C, and the reaction mixture stirred at room temperature for 5 hours. The solvents were removed in vacuo, and the residues were dissolved in diethyl ether (10 ml). The reaction mixture was cooled to −20°C, and ammonia gas was bubbled through for 5 minutes. The solvents were removed in vacuo, and the crude product was purified by column chromatography (normal phase silica, 10–20% ethyl acetate in hexane) to give 4-(benzyloxy)-7-methyl-1H-indole-2-carboxamide (0.6 g, 67% yield) as a white solid. LCMS (Method A): m/z 281 [M+H]+ (ES+) at 3.04 minutes, 99.4%. 1H NMR: (400 MHz, DMSO-d6) δ: 2.42 (s, 3H), 5.18 (s, 2H), 6.52 (d, J = 7.5, 1H), 6.85 (d, J = 7.5, 1H), 7.25–7.27 (m, 2H), 7.32–7.35 (m, 1H), 7.42 (t, J = 7.5, 2H), 7.50–7.52 (m, 2H), 7.91 (br second, 1H), and 11.34 (br second, 1H).
Step 5: Synthesis of 4-hydroxy-7-methyl-1H-indole-2-carboxamide.
4-(Benzyloxy)-7-methyl-1H-indole-2-carboxamide (0.6 g, 2.4 mmol) was dissolved in methanol (20 ml), and 10% Pd/C (0.2 g, 1.8 mmol) was added. The reaction mixture was stirred at room temperature under 1 atmosphere of H2 gas for 8 hours. The reaction mixture was filter through celite, and the solvents were removed in vacuo to give 4-hydroxy-7-methyl-1H-indole-2-carboxamide (0.4 g, 86% yield) as a white solid. LCMS (Method A): m/z 191 [M+H]+ (ES+) at 0.59 minutes, 80.5%. 1H NMR: (400 MHz, DMSO-d6) δ: 2.37 (s, 3H), 6.28 (d, J = 7.5, 1H), 6.73 (d, J = 7.5, 1H), 7.15–7.17 (m, 1H), 7.27 (br second, 1H), 7.87 (br second, 1H), 9.38 (br second, 1H), and 11.09 (br second, 1H).
Step 6: Synthesis of 4-hydroxy-7-methyl-1H-indole-2-carbonitrile.
4-Hydroxy-7-methyl-1H-indole-2-carboxamide (0.4 g, 2.1 mmol) was dissolved in 1,4-dioxane (4 ml). POCl3 (0.5 ml) was added, and the reaction mixture was stirred at 75°C for 15 minutes. The reaction mixture was cooled to room temperature, partitioned and quenched with ammonia solution (5 ml), partitioned between H2O (50 ml) and EtOAc (100 ml), the aqueous layer was further extracted with EtOAc (3 × 100 ml), and the organic layers were combined and dried (Na2SO4). The solvent was removed in vacuo, and the crude product was purified by column chromatography (normal phase silica, 15–20% ethyl acetate in hexane) to give 4-hydroxy-7-methyl-1H-indole-2-carbonitrile (0.2 g, 55% yield) as a yellow solid. LCMS (Method A): m/z 171 [M−H]+ (ES−), at 1.56 minutes, 92.1%. 1H NMR: (400 MHz, DMSO-d6) δ: 2.34 (s, 3H), 6.37 (d, J = 7.5, 1H), 6.90 (d, J = 7.5, 1H), 7.32–7.33 (m, 1H), 9.71 (br second, 1H), and 12.11 (br second, 1H).
Step 7: Synthesis of 7-methyl-4-[(2S)-oxiran-2-ylmethoxy]-1H-indole-2-carbonitrile.
4-Hydroxy-7-methyl-1H-indole-2-carbonitrile (0.25 g, 1.4 mmol) was dissolved in water (2.5 ml) and 1,4-dioxane (1 ml). NaOH (0.058 g, 1.4 mmol) was added, followed by (S)-epichlorohydrine (1.25 ml, 0.58 mmol). The reaction mixture was stirred at room temperature for 16 hours. The reaction mixture was partitioned between H2O (50 ml) and ethyl acetate (50 ml), the aqueous layer was further extracted with ethyl acetate (2 × 50 ml), and the organic layers were combined and dried (Na2SO4). Solvent was removed in vacuo to give 7-methyl-4-[(2S)-oxiran-2-ylmethoxy]-1H-indole-2-carbonitrile (0.2 g, 60% yield) as a yellow gum, which was used in the next step without further purification. LCMS (Method A): m/z 229 [M+H]+ (ES+) at 1.94 minutes, 70%.
Step 8: Synthesis of 4-[(2S)-3-(tert-butylamino)-2-hydroxypropoxy]-7-methyl-1H-indole-2-carbonitrile.
