

Abstract
The search for biomarkers that characterize specific aspects of inflammatory bowel disease (IBD), has received substantial interest in the past years and is moving forward rapidly with the help of modern technologies. Nevertheless, there is a direct demand to identify adequate biomarkers for predicting and evaluating therapeutic response to different therapies. In this subset, pharmacogenetics deserves more attention as part of the endeavor to provide personalized medicine. The ultimate goal in this area is the adjustment of medication for a patient’s specific genetic background and thereby to improve drug efficacy and safety rates. The aim of the following review is to utilize the latest knowledge on immunopathogenesis of IBD and update the findings on the field of Immunology and Genetics, to evaluate the response to the different therapies. In the present article, more than 400 publications were reviewed but finally 287 included based on design, reproducibility (or expectancy to be reproducible and translationable into humans) or already measured in humans. A few tests have shown clinical applicability. Other, i.e., genetic associations for the different therapies in IBD have not yet shown consistent or robust results. In the close future it is anticipated that this, cellular and genetic material, as well as the determination of biomarkers will be implemented in an integrated molecular diagnostic and prognostic approach to manage IBD patients.
Keywords: Mucosal immunology, Biomarkers, Pharmacology, Mode of action, Therapeutic drug monitoring
Core tip: The following article is an update on the latest findings on the pathogenesis of inflammatory bowel disease (IBD) and its correlation with genetic and non-genetic predictors of the efficacy of the different strategies of treatment. Although many therapies have been used for decades, this is a completely new approach that has become even more complicated with new therapies like biologics. While most of these strategies are still in a very early stage, and have not been validated in clinical practice, they have begun suggesting the direction in which physicians should start looking to establish the most adequate therapeutic strategy for each individual patient.
INTRODUCTION
The search for biomarkers that characterize specific aspects of inflammatory bowel disease (IBD), has received substantial interest in the past years and is moving forward rapidly with the help of modern technologies. Currently, biomarkers are more progressively used in routine clinical care of patients with IBD. Most biomarkers used are not disease specific, but in general reflect inflammation. The last decade has brought significant gains in insight to IBD genetics and pathogenesis. These insights have the potential to improve the utility of biomarkers currently in use in clinical practice or are under investigation in clinical trials[1].
Although some reviews have been recently published on biomarkers[1], the most lacking topic is possibly is to identify adequate biomarkers for predicting and evaluating therapeutic response to different therapies which is less developed. With the progress in genetics research in IBD, genetic markers are increasingly being proposed to improve stratification of patients. Nevertheless, none of the genetic variants associated with particular outcomes have shown sufficient sensitivity or specificity to be implemented in daily management, maybe with the exception on those related to thiopurine metabolism.
Along a same line of thinking, pharmacogenetics, the study of association between variability in drug response and genetic variation, has also received more attention as part of the endeavor for personalized medicine. The ultimate goal in this area of medicine is the adaptation of medication for a patient’s specific genetic background and therefore to improve drug efficacy and safety.
The aim of the following review is to utilize the latest knowledge on immunopathogenesis of IBD and update the findings in the field of Immunology and Genetics, to evaluate the response to the different therapies with the intent to predict the outcome within the diverse therapeutic strategies.
IMMUNOPATHOGENESIS OF IBD
The exact cause of IBD is still unknown, but is thought to be due to a combination of a patient’s microbiome, immune response, and the environment that result in an excessive and abnormal immune response against commensal flora in genetically susceptible individuals (Figure 1).
Figure 1.
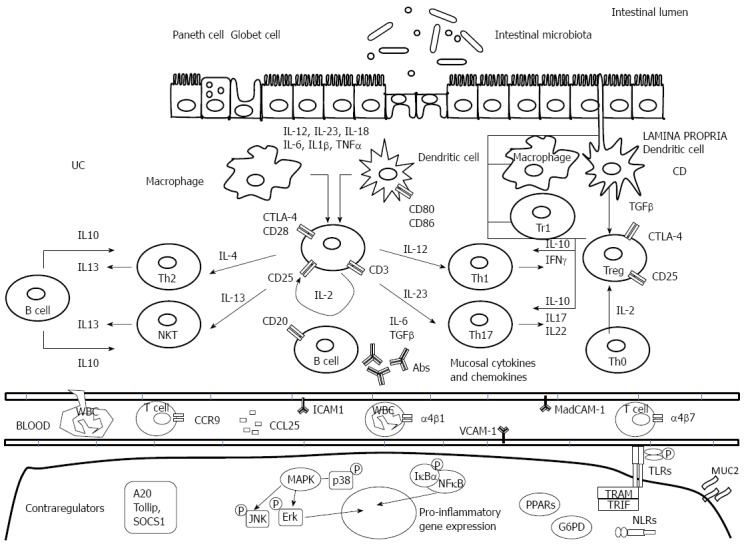
Inflammatory and regulatory pathways involved in inflammatory bowel disease pathogenesis. Crohn’s disease (CD) is characterized by the generation of Th1- and Th17 T cell responses driven by the production of interleukin (IL)-12, IL-18, IL-23, IL-6 and tumor necrosis factor (TNF)-α by dendritic cells and macrophages. Th1-cells secrete IL-2, IL-17, interferon (IFN)-γ, and TNF-α. Ulcerative colitis (UC) is characterized by a Th2- T cell, and NKT response mediated by IL-5 and IL-13. T cell responses initiate an inflammatory cascade that involves endothelial activation, chemokine production, and white blood cell recruitment. Inappropriate triggering and maintenance of these pathogenic responses has been associated with innate immunity defects, e.g., lack of efficient control by anti-inflammatory cytokines such as IL-10 and transforming growth factor (TGF)-β. In the bottom of the figure, intracellular markers of activation are represented. Ab: antibody; CTLA-4: Cytotoxic T lymphocyte antigen-4; ICAM-1: Intercellular adhesion molecule-1; MAdCAM-1: Mucosal addressin cell adhesion molecule-1; VCAM-1: Vascular cell adhesion molecule-1; TLRs: Toll-like receptors; NLR: NOD-like receptor; NKT: Natural killer T.