7-Methyl-4-[(2S)-oxiran-2-ylmethoxy]-1H-indole-2-carbonitrile (0.19 g, 0.8 mmol) was dissolved in tert-butylamine (2.85 ml, 26.9 mmol), and the reaction mixture was stirred at 70°C for 8 hours. The solvent was removed in vacuo, and the residue was purified by reverse phase prep high-performance liquid chromatography [X Bridge, C-18, 150 × 30 mm, 5 μm, 30 ml per min, gradient 50–100% (over 10 minutes) and then 100% (2 minutes) acetonitrile in 10% acetonitrile/water] to give 4-[(2S)-3-(tert-butylamino)-2-hydroxypropoxy]-7-methyl-1H-indole-2-carbonitrile (0.02 g, 8% yield) as a light yellow solid. LCMS (Method B): m/z 302 [M+H]+ (ES+) at 3.95 minutes, 100%. 1H NMR: (400 MHz, DMSO-d6) δ: 1.09 (s, 9H), 2.36 (s, 3H), 2.56–2.68 (m, 2H), 3.83–3.86 (m, 1H), 3.93–3.97 (m, 1H), 4.02–4.06 (m, 1H), 6.48 (d, J = 7.8, 1H), 6.99 (d, J = 7.8, 1H), 7.30 (s, 1H), and 12.26 (br second, 1H). Two exchangeable protons were not observed. 13C NMR: (400 MHz, DMSO-d6) δ: 16.45, 29.25, 45.63, 50.17, 69.38, 71.22, 101.62, 104.73, 111.47, 114.42, 115.24, 117.72, 126.70, 138.40, and 151.32.
Expression, Purification, and Crystallization of β1AR
The β44-TS construct was used, which contained additional thermostabilizing mutations, I1293.40V, E1303.41W, and Y3437.53L (Miller and Tate, 2011), to the previously published turkey (Meleagris gallopavo) β1AR construct β44-m23 (Warne et al., 2009, 2011). The construct used here is identical to that used for the structure determination of β1AR at 2.1-Å resolution (Miller-Gallacher et al., 2014), except that it contains the E1303.41W mutation to improve the amount of functional receptor expressed and Asp322 is identical to the wild-type receptor instead of being mutated to Lys to form a salt bridge in the extracellular region. None of these mutations affect the structure of the binding pocket of the receptor. Ballesteros-Weinstein numbers are shown as superscripts (Ballesteros and Weinstein, 1995).
Expression of β44-TS using recombinant baculovirus and receptor purification were all performed as described previously (Warne et al., 2003, 2009), although expression was performed in High Five cells grown in ESF921 medium (Expression Systems, Davis, CA). A 2-fold increase in functional expression to 8 mg/l culture was observed with this construct than with β44-m23, which is most likely attributable to the inclusion of the mutation E1303.41W (Roth et al., 2008). Solubilization of the receptor from the membrane fraction was performed using 1.5% DM, and all buffers used in the purification contained 0.1% DM. The purified receptor was competitively eluted from the final alprenolol Sepharose affinity column with 100 μM 7-methyl-cyanopindolol in buffer [20 mM Tris-HCl (pH 7.6), 0.35 M NaCl, 0.1% DM, and 1 mM EDTA (pH 8.0)]. For desalting and concentration, the sample (15–20 ml) was concentrated to 0.1 ml using an Amicon-ultra concentrator (Ultracel-50K; Millipore Darmstadt, Germany), diluted 5- to 10-fold in dilution buffer [20 mM Tris-HCl (pH 7.6), 0.1 M NaCl, 0.1% DM, 0.1 mM EDTA, and 1 mM 7-methyl-cyanopindolol] and concentrated down again to 0.1 ml. This step was repeated twice, and finally the receptor was concentrated to 30–50 mg/ml. Before crystallization, CHS was added from a concentrated stock (10 mg/ml in 2% DM) to give a final concentration of 2 mg/ml CHS and 0.4% DM. Protein determination was performed using the amido black assay (Schaffner and Weissmann, 1973).
Crystals were generated using the lipidic cubic phase (LCP) method in monoolein. Monoolein and the purified receptor were mixed using two separate Hamilton syringes in a 3:2 (v/v) ratio of monoolein to protein solution. One hundred nanoliters of the protein/lipid mixture was dispensed on a plastic plate, overlaid by 500 nl of precipitant solution (18–36% polyethylene glycol 600 and 0.1 M N-(2-acetamido)iminodiacetic acid, pH 7.0) in each well with a mosquito-LCP (TTP Labtech, Melbourn, UK), sealed with a plastic cover, and stored in a humidified 23°C incubator. Crystals grew as thin plates in LCP and reached maximum dimensions of 200 × 200 × ∼10 μM. Crystals were directly picked from LCP and cryo-cooled in liquid nitrogen.
Data Collection, Structure Solution, and Refinement.
Diffraction data were collected from four cryo-cooled crystals on beamline ID29 (wavelength: 0.9723Å) at the European Synchrotron Radiation Facility (Grenoble, France) and on beamline I24 (wavelength: 0.9687Å) at the Diamond Light Source (Oxfordshire, UK). Thirteen wedges of data from four crystals were merged. Images were processed with MOSFLM (Leslie, 2006) and AIMLESS (Evans, 2006, 2011). The structure was solved by molecular replacement with PHASER (McCoy et al., 2007) using the β1AR-LCP structure with cyanopindolol bound (Miller-Gallacher et al., 2014) as a starting model. Refinement, rebuilding, and validation were carried out with REFMAC5 (Murshudov et al., 1997, 2011), COOT (Emsley et al., 2010), and MOLPROBITY (Davis et al., 2007; Chen et al., 2010).
Results
Affinity of 7-Methylcyanopindolol for Turkey β1AR and Human β2AR.
7-Methylcyanopindolol was synthesized in an eight-step process to yield 20 mg of pure product, as defined by LCMS, and the structure was confirmed by 1H and 13C NMR (see Materials and Methods). The affinity of 7-methylcyanopindolol for turkey β1AR and human β2AR was compared with other well characterized antagonists through competition binding assays using 3H-CGP12177 and performed on whole cells (Fig. 3; Table 1). The affinity of turkey β1AR for 7-methylcyanopindolol was 43 ± 3 pM (n = 6), which is very similar to its affinity for cyanopindolol (35 ± 3 pM; n = 6). Human β2AR bound both ligands with similar affinities to turkey β1AR. The affinities of the receptors for two other antagonists (used in the efficacy studies below), carazolol and ICI118551, were also determined and found to be similar to previous determinations (Baker, 2010a,b).
Fig. 3.

Competition binding curves of ligands to turkey β1AR and human β2AR. Inhibition of 3H-CGP12177 binding to (A) CHO cells stably expressing the turkey β1AR (CHO-tβ1) and (B) CHO-hβ2 cells was measured in response to cyanopindolol (filled circles), 7-methylcyanopindolol (open circles), carazolol (filled triangles), 2-hydroxy-5-(2-[(hydroxy-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]propyl)amino]ethoxy)benzamide (open inverted triangles), and ICI118551 (stars). Log KD values are given in Table 1. Bars to the left of the graphs represent total 3H-CGP12177 binding (filled bar) and nonspecific binding (hatched bar) as determined in the presence of 10 μM of propranolol. The concentrations of 3H-CGP12177 present were (A) 0.98 nM and (B) 0.53 nM. Data points are the mean ± S.E.M. of triplicate determinations, and these single experiments are representative of (A) five and (B) 10 separate experiments.
TABLE 1.
Log KD values obtained from 3H-CGP12177 whole cell binding in CHO-tβ1 and CHO-hβ2 cells
Values represent mean ± S.E.M. of n separate experiments.
| Ligand |
β1AR |
β2AR |
||
|---|---|---|---|---|
| Log KD | n | Log KD | n | |
| Cyanopindolol | −10.46 ± 0.04 | 6 | −10.49 ± 0.01 | 12 |
| 7-Methylcyanopindolol | −10.37 ± 0.03 | 6 | −10.42 ± 0.04 | 14 |
| Carazolol | −10.23 ± 0.04 | 5 | −10.54 ± 0.04 | 10 |
| (±)-Isoprenaline | −7.34 ± 0.11 | 5 | −6.57 ± 0.08 | 8 |
| ICI118551 | −7.17 ± 0.06 | 5 | −9.36 ± 0.07 | 10 |
| CGP20712A | −7.76 ± 0.05 | 5 | −5.76 ± 0.02 | 10 |
CGP20712A, 2-hydroxy-5-(2-[(hydroxy-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]propyl)amino]ethoxy)benzamide; CHO-tβ1, CHO cells stably expressing the turkey β1AR.
Efficacy of 7-Methylcyanopindolol at Turkey β1AR.
Previous studies have shown a large response window in 3H-cAMP accumulation measurements in a CHO cell line stably expressing turkey β1AR (148 fmol of receptor per mg protein) to measure small responses in ligand efficacy (Baker, 2010b). Isoprenaline stimulated an increase in 3H-cAMP accumulation that was 22.5 ± 1.3-fold over basal, confirming a large response window (n = 4; Fig. 4). As previously observed (Baker, 2010a), cyanopindolol elicited a biphasic response curve, with the maximum response reaching 31.8 ± 1.1% (n = 4) of the response elicited by the full agonist isoprenaline (Table 2). In contrast, 7-methylcyanopindolol gave a response that reached a maximum of 2.3 ± 0.3% (n = 4) of that attained by isoprenaline. While it appeared that this response was best described by a single site sigmoidal response curve, a second component cannot be excluded because the increase in 3H-cAMP was so small. ICI118551, a potent inverse agonist of β2AR (Bond et al., 1995; Azzi et al., 2001), gave no response. Carazolol was previously described as a partial inverse agonist of the human β2AR (Rosenbaum et al., 2007), although other studies have shown it to stimulate a partial agonist response at the human β1AR and β2AR (Baker, 2010b) and turkey β1AR. Here, carazolol was again shown to be a weak partial agonist of turkey β1AR, and it elicited a biphasic response similar to that seen for cyanopindolol (Fig. 4; Table 2) and the human β1AR (Baker, 2010b).
Fig. 4.

3H-cAMP accumulation in whole cells expressing either turkey β1AR or human β2AR. The responses to either cyanopindolol (filled circles), 7-methylcyanopindolol (open circles), carazolol (filled triangles), or ICI118551 (stars) was measured either in (A and B) CHO cells stably expressing the turkey β1AR (CHO-tβ1) or (C and D) CHO-hβ2 cells. Log EC50 values are given in Tables 2 and 3. The inset graph in (A) is a magnified portion of the main graph, with the y-axis altered to see only the bottom 2000 dpm. Bars to the left of the graphs represent basal 3H-cAMP accumulation (filled bar) and the response to 10 µM of isoprenaline (hatched bar). Data points are the mean ± S.E.M. of triplicate determinations, and these single experiments are representative of four separate experiments in each case.