Epithelial cells are able to identify bacterial components via extracellular receptors like toll-like receptors (TLRs) on the cell surface or intracellular NOD-like receptors in the cytoplasm - NOD2 (nucleotide-binding oligomerization domain containing 2)/CARD15 (caspase-activating recruitment domain 15 receptor). NOD2 receptor, recognizes the muramyl dipeptide (MDP), the minimal bioactive peptidoglycan motif common to all bacteria[2]. MDP stimulation induces autophagy which controls bacterial replication and antigen presentation, and modulates both innate and adaptive immune responses[3-5]. Autophagy is involved in intracellular homeostasis, contributing to the degradation and recycling of cytosolic contents and organelles, as well as to the resistance against infection and removal of intracellular microbes[6-8]. In the innate immune arm, the association of IBD [specifically, Crohn’s disease (CD)] with NOD2 mutations and the two-autophagy-related genes ATG16L1 and IRGM suggests that alterations in the recognition and intracellular processing of bacterial components may have a role in the immunopathogenesis of the disease[9-11]. The unfolded protein response has been identified as a critical pathway in the maintenance of cellular homeostasis[12].
Barriers of protection
Upon penetration of luminal contents into underlying tissues due to leakage in the mucosal barrier, impaired clearance of foreign material from the lumen leads to a compensatory acquired immune response that can result in a chronic inflammatory state. Recently, a immunoregulatory dysfunction of hyperglycosylated mucin (MUC2) has been related to aggravation of IBD. Mucus does not seem to merely form a nonspecific physical barrier, but also constrains the immunogenicity of gut antigens by delivering tolerogenic signals[13].
Dendritic cells, as a part of the innate immune response, present antigens to naïve CD4+ helper T-cells and ensure tolerance to commensal flora by promoting their differentiation into regulatory T-cells. In response to over-activation of dendritic cells, there is a production of pro-inflammatory cytokines and a promotion of the differentiation of effector T-cells Th1, Th2 and Th17 (CD4+); moreover, over-activation induces a strong differentiation of CD8+ lymphocytes and other effector cells such as natural killer (NK) and NK T-cells while abolishing the production of regulatory cells[14].
Innate and adaptive immunity
Th1 cells, whose differentiation is induced by IL-12, produce a high amount of IFN-γ, TNF-α and IL-12, whereas Th2 cells release IL-4, IL-5 and IL-13[15]. An abnormal Th1 immune response is thought to predominate the intestinal inflammation in CD[16]. It has also been observed that in Ulcerative Colitis (UC), atypical NKT cells release higher amounts of the Th2 cytokine IL-13 than T cells from controls or CD patients[17,18]. However, recent data suggest that the CD-Th1 and UC-Th2 paradigms are not so straight forward[19,20].
The differentiation into Th17 cells, a subset of helper T-cells, is induced by IL-6 and TGF-β, acting in concert, and their expansion is promoted by IL-23. There is a delicate balance between Th17 and Treg. The absence of IL-6 drives Treg differentiation[21]. Mature Th17 cells are characterized by the secretion of copious amounts of IL-17A, IL-17F, IL-21, and IL-22[22-24]. The involvement of Th17 cells and, in particular, their signature cytokine IL-17A in intestinal inflammation has been extensively studied[25,26]. Only when the Th17 cells are exposed to IL-23 they cease IL-10 production and attain their full pathogenic function[27].
TGF-β is produced by Treg cells and suppresses T-cell-mediated colitis in animal models[28]. TGF-β effects in IBD T cells are inhibited by the protein Smad7 and Smad7 is markedly overexpressed in IBD patients[29]. Inhibition of Smad7 via antisense DNA restored TGF-β sensitivity in IBD T cells has shown to be effective in murine models of experimental colitis[30,31]. Active IBD is dependent on the recruitment of mononuclear cells and leukocyte populations from the blood stream into the bowel wall. Recruitment is dependent on a series of steps known as rolling, tight binding/adhesion to endothelial cells, diapedesis, and migration of immune cells. This process is coordinated by selective adhesion molecules on the surface of immune cells and mucosal addressins on endothelial cells[32]. Selective adhesion molecules include cell-surface integrins that form heterodimers by various combinations of α- and β subunits. For gut homing of leukocytes, the interaction between α4/β7-integrins on T cells and the mucosal vascular addressing cell adhesion molecule 1 (MAdCAM-1) addressing on endothelial cells appears to be of crucial relevance.
Recent developments have classified NK cells as a subset of a new family of hematopoietic effector cells called innate lymphoid cells (ILCs). ILCs derive from an Id2 (inhibitors of DNA binding) expressing progenitor and the key cytokines secreted by ILCs tend to mirror those secreted by the T-helper cells of the adaptive immune system. Recent data has implicated ILCs, in particular group 3 ILCs in the development of IBD (ILCs IL-23 dependent with retinoid-related orphan receptor were found to be increased in the lamina propria of CD patients[33].