TABLE 2.
CRE-SPAP gene transcription response and 3H-cAMP accumulation in ligand-stimulated tβ1AR-CHO cell lines
Cyanopindolol and carazolol stimulated concentration responses that were best described by a two-component curve. Here, the log EC50 values are given for each component. The percentage of the response represented by the first component and the percentage of the total response in relation to the isoprenaline stimulation are given. Values represent the mean ± S.E.M. of n separate experiments.
| Log EC150 |
Log EC250 |
Site 1 |
Iso Maximum |
n | |
|---|---|---|---|---|---|
| % | % | ||||
| 3H-cAMP accumulation | |||||
| Cyanopindolol | −10.25 ± 0.02 | −7.31 ± 0.09 | 64.8 ± 2.4 | 31.8 ± 1.1 | 4 |
| 7-Methylcyanopindolol | −9.76 ± 0.14 | 2.3 ± 0.3 | 4 | ||
| Carazolol | −9.95 ± 0.10 | −6.76 ± 0.06 | 40.2 ± 1.2 | 22.4 ± 0.9 | 4 |
| ICI118551 | No response | 4 | |||
| CRE-SPAP production | |||||
| Cyanopindolol | −10.22 ± 0.06 | −7.19 ± 0.22 | 56.1 ± 5.2 | 40.2 ± 2.4 | 8 |
| 7-Methylcyanopindolol | No response | 8 | |||
| Carazolol | −9.74 ± 0.22 | −6.66 ± 0.25 | 28.7 ± 3.2 | 31.8 ± 3.2 | 4 |
ICI118551, (–)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)-amino]-2-butanol. Iso Max, maximum response compared to the response of isoprenaline.
Efficacy of 7-Methylcyanopindolol at Human β2AR.
The results from the previous section indicated that 7-methylcyanopindolol was an exceedingly weak partial agonist of turkey β1AR, but as no convincing inverse agonism was detected in this assay with ICI118551, the experiments were repeated on human β2AR (cell line CHO-hβ2) (receptor expression level of 466 fmol receptor per mg protein), which has higher basal activity than β1AR (Engelhardt et al., 2001). Isoprenaline stimulated a large increase in 3H-cAMP accumulation in CHO-β2 cells (61.4 ± 5.4-fold over basal; n = 8; Fig. 4). Cyanopindolol was found to stimulate an agonist response (with a maximum stimulation of 8.0% that of isoprenaline; Fig. 4; Table 3), and a small stimulation was also seen in response to carazolol. When 7-methylcyanopindolol was examined, there was no increase in 3H-cAMP accumulation, but rather a very small decrease in basal activity. As inverse agonism has previously been reported in this cell line (Baker et al., 2003), the response to 7-methylcyanopindolol was examined alongside that of the known inverse agonist ICI118551 (Fig. 4). ICI118551 was confirmed to have inverse agonist activity resulting in a decrease of basal activity by 55%. The inverse agonist activity of 7-methylcyanopindolol was found to be less, causing a decrease in basal activity by 25%.
TABLE 3.
CRE-SPAP and 3H-cAMP accumulation assays for β2AR-CHO cell lines
Log EC50 values and percent isoprenaline maximal responses obtained from 3H-cAMP accumulation and CRE-SPAP gene transcription responses in CHO-hβ2 cells. 7-methylcyanopindolol and ICI118551 stimulated inverse agonist concentration responses; thus, IC50 values are given with a comparison with the response in relation to the basal activity, where basal activity = 100%. Values represent mean ± S.E.M. of n separate experiments.
| Log EC50/IC50 |
Iso Maximum |
Basal |
n |
|
|---|---|---|---|---|
| % | % | |||
| 3H-cAMP accumulation | ||||
| Cyanopindolol | −9.86 ± 0.04 | 8.0 ± 0.9 | — | 8 |
| 7-Methylcyanopindolol | −9.99 ± 0.10 | 74.5 ± 9.2 | 8 | |
| Carazolol | −10.14 ± 0.09 | 1.3 ± 0.2 | — | 8 |
| ICI118551 | −9.48 ± 0.17 | 45.3 ± 3.3 | 7 | |
| CRE-SPAP production | ||||
| Cyanopindolol | −9.97 ± 0.05 | 50.8 ± 3.7 | — | 8 |
| 7-Methylcyanopindolol | No response | — | — | 8 |
| Carazolol | −10.30 ± 0.13 | 6.0 ± 1.4 | — | 4 |
| ICI 118551 | No response | — | — | 6 |
ICI118551, (–)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)-amino]-2-butanol. Iso Max, maximum response compared to the response of isoprenaline.
Efficacy of 7-Methylcyanopindolol in CRE-Gene Transcription Responses.
The β-antagonist propranolol acts as an inverse agonist of the G protein–coupled pathway, causing a decrease in cAMP production, but it stimulates CRE-gene transcription in the CHO-hβ2 cell line through signaling via the G protein–independent mitogen-activated protein kinase pathway through β-arrestins (Baker et al., 2003). As 7-methylcyanopindolol was found to be a weak inverse agonist, its response at the level of CRE-gene transcription was also investigated. For turkey β1AR, the CRE-gene transcription responses observed for cyanopindolol was similar to the response in the 3H-cAMP accumulation studies (Fig. 5; Table 2), but no response was detected for 7-methylcyanopindolol. For human β2AR, an agonist response was observed for cyanopindolol, but 7-methylcyanopindolol did not stimulate an increase in CRE-SPAP production (Fig. 5; Table 3). There is thus no evidence for biased signaling through β-arrestins by 7-methylcyanopindolol.
Fig. 5.

CRE-SPAP production in response to 7-methylcyanopindolol and cyanopindolol. (A) CHO cells stably expressing the turkey β1AR (CHO-tβ1), (B) CHO-hβ2 cells, and (C) CHO-CRE-SPAP cells (parental cells without the transfected receptor) were treated with either cyanopindolol (filled circles) or 7-methylcyanopindolol (open circles), and the amount of secreted placental alkaline phosphatase was measured. Log EC50 values are given in Tables 2 and 3. Bars to the left of the graphs represent either basal CRE-SPAP production in the absence of the ligand (filled bar) or the response to 10 µM of isoprenaline (hatched bar). In the parental cell line (C), forskolin was used as the positive control (diagonal hatching of the bar). Data points are mean ± S.E.M. of triplicate determinations. These single experiments are representative of (A) eight, (B) eight, and (C) four separate experiments. Error bars are shown, but are smaller than the size of the symbols.
Negative control experiments in CHO-CRE-SPAP cells (i.e., with no transfected receptor) showed that 10 μM of forskolin stimulated a response that was 18.3 ± 4.7-fold over basal (n = 4) in the 3H-cAMP accumulation assay (not shown) and 5.9 ± 0.5-fold over basal (n = 4) in the CRE-gene transcription assays. No response was observed in CHO-CRE-SPAP cells upon addition of either cyanopindolol, 7-methyl-cyanopindolol, carazolol, or isoprenaline (n = 4 in each case; Fig. 5).
Structure of 7-Methylcyanopindolol–Bound β1AR.
To produce high-quality crystals of β1AR, a thermostabilized version of the receptor was used, which contained nine thermostabilizing mutations in addition to truncations at the N terminus, C terminus, and cytoplasmic loop 3 (see Materials and Methods). Thermostabilized β1AR was expressed using the baculovirus expression system, purified by Ni2+-affinity chromatography and alprenolol sepharose affinity chromatography (Warne et al., 2003, 2009), and crystallized using the lipidic cubic phase technique (Miller-Gallacher et al., 2014). Data were collected at synchrotron microfocus beamlines, and the structure was determined by molecular replacement and refined to a final resolution of 2.4 Å (Table 4). The final model contained one receptor molecule per asymmetric unit, which was associated with two Na+ ions, five lipids, and 26 water molecules, with good density for 7-methylcyanopindolol in the ligand-binding pocket (Fig. 6). The receptor is in the inactive state and is virtually identical to the structure of the cyanopindolol-bound receptor (PDB code 4BVN; root-mean-square deviation (RMSD) 0.19 Å over 2062 atoms) (Miller-Gallacher et al., 2014). The major difference in the ligand-binding pocket between β1AR bound to either cyanopindolol or 7-methylcyanopindolol is that the hydroxyl group of Ser2155.46 is displaced 0.8 Å from its position when cyanopindolol is bound in a direction away from the center of the receptor.
TABLE 4.
Data collection and refinement statistics
| β1AR–7-Methylcyanopindolol | |
|---|---|
| Number of crystals | 4 |
| Space group | P21221 |
| Unit cell parameters | |
| a, b, and c (Å) | 53.0, 61.8, 95.5 |
| α, β, and γ (°) | 90, 90, 90 |
| Data processing | |
| Resolution (Å) | 37.8–2.4 |
| Rmergea | 0.161 (0.704) |
| < I/σ(I) >a | 8.2 (1.9) |
| Completeness (%)a | 98.4 (98.3) |
| Multiplicitya | 4.8 (4.9) |
| Wilson B factor (Å2) | 27.7 |
| Refinement | |
| Total number of reflections | 11,942 |
| Total number of atoms | 2436 |
| Number of waters | 26 |
| Number of lipid molecules | 5 |
| Number of sodium ions | 2 |
| Rworkb,d | 0.217 (0.294) |
| Rfreec,d | 0.248 (0.285) |
| RMSD bonds (Å) | 0.007 |
| RMSD angles (o) | 1.29 |
| Mean atomic B factor (Å2) | 39.35 |
| Estimated coordinate error (Å) | 0.223 |
| Ramachandran plot favored (%)e | 98.94 |
| Ramachandran plot outliers (%)e | 0 |
The values in parentheses are for the highest resolution bin (2.53–2.4 Å).
The number of reflections used to calculate Rwork is 11,942.
The number of reflections from a randomly selected subset used to calculate Rfree is 602.
The values in parentheses are for the highest resolution bin for refinement (2.46–2.4 Å)
The figures were obtained using MolProbity.