Based on this very recent knowledge, several of these molecules have been investigated as possible biomarkers/indicators of the immune response to therapies, however the results in sensitivity and specificity were moderate and validation was difficult. Here starts a review on the most promising ones[34].
PHARMACOTHERAPEUTIC OPTIONS
Besides nutritional and hygienic measures (smoking cessation), and the use of antibiotics to control symptoms there are several categories of medications used in the treatment of IBD: aminosalicylates (mesalazine), which are effective in treating mild-to-moderate episodes of UC and CD, as well as preventing relapses and maintaining remission[35-37], corticosteroids, recommended only for short-term use in order to achieve remission[38-40], thiopurines [azathioprine (AZA), mercaptopurine (MP)], effective at maintaining of clinical remission in steroid dependent IBD[41-43], methotrexate (MTX), positioned as an alternative immunosuppressive agent in patients with CD resistant or intolerant to AZA or MP[44-48], calcineurin inhibitors [cyclosporine (CsA), tacrolimus (Tac)], effective in the management of steroid refractory UC[49-51]; and, finally, the biologic therapies (adalimumab, certolizumab pegol, infliximab, golimumab, ustekinumab and vedolizumab) that interfere with the body's inflammatory response in IBD by targeting specific molecular players in the process such as cytokines and adhesion molecules[52-54].
Mesalazine
Mechanism of action: Mesalazine [5-aminosalicylic acid (5-ASA)], also in the form of the pro-drug sulfasalazine, has been used for the treatment of UC for decades. It appears to act locally on colonic mucosa and reduces inflammation through a variety of anti-inflammatory processes (Figure 2). The current hypothesis is that 5-ASA activates a synthetic class of nuclear receptor. The anti-inflammatory actions of 5-ASA produce effects similar to activation of the γ-form peroxisome proliferator-activated receptors (PPAR-γ). PPAR-γ is a key receptor that mediates the effect of 5-ASA therapy in IBD by trans-repressing several key target genes such as nuclear factor B, signal transducers and activators of transcription: modulation of inflammatory cytokine production, modulation of RelA/p65 dephosphorylation, leading to decreased transcriptional activity of nuclear factor (NF)-κB, and reduced synthesis of prostaglandins and leukotrienes[55]. Activation of PPAR-γ also has anti-tumorigenic effects. PPAR-γ has a role in the regulation of intestinal inflammation and is highly expressed in the colon, where epithelial cells and macrophages are the main cellular sources of this nuclear receptor[56]. However, additional levels of activity at which the mechanism of action of mesalazine becomes apparent have been described. These include the inhibition of mediators of lipoxygenase and cyclooxygenase, IL-1, IL-2 and TNF-α. 5-ASA has also been recognized as a potent antioxidant and free-radical scavenger[55,57-61].
Figure 2.
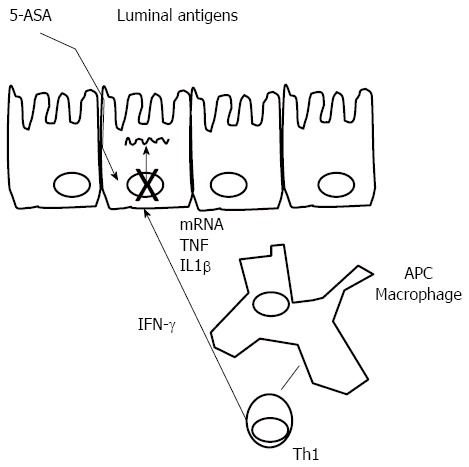
Mesalazine mode of action. Antigen presenting cells (APCs) recognize luminal antigens penetrating the colonic wall and through interaction with Th1 cells induce the production of interferon (IFN)-γ. IFN-γ activates epithelial cells. 5-aminosalicylic acid (5-ASA) is able to block transcription of inflammatory cytokines in colonic epithelial cells. IL: Interleukin; TNF: Tumor necrosis factor.
Measuring response to aminosalicylates in IBD: Heat shock proteins (Hsps) are a family of molecules that are typically involved in folding, refolding, translocation and degradation of intracellular proteins under normal and stress conditions[55,62]. Hsps can stimulate innate and adaptive immune responses and can also, by virtue of the sequence similarity between bacterial and human orthologs, become primary targets of autoimmunity due to a phenomenon known as molecular mimicry[63]. Thus, Hsps have been implicated in the pathogenesis of a number of chronic inflammatory and autoimmune diseases. Hsp60 and Hsp10 (Hsp60 co-chaperonin) are increased in the affected intestinal mucosa from patients with CD or UC[64]. Hsp60 and Hsp10 are increased in the cytoplasm of epithelial cells in CD and UC and also co-localised to epithelial cells of mucosal glands but not always in connective tissue cells of lamina propria, where only Hsp60 or, less often, Hsp10 is found[65]. Tomasello et al[66], demonstrated that mucosal Hsp60 levels in UC patients decrease after therapy with either mesalazine alone or mesalazine plus probiotics, with the decrease in the latter being more pronounced. This same group has demonstrated that Hsp90 levels are high in UC mucosa, both in epithelium and lamina propria. Treatment with 5-ASA plus probiotics reduces Hsp90 levels in the lamina propria, while 5-ASA alone does not have any effect. However, Hsp90 levels within the epithelium were not affected by any of the treatment regimens. In fact, authors have found a linear correlation between Hsp90 and CD4 levels in lamina propria in both UC patients at diagnosis and 6 mo after 5-ASA alone therapy[67].