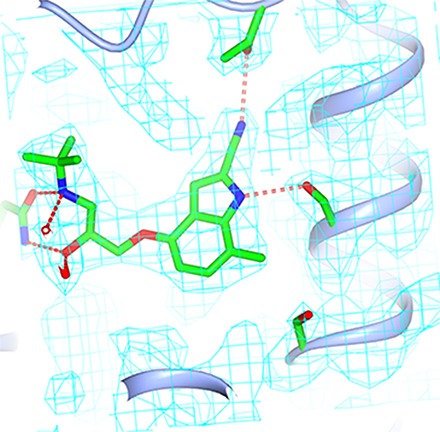
Fig. 6.

Structure of 7-methylcyanopindolol–bound β1AR. (A) Omit map for the ligand-binding site. A 2Fo-Fc map was generated where the ligand and side chains of Ser211 and Ser215 were omitted from the model. The contour level is 1.0 sigma, and the figure was produced using CCP4MG. (B and C) Superposition of the structures of β1AR bound to 7-methylcyanopindol (receptor in rainbow coloration and the ligand in pink) and cyanopindol (receptor and ligand are both gray). The views are either within the membrane plane (A and B) or from the extracellular surface (C), with portions of H3 removed for clarity. The red arrows highlight the different positions of Ser215 in the two β1AR structures. The Ballesteros-Weinstein numbers for the residues depicted are as follows: D121, 3.32; S211, 5.42; S215, 5.46; and N329, 7.39.
Discussion
The synthesis of 7-methylcyanopindolol has allowed further understanding of the components that affect ligand efficacy in β1AR. Previous data suggested that the rotamer change of Ser2155.46 and a contraction of the ligand-binding pocket were sufficient to increase the probability of formation of the activated state of the receptor (Warne et al., 2011; Warne and Tate, 2013). Therefore, ligands that prevented rotamer changes of Ser2155.46 and prevented contraction of the orthosteric binding pocket should, in theory, have greatly reduced efficacy and may even become inverse agonists. Cyanopindolol was described previously as a weak partial agonist of both human and turkey β1AR (Baker, 2010a,b) and we therefore synthesized 7-methylcyanopindolol with the methyl group in a position predicted to dramatically decrease its efficacy. Pharmacological analysis of 7-methylcyanopindolol showed that it bound with similar high affinity to both turkey β1AR and human β2AR, and that there was a marked reduction in efficacy at both receptors. However, at the turkey β1AR, 7-methylcyanopindol was an exceedingly weak partial agonist, whereas for β2AR, the ligand acted as a weak partial inverse agonist. Thus, the goal was achieved in reducing efficacy, but raised the question of why a tiny amount of agonist activity could still be detected at β1AR, whereas ICI118551 did not elicit any response.
In efforts to further understand the molecular mechanism of 7-methylcyanopindolol, the structure of 7-methylcyanopindolol–bound β1AR was determined at 2.4-Å resolution, which is sufficient to unambiguously determine the rotamer conformation and precise positioning of serine side chains. Comparison of this structure with structures of β1AR bound either to cyanopindolol (Miller-Gallacher et al., 2014) or carazolol (Moukhametzianov et al., 2011), both weak partial agonists, showed that the binding pocket was actually slightly larger than expected by 0.3–0.5 Å, as measured between the Cα atoms of Ser2115.42 and Asn3297.39. In addition, there was a difference in position of the hydroxyl group of Ser2155.46 in the 7-methylcyanopindolol–bound structure compared with the cyanopindolol-bound structure of 0.8 Å in a direction away from the ligand, presumably due to the proximity of the methyl group of 7-methylcyanopindolol. Both the increase in size of the binding pocket and the outward shift of the hydroxyl group of Ser2155.46 are consistent with the dramatic reduction in efficacy of 7-methylcyanopindolol compared with cyanopindolol. If an analogous comparison is performed in the structures of β2AR bound to the weak partial agonist carazolol (Cherezov et al., 2007) and the inverse agonist ICI118551 (Wacker et al., 2010), exactly the same differences are observed, i.e., an expansion of the binding pocket of 0.4 Å (measured as above) and a 0.7-Å shift of the hydroxyl group of Ser2075.46 (Fig. 7).
Fig. 7.

Comparison of the orthosteric binding site of β1AR and β2AR bound to either carazolol, ICI118551, or 7-methylcyanopindolol. (A) The structure of 7-methylcyanopindolol–bound β1AR (rainbow-colored cartoon) was aligned with the structure of carazolol-bound β1AR (gray cartoon; PDB code 2YCW, chain A) using PyMol (RMSD 0.5 Å, 1734 atoms). A stick format is used to depict the ligands and side chains (labeled) that interact with the ligands via hydrogen bonds: 7-methylcyanopindolol–bound β1AR (carbon atoms are in green and hydrogen bonds are depicted as red dashed lines) and carazolol-bound β1AR (carbon atoms are in gray and hydrogen bonds are depicted as blue dashed lines). (B) The structure of ICI118551-bound β2AR (green-colored cartoon; PDB code 3NY8) was aligned with the structure of carazolol-bound β2AR (brown cartoon; PDB code 2RH1) using PyMol (RMSD 0.4 Å, 1810 atoms). A stick format is used to depict the ligands and side chains (labeled) that interact with ligands via hydrogen bonds: ICI118551-bound β2AR (carbon atoms are in green and hydrogen bonds are depicted as red dashed lines) and carazolol-bound β2AR (carbon atoms are in gray and hydrogen bonds are depicted as blue dashed lines). In both panels, oxygen atoms are red and nitrogen atoms are blue. The Ballesteros-Weinstein numbers for the residues depicted are as follows (β1AR and β2AR residues are given respectively): D121 and D113, 3.32; S211 and S203, 5.42; S215 and S207, 5.46; N329 and N312, 7.39; and Y333 and Y316, 7.43.
Although Ser2155.46 appears to be a major determinant in defining ligand efficacy, two other serine residues have also been implicated by mutagenesis in the activation of β-receptors, namely, Ser2115.42 and Ser2125.43 (Strader et al., 1989; Liapakis et al., 2000). The orientation of Ser2115.42 has been suggested to correlate with decreased efficacy of ligands based on the comparison of β1AR structures bound to eight different ligands (Warne and Tate, 2013). Structures of β1AR bound to agonists and partial agonists, including β1AR bound to cyanopindolol and 7-methylcyanopindol, invariably have the side chain of Ser2115.42 in a gauche+ rotamer, whereas β1AR bound to carazolol and carvedilol has Ser2115.42 in a trans-rotamer (see Fig. 7A). Thus, one possibility to explain the residual agonist activity of 7-methylcyanopindolol is that the slight destabilization of the H5 interface between H3 and H4 caused by the gauche+ orientation of the Ser2115.42 side chain is sufficient to make the ligand an extremely weak partial agonist. The role of Ser2125.43 in determining ligand efficacy is less clear, as it does not make direct contact to the ligand, but instead forms a hydrogen bond with Asn3106.55, which may assist in orienting the side chain for optimal hydrogen bond formation with full agonists (Warne et al., 2011). As Asn3106.55 forms van der Waals contacts with the weak partial agonists studied here, as opposed to a hydrogen bond with full agonists, then its effect may be minimal in determining the efficacy of the weak partial agonists given that different rotamers of Asn3106.55 would presumably always be in weak van der Waals contact with the ligand. This view is supported by the absence of the Ser2125.43-Asn3106.55 hydrogen bond in one structure of cyanopindolol-bound β1AR (Miller-Gallacher et al., 2014), although it is clearly present in other cyanopindolol-bound structures (Warne et al., 2008).
In considering the factors that dictate the efficacy of a ligand for β1AR, there appears to be a hierarchy of factors that have a diminishing effect as you go down the list. First, the most important factor is probably the contraction of the binding pocket upon agonist binding, which has been observed in all structures bound to full or partial agonists (Warne et al., 2011). Without this contraction, it is unlikely that hydrogen bonds would be able to form between the catecholamine moiety of the agonist and the side chains of Ser2155.46 and Ser2115.42. Second, the rotamer change of Ser2155.46 is important as this dictates the difference in binding between a full and partial agonist and allows hydrogen bond formation to the ligand (Warne et al., 2011). Third, the rotamer change of Ser2115.42, in addition to its defining role in the binding of partial agonists, may promote lower levels of activity in antagonists when the binding pocket is less likely to contract and Ser2155.46 is less likely to rotate due to steric clashes with the ligand (Warne and Tate, 2013). Currently, we do not know where in this hierarchy the importance of Ser2125.43 falls. Mutagenesis suggests that this is an important residue in determining agonist affinity in β2AR (Strader et al., 1989), and in the structures of β1AR, it usually forms a hydrogen bond with Asn310, which in turn forms a strong hydrogen bond to the catecholamine headgroup (or equivalent) when agonists bind to the receptor (Warne et al., 2011). As it appears that the hydrogen bond is present in the activated state of the β2AR bound to the G protein mimic Nb80 (Rasmussen et al., 2011a), it seems likely that ligands that reduce the probability of this bond forming will have poor efficacy, although no such ligands have been described so far.
In conclusion, we have demonstrated that a single modification of cyanopindolol to make 7-methylcyanopindolol was sufficient to dramatically reduce the efficacy of the ligand and the structure of the ligand-receptor complex has confirmed the importance of the rotamer conformation of Ser2155.46 in receptor activation and inverse agonist activity. The unexpected residual agonist activity of the ligand highlights the role of Ser2115.42 in ligand-induced activation of β1AR and that it is not just playing a passive role in increasing ligand affinity. Further work will therefore be needed to finally make a high-affinity inverse agonist for β1AR.
Coordinates and structure factors for 7-methylcyanopindolol–bound β1AR have been deposited with the Protein Data Bank (PDB ID 5A8E).
Acknowledgments
Diffraction data were collected at the European Synchotron Radiation Facility (ID29; Grenoble, France) and Diamond Light Source (I24; Harwell, UK). We would like to thank Beamline staff there for their continued assistance.