According to these and previously published results, it has been proposed that a synthetic Hsp90 inhibitor, able to block LPS-induced TLR4 signaling of CD4+ cells, could be applicable to treatment of autoimmune diseases involving inflammation and activation of the adaptive immune response[68]. The latest results show that Hsp10 levels in UC mucosa decrease after therapy. This decline is similar to what is previously described for Hsp60; however, in contrast to Hsp60, Hsp10 has been described as an anti-inflammatory agent. In conclusion, these results altogether indicate that determination of Hsp levels in intestinal mucosa as done in this study has a promising potential for monitoring response to treatment in UC.
Corticosteroids
Glucocorticoids (GC) are potent inhibitors of T cell activation and pro-inflammatory cytokines. However, failure to respond to glucocorticoid therapy is a risk factor for a progressive course of IBD[69,70]. In these patients reduced peripheral T lymphocyte GR binding affinity and abnormalities of glucocorticoid receptor activator protein (GR-AP)-1 interaction and increased expression of GRβ (a truncated splice variant of the normal isoform GRα that does not bind to glucocorticoid ligands) are observed[71].
Mechanism of action: Glucocorticoids mediate their anti-inflammatory responses by binding the intracellular glucocorticoid receptor (GR), a phosphorylated 92-kDa protein, which is a member of the nuclear receptor superfamily[72] (Figure 3). The unliganded receptor is sequestered in the cytoplasm, bound to heatshock proteins Hsp90 and Hsp70 and immunophilin FKBP59, a 59-kDa protein. Upon GC binding and dissociation from heterocomplex proteins, GR translocates into the nucleus; translocation is mediated by specific nuclear transport factors that belong to the importin β family of nuclear transporters, and in particular by importin 13[73]. The activated receptor then binds as homodimer to palindromic DNA-binding sites, the so-called glucocorticoid responsive elements (GREs), localized in the promoter region of target genes[74-76]. Although some GC anti-inflammatory effects are achieved through induction of anti-inflammatory genes, such as interleukin (IL)-10, annexin 1 and the inhibitor of NF-κB[77,78], transactivation enhances mainly the expression of genes involved in metabolic processes[79,80], and is therefore, responsible for the majority of unwanted side effects[81,82]. Indeed, the presence of GR on GRE might competitively prevent the binding of activator protein (AP)-1 and NF-κB on the same promoter regions or might trans-activate their inhibitor proteins. Furthermore, GRE-independent mechanisms of trans-repression also exist: the GR physically interacts with AP-1[83], NF-κB[84] and signal transducers and activators of transcription[85]. Trans-repression is believed to be responsible for the majority of the beneficial, anti-inflammatory effects of GCs[79,86-88].
Figure 3.
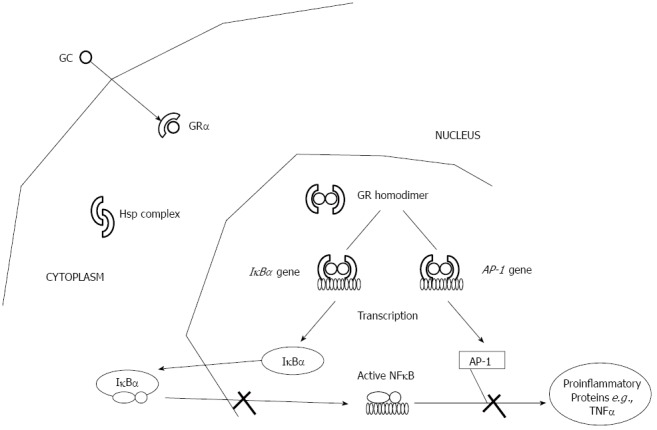
Glucocorticoids mode of action. AP: Activator protein; GC: Glucocorticoids; GR: Glucocorticoid receptor; TNF: Tumor necrosis factor; NF: Nuclear factor.
Measuring response to corticosteroids in IBD: Research in impaired sensitivity to glucocorticoid inhibition in IBD has highlighted three potential molecular mechanisms: (1) decreased cytoplasmic glucocorticoid concentration secondary to increased P-glycoprotein-mediated efflux of glucocorticoid from target cells due to overexpression of the multidrug resistance gene (MDR1)[89-91]; (2) impaired glucocorticoid signaling because of dysfunction at the level of the glucocorticoid receptor[92,93]; and (3) constitutive epithelial activation of pro-inflammatory mediators, including NFκB, resulting in inhibition of glucocorticoid receptor transcriptional activity[94,95].
The multi-drug resistant (MDR1) gene codes for a drug efflux pump P-glycoprotein-170 (permeability-glycoprotein or Pgp), which is expressed on the apical surface of lymphocytes and intestinal epithelial cells and actively transports toxins and drugs out of target cells, thereby removing toxic metabolites and xenobiotics from cells into urine, bile, and the intestinal lumen. This efflux pump also regulates the distribution and bioavailability of drugs, and in conclusion reduces their efficacy. To date, 15 MDR1 polymorphisms have been identified and a polymorphism in exon 26 (C3435T) of the MDR1 gene has been shown to be significantly correlated with levels of expression and function of P-gp-170 in healthy individuals. Healthy individuals are classified as: homozygous (C/C or“resistant” genotype and T/T or “responsive” genotype) or heterozygous (C/T). The C/C genotype is highly prevalent in West Africans (83%) and African Americans (61%) compared with 26% and 34% in caucasians and Japanese populations, respectively[90,91].
Pgp and MDR expression have been shown to be significantly higher in CD and UC patients requiring surgery due to failure of medical therapy[92]. In MDR1 knockout mice associations between C3435T and UC and G2677C/T and IBD have been described. Several other associations with SNPs in the TNF (tumor necrosis factor) gene and the macrophage MIF (migration inhibitory factor) gene and GC dependency or sensitivity have also been reported. According to these results a protective role for the MDR1 3435 C/C versus MDR1 3435 T/T genotype and C versus T allele for the progression of IBD is suggested[93-112].
Matrixmetalloproteinases (MMPs) make up a family of 24 human zinc-dependent endopeptidases and degrade practically all extracellular matrix components[102,103]. They are fundamental for tissue damage[103] and expressions correlate with the degree of inflammation in the gut[104,105]. MMP activity is inhibited by tissue inhibitors of MMPs (TIMPs), as well as nonspecific inhibitors such as α2-macroglobulin (α2M)[106]. TIMPs modulate the activity of soluble, matrix-bound, and cell-associated MMPs[106] and are upregulated in IBD[107,108]. α2M is a serum anti-proteinase, capable of almost universally inhibiting endoproteinases, and is thought to be the major plasma inhibitor of MMPs[106]. Pro-inflammatory cytokines, such as TNF-α, increase MMP production[109], and production of TNF-α correlates with both MMP and TIMP production in IBD[110]. Serum levels of MMP-7, -8, and -9, TIMP-1, and α2M, are elevated in active IBD. Both, GC and anti-TNF-α therapies reduce MMP-7 levels, but only in GC treated patients, the levels decline corresponding to levels of control patients. Interestingly, no significant changes in α2M are associated to GC treated group. MMP-7 and TIMP-1 seem promising in monitoring the effect of GC treatment. GCs inhibit MMP synthesis by controlling gene expression as well as by inducing the transcription of TIMPs[83]. While MMP-7/TIMP-2 ratio is associated with greater severity of UC[86], the decrease in MMP-7/TIMP-2 ratio in GC-treated patients is more likely a result of decrease in MMP-7 itself as TIMP-2 is not affected.
Several investigations have also identified GR abnormalities as potential mechanisms influencing response to glucocorticoid treatment in several inflammatory conditions as: (1) reduced peripheral T-lymphocyte GR binding affinity[91]; (2) abnormalities of GR-AP-1 binding in glucocorticoid resistant asthma, suggesting a post-receptor mechanism[79]; and (3) increased expression of glucocorticoid receptor β (GRβ), a truncated splice variant of the normal isoform GRα that does not bind glucocorticoid ligands. GRβ is unable to transactivate glucocorticoid-responsive genes, and has therefore been suggested to act as a dominant-negative inhibitor of glucocorticoid action[92].
Honda et al[111] reported GRβ mRNA expression in 83% of the patients with steroid-resistant UC compared to only 9% in steroid-responsive patients, and 10% in healthy controls and chronic active CD patients. These results were confirmed in a recent study from Japan, where the authors looked at the frequency of GRα and β positive cells in colonic biopsies of GC-sensitive (n = 6) and GC-resistant (n = 8) UC patients[112]. They also found that there were significantly more GRβ-positive cells in the GC-resistant group than in the GC-sensitive and the control groups.
miRNAs are small (18-24 nucleotides) non-coding RNAs, which bind the 3’UTRs (mRNA that immediately follows the translation termination codon) and the coding exons of their target genes and inhibit gene expression. By affecting gene regulation, miRNAs are likely to be implicated in the control of diverse biological processes Moreover, miRNAs have important regulatory roles in the innate and adaptive immune system, and characteristic miRNA expression profiles have been demonstrated even in IBD[113]. A number of studies have shown that GCs can modify the expression profile of different miRNAs but to date it is not possible to recognize a specific miRNA pattern regulated by GCs. It has been demonstrated that activation of GR by GCs might induce or repress specific miRNAs in various target genes. The majority of studies have evaluated the effect of GCs on miRNA expression levels in tumor leukemic cells, during GC induced apoptosis[114]. Of interest, miRNA could target mRNAs encoded by genes involved in the importin pathway, or appear like potential regulators of components of the inflammasome pathway (key signalling platforms that detect pathogenic microorganisms and sterile stressors, and that activate the highly pro-inflammatory cytokines IL-1β and IL-18). Both importins and the inflammasome are involved in molecular mechanism of GC signaling: importin is a nuclear transport protein responsible for the translocation of the complex GR-GC into the nucleus[115], and variants of the inflammasome gene have been correlated with steroid resistance in pediatric IBD patients[116].
Conversely, NF-κB and GRα can mutually repress each other’s transcriptional activity. Consequently, the debate as to whether inflammation drives glucocorticoid resistance or vice versa has refocused investigators’ efforts into the critical role played by NF-κB[93]. Further investigations have shown that while the activation of AP-1 and the upstream kinases p38 and c-Jun N-terminal kinase (JNK) in steroid-sensitive patients with CD was mainly found in lamina propria macrophages, steroid-resistant patients revealed activation of all these mediators mostly in epithelial cells[94].
Gene expression profiling can be successfully used to stratify patients and identify transcriptional signatures associated with clinical parameters. Several predictor gene panels containing genes involved in immune mechanisms (PTN, OLFM4, LILRA2, CD36), autophagy or GC response (STS, MDM2) have been identified. This represents, the first biomarker discovery [predictor gene panels that contain genes involved in immune mechanisms (PTN, OLFM4, LILRA2, CD36), autophagy or GC response (STS, MDM2)] based on specifically designed analytical algorithms with potential value to predict GC response and need of surgery as well as with diagnostic value for IBD patients[117].
Thiopurines
Mechanism of action: In vitro studies have shown that AZA and 6MP exert their effect by controlling T cell apoptosis through modulation of Rac1 activation upon CD28 co-stimulation(118). Apoptosis induction required co-stimulation with CD28 and was mediated by specific blockade of Rac1 activation through binding of 6-thioguanine nucleotide (6-TGN) to Rac1 instead of guanine triphosphate (GTP). Activation of the Rac1 gene in turn leads to activation of mitogen-activated protein kinase (MAP kinase), NF-κB, bcl-x(L)(B cell lymphoma) and finally to a mitochondrial pathway of apoptosis. Thus AZA and 6MP convert a co-stimulatory signal into an apoptotic signal by modulating Rac1 activity. In Figure 4 the thiopurines biotransformation pathway is represented.
Figure 4.
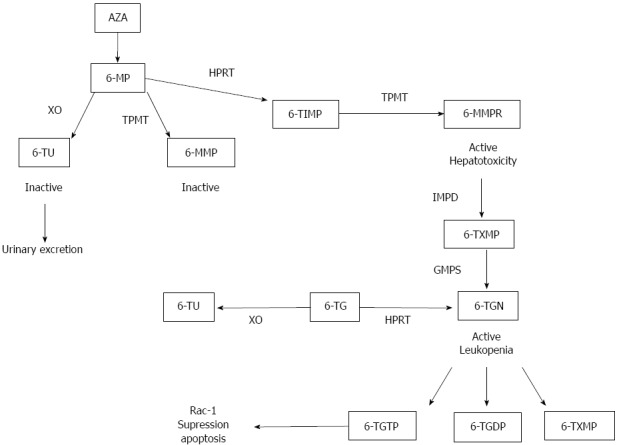
Thiopurines metabolic pathway. XO: Xanthin oxidase; 6-TU: 6-thiouracil; TPMT: Thiopurine methyltransferase; HPRT: Hypoxanthine phosporybosil transferase; 6-MMP: 6-methyl mercaptopurine; 6-TIMP: 6-thiosine 5’ monophosphate; 6-MMPR: 6-methyl mercaptopurine ribonucleotide; IMPD: Inosine monophosphate dehydrogenase; 6-TXMP: 6-Thioxanthosine monophosphate; GMPS: Guanosine monophosphate synthetase; 6-TGN: 6-thioguanine nucleotide; 6-TG: 6-thioguanine; 6-TGDP: 6-thioguanine diphosphate; 6-TGTP: 6-thioguanine triphosphate.
Measuring thiopurines derivatives: TPMT enzyme activity (measured by radioimmunoassay) is genetically determined and has been extensively reviewed[118]. In summary, TPMT enzyme activity can identify patients with high TPMT activity that metabolize 6-MP to 6-methyl-MP and therefore may be resistant to treatment with thiopurine drugs. It is estimated that TPMT deficiency is responsible for up to 30% of all adverse drug reactions (ADRs) experienced on AZA, but whilst TPMT deficiency strongly predicts the development of myelotoxicity, the most serious ADR of AZA therapy, it fails to account for over 70% of cases of myelotoxicity[119,120]. Another candidate enzyme for further study is xanthine oxidase ⁄dehydrogenase (XDH)[121,122]. Blocking XDH activity using allopurinol (which, as recently described, also inhibits TPMT due to skewed drug metabolism)[123] is known to cause severe toxicity with conventional doses of AZA and safe co-prescription of allopurinol requires an AZA dose-reduction of approximately 80%[124]. A molybdenum cofactor[125] is essential for the action of three oxidases, XDH, aldehyde oxidase (AO) and sulphite oxidase. This molybdenum cofactor requires the action of molybdenum cofactor sulfurase (MOCOS). MOCOS deficiency (which results in the deficiencies of both XDH and AO, but not sulphite oxidase) is, in contrast, relatively benign, causing only a predisposition to renal stones (Type II Xanthinuria)[126]. AO has been considered of minimal clinical significance and so it has not been carefully examined until recently.
Monitoring of 6-MP metabolites is a helpful, but not an indispensable tool in thiopurine non-responders to discriminate poor adherence and under-dosing from pharmacogenetic thiopurine resistance and thiopurine refractory disease. Several studies have reported that patients with IBD treated with AZA or 6-MP who respond to therapy have higher median concentrations of 6-TGN than patients who fail to respond to therapy[127-129]. One study in 93 patients with IBD reported that the median concentration of 6-TGN in responding patients was 312 pmol/8 × 108 RBCs compared with a median concentration of 199 in patients who failed to respond[129]. There was no difference in the median concentrations of 6-MMP between the 2 patient groups. The breakpoint between the lower two quartiles and the higher two quartiles of 6-TGN concentrations was 235 pmol/8 × 108 RBCs. Sixty-five percent of responding patients had an erythrocyte 6-TGN concentration > 235 as compared with only 27% of patients failing therapy. Thus, the authors suggested that clinicians should adjust AZA or 6MP doses to achieve 6-TGN concentrations > 235pmol/8 × 108 RBCs. The authors also reported that hepatotoxicity (defined as liver enzymes more than twice normal) occurred in 16 patients, and that the median 6-MMP concentrations were 5463 pmol/8 × 108 RBC’s in patients with hepatotoxicity compared with only 2213 pmol/8 × 108 RBCs in patients without hepatotoxicity.
Unfortunately, dose escalation of thiopurines does not necessarily result in higher efficacy. Instead of increasing 6-TGN concentration, following increasing the thiopurine dose, some patients shift their metabolism towards the production of 6-MMP resulting in hepatotoxicity[130]. Another approach to bypass the influence of TPMT has been by direct administration of 6-TGN[131-133]. High concentrations of 6-TGN were achieved but the drug had to be stopped because of nodular regenerative hyperplasia in the liver[134].
Smith et al[135] have recently published the impact of introducing nucleotide monitoring into clinic. They obtained 608 TGN results from 189 patients with IBD. In non-responders, TGNs directed treatment change in 39/53 patients. When treatment was changed as directed by TGN, 18/20 (90%) improved vs 7/21 (33%) where the treatment decision was not TGN-directed (P < 0.001). Where treatment change was directed at optimization of thiopurine therapy, 14/20 achieved steroid-free remission at 6 mo vs 3/10 where the TGN was ignored (P = 0.037). Six per cent of patients were non-adherent, 25% under-dosed and 29% over-dosed by TGN. Twelve per cent of patients demonstrated preferential thiopurine methylation; this group had low TGN levels and high risk of hepatotoxicity. In responders, adherence and dosing issues were identified and TGN-guided dose-reduction was possible without precipitating relapse. Mean cell volume (MCV), white blood cell count (WBC) and lymphocyte counts were not adequate surrogate markers. MCV/WBC ratio correlated with clinical response, but was less useful than TGN for guiding clinical decisions.
In a previous study, the same group identified the presence of the coding region SNP AOX1 3404G as a predictor of non-responsiveness to AZA therapy[136]. The authors suggested that those with a poor chance of responding to AZA (high TPMT activity and AOX13404G variant) should be offered an alternative treatment as first-line therapy, which might include reduced dose azathioprine in combination with allopurinol, a combination which has been shown to circumvent the problem of hyper-methylation in some patients[137]. In patients with CD, the next immunomodulator considered for treatment would usually be MTX. It is possible that the same polymorphism AOX1 c.3404A > G could also affect an individual’s chance of response to MTX, as AO is known to metabolize MTX producing a 7-hydroxymetabolite, which is considered inactive.
Genotype variants, which have a functional impact, most commonly decrease the activity of the affected enzyme. If this is true for the AOX1 3404G variant, then the association with lack of clinical response would suggest that AO metabolites of AZA have immunosuppressive activity: 8-hydroxy-6MP did not slow the growth of rat sarcoma[138]; however, AO produces several other AZA metabolites on which no functional work has been carried out. Another possibility is that AO activity is increased in the presence of the AOX1 3404G variant. In this case, it is possible that overactive AO removes and inactivates a higher proportion of the ingested drug, resulting in decreased efficacy; but in this case, one would expect carriers of the AOX1 sequence variant to have lower TGN levels.
Purinergic signaling and associated ectonucleotidases, such as CD39 and CD73, have been implicated in the pathogenesis of IBD. Adenosine generated by CD73 and CD39 components might play an important role in the resolution of inflammation and in the promotion of healing. The anti-inflammatory effects of AZA, have been ascribed to induce apoptosis of predominantly CD45RO+ memory T cells (within CD73+CD4+ T cells)[139].
Methotrexate
Mechanism of action: MTX, like folic acid, is a substrate for the enzyme folylpolyglutamate synthetase, which adds glutamic acid residues to these compounds. Parent MTX, polyglutamated MTX metabolites (MTXPG), and another major metabolite, 7-hydroxymethotrexate (7OH-MTX), are all folic acid analogues with inhibitory activity against many of the enzymes in the metabolic pathway of folic acid[140]. The principal cellular action of MTX, is competitive inhibition of the enzyme dihydrofolatereductase (DHFR)[141]. The metabolites of MTX have considerable importance as inhibitors of the folate-dependent enzymes distal to DHFR. MTXPG are preferentially retained intracellularly in a non-effluxable form in proportion to the length of the polyglutamate chain, and they account for more than 50% of intracellular drug 24 h after exposure[142]. 7-0H-MTX, which is the major circulating variant of MTX 24 h after a dose of the drug, undergoes polyglutamation 2.7-fold faster than does MTX, and it is also a 4.5-fold more potent inhibitor of 5-aminoimidazole-4-carboxamide ribotide (AICAR) transformylase, and possibly other distal folate-dependent enzymes, than is MTX[143,144]. MTX is effective in the treatment of inflammatory diseases such as CD in low doses. The question is whether inhibition of T cell proliferation is a major mechanism of action in low-dose MTX treatment and if it is related to inhibition of DHFR or not. Some studies suggest that low-dose MTX is indeed able to inhibit lymphocyte proliferation through DNA synthesis inhibition[145]. However, anti-inflammatory properties of MTX were not always found to depend on lymphocyte proliferation inhibition. On the other hand, overt inhibition of cellular proliferation produced by inhibition of DHFR is not a requirement for efficacy but rather is a sign of toxicity of low-dose MTX therapy. The coadministration of folinic acid or leucovorin (fully reduced tetrahydrofolate), which bypasses blockage of DHFR, ameliorates many of the side effects of MTX. However, if given in excess quantities, it also retards the efficacy of the drug[146-148] (Figure 5).
Figure 5.
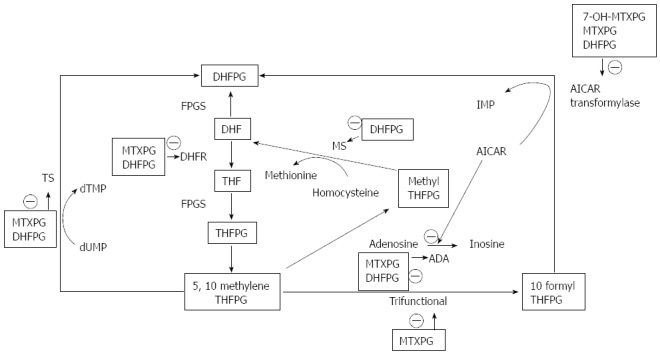
Methotrexate mode of action. DHF: Dihydrofolate; FPGS: Folil polyglutamate synthetase; DHFPG: Dihydrofolate polyglutamates; DHFR: Dihydrofolate reductase; THF: Tetrahydrofolate; THFPG: Tetrahydrofolate polyglutamates; TS: Thymidylate synthetase; MS: Methionine synthetase; ADA: Adenosine deaminase; IMP: Inosine monophosphate; AICAR: 5-aminoimidazole-4-carbox-amide ribonucleotide.
Proliferation of T cells can also be inhibited by MTX through its inhibition of cytokines, such as IL-2, that promote proliferation. MTX generally inhibits Th1 cytokines and up-regulates or does not affect Th2 cytokines[149-151]. TNF-α, in particular, was found to be suppressed in both ex vivo stimulated T cells of patients with rheumatoid arthritis (RA) treated with MTX and in vivo and in vitro experiments in T cells and macrophages[152,153]. Also, the number of CD4+ T cells that secrete TNF-α was significantly lower in MTX-treated patients with RA than in untreated patients[154]. The inhibitory effect of MTX on TNF-α can be attributed to several mechanisms: high levels of adenosine[155]; inhibition by MTX of TNF-α promoter activity in lymphocytes[154]; inhibition of NF-κB activation, indirectly inhibiting subsequent TNF-α transcription[156]. IFN-γ, has also been shown to be inhibited by MTX.
In vivo, MTX enhances IL-10 production. Patients with RA treated with MTX showed increased numbers of IL-10-producing T cells, and ex vivo stimulated monocytes of patients with RA treated with MTX showed increased IL-10 production[154]. Interestingly, upregulation of IL-10-producing monocytes was observed only in patients who responded to therapy. In summary, pro-inflammatory cytokines and cytokines that promote proliferation are inhibited by MTX, whereas the anti-inflammatory cytokine IL-10 is upregulated. This may suggest that induction of IL-10 by MTX is one of its important anti-inflammatory mechanisms[157].
Genetic polymorphisms and treatment response: The uptake of MTX into cells is mainly controlled by the reduced folate carrier (RFC). A non-conservative polymorphism G80A (R27H) has been associated with higher plasma concentrations of MTX and a worse prognosis (event-free survival estimates) in children with acute lymphoblastic leukaemia treated with this drug[158].
The folypolyglutamase hydrolase, also known as c-glutamyl hydrolase (GGH), converts long-chain MTXPG into short-chain MTXPG and ultimately back to MTX resulting in higher efflux of the drug. Also, in acute lymphoblastic leukaemia patients, several polymorphisms in this gene have been identified including a putatively functional non-conservative SNP C452T (T127I) associated with low enzyme activity and higher accumulation of long-chain MTX-PG experiencing a better event-free survival[159,160].
Therapy with MTX results in a reduction of the reduced folate pool by inhibiting dehydrofolate reductase. Another enzyme, the methylenetetrahydrofolate reductase (MTHFR) is crucial for folate homeostasis by converting 5,10-methylene-tetrahydrofolate, the methyl-donor in dTMP synthesis, into 5-methyltetrahydrofolate, the carbon-donor required for methionine synthesis. Two common, non-synonymous polymorphisms in this gene have been found to influence MTX toxicity and efficacy. The SNP C677T results in a more thermolabile variant of the protein and has been associated with increased drug toxicity[161]. Interestingly, this SNP has also been associated with overall susceptibility to IBD in an Irish cohort of patients[162]. The second SNP, A1298C, also leads to a reduced activity of the MTHFR[163,164] and has been associated with increased efficacy in patients with RA[165].
There is also evidence from in vitro experiments that impaired folylpolyglutamasesynthase (VMcN1) activity may play a crucial role in MTX resistance[166,167].
A small clinical trial in 18 CD patients has de monstrated that individual RBC MTXGlu1-5 concentrations can be measured accurately and have low intra-patient variation at steady state. Unexpectedly, the study suggested that RBC MTXGlu4 and 5 concentrations correlated inversely with efficacy in patients with CD. In addition, high RBC MTXGlu4&5 concentrations were associated with an increased incidence of adverse effects. Although the findings were statistically significant, the number of subjects in this pilot study was small and, therefore, as stated by the authors, there is a high possibility of a type I statistical error[168].
Calcineurin inhibitors
Mechanism of action: Calcineurin inhibitors exert their cellular effects through binding to proteins called immunophilins[169]. Cyclophilins (CP) bind CsA and FK-binding proteins (FKBPs) bind Tac. Cyclophilin A is the most abundant cyclophilin in T lymphocytes, and the predominant Tac-binding immunophilin is the FKBP12. The CPs and FKBPs are structurally unrelated but both families have a cis-trans prolyl-peptidyl isomerase activity. The binding of CsA or Tac to its respective immunophilin enhances the immunophilin's affinity to calcineurin. Formation