Abbreviations
- 7-methylcyanopindolol
4-[(2S)-3-(tert-butylamino)-2-hydroxypropoxy]-7-methyl-1H-indole-2-carbonitrile
- AR
adrenergic receptor
- carazolol
1-(9H-carbazol-4-yloxy)-3-(propan-2-ylamino)propan-2-ol
- CGP12177
(–)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one
- CHO
Chinese hamster ovary
- CHO-CRE-SPAP
Chinese hamster ovary cells with the stably transfected reporter but without any transfected receptor
- CHO-hβ2
Chinese hamster ovary cells stably expressing the human β2-adrenoceptor
- CHS
cholesteryl hemisuccinate
- CRE
cAMP response element
- CRE-SPAP
reporter gene containing six cAMP response elements upstream of a secreted placental alkaline phosphatase reporter gene
- cyanopindolol
(RS)-4-[3-(tert-butylamino)-2-hydroxypropoxy]-1H-indole-2-carbonitrile
- DM
n-decyl-β-D-maltopyranoside
- LCP
lipidic cubic phase
- RMSD
root-mean-square deviation
Authorship Contributions
Participated in research design: Sato, Baker, Warne, Tate.
Conducted experiments: Sato, Baker, Brown, Warne.
Performed data analysis: Sato, Baker, Warne, Leslie.
Wrote or contributed to the writing of the manuscript: Sato, Baker, Brown, Warne, Congreve, Leslie, Tate.
Footnotes
C.G.T. is a consultant for Heptares Therapeutics Ltd. This work was funded by a core grant from the Medical Research Council to C.G.T. [Grant MRC U105197215] and A.G.W.L. [Grant MRC U105184325] and a Wellcome Trust Clinician Scientist Fellowship awarded to J.G.B. [Grant number 073377/Z/03/Z].
References
- Azzi M, Piñeyro G, Pontier S, Parent S, Ansanay H, Bouvier M. (2001) Allosteric effects of G protein overexpression on the binding of beta-adrenergic ligands with distinct inverse efficacies. Mol Pharmacol 60:999–1007. [DOI] [PubMed] [Google Scholar]
- Baker JG. (2010a) A full pharmacological analysis of the three turkey β-adrenoceptors and comparison with the human β-adrenoceptors. PLoS One 5:e15487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG. (2010b) The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br J Pharmacol 160:1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. (2003) Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol 64:1357–1369. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. (1995) Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors. Methods Neurosci 25:366–428. [Google Scholar]
- Bond RA, Leff P, Johnson TD, Milano CA, Rockman HA, McMinn TR, Apparsundaram S, Hyek MF, Kenakin TP, Allen LF, et al. (1995) Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the beta 2-adrenoceptor. Nature 374:272–276. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher JA, Brown J, Doré AS, Errey JC, Koglin M, Marshall FH, Myszka DG, Rich RL, Tate CG, Tehan B, et al. (2013) Biophysical fragment screening of the β1-adrenergic receptor: identification of high affinity arylpiperazine leads using structure-based drug design. J Med Chem 56:3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375-W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt S, Grimmer Y, Fan GH, Lohse MJ. (2001) Constitutive activity of the human beta(1)-adrenergic receptor in beta(1)-receptor transgenic mice. Mol Pharmacol 60:712–717. [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. [DOI] [PubMed] [Google Scholar]
- Evans PR. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 67:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Tate CG. (2012) Agonist-bound structures of G protein-coupled receptors. Curr Opin Struct Biol 22:482–490. [DOI] [PubMed] [Google Scholar]
- Leslie AG. (2006) The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 62:48–57. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. (2000) The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptor. J Biol Chem 275:37779–37788. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JL, Tate CG. (2011) Engineering an ultra-thermostable β(1)-adrenoceptor. J Mol Biol 413:628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Gallacher JL, Nehmé R, Warne T, Edwards PC, Schertler GF, Leslie AG, Tate CG. (2014) The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One 9:e92727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moukhametzianov R, Warne T, Edwards PC, Serrano-Vega MJ, Leslie AG, Tate CG, Schertler GF. (2011) Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc Natl Acad Sci USA 108:8228–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. (2011a) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011b) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318:1266–1273. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, et al. (2011) Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature 469:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth CB, Hanson MA, Stevens RC. (2008) Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. J Mol Biol 376:1305–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner W, Weissmann C. (1973) A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 56:502–514. [DOI] [PubMed] [Google Scholar]
- Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. (1989) Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. J Biol Chem 264:13572–13578. [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194. [DOI] [PubMed] [Google Scholar]
- Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. (2010) Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc 132:11443–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Chirnside J, Schertler GF. (2003) Expression and purification of truncated, non-glycosylated turkey beta-adrenergic receptors for crystallization. Biochim Biophys Acta 1610:133–140. [DOI] [PubMed] [Google Scholar]
- Warne T, Edwards PC, Leslie AG, Tate CG. (2012) Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 20:841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AG, Schertler GF, Tate CG. (2011) The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature 469:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Tate CG, Schertler GF. (2009) Development and crystallization of a minimal thermostabilised G protein-coupled receptor. Protein Expr Purif 65:204–213. [DOI] [PubMed] [Google Scholar]
- Warne T, Tate CG. (2013) The importance of interactions with helix 5 in determining the efficacy of β-adrenoceptor ligands. Biochem Soc Trans 41:159–165. [DOI] [PubMed] [Google Scholar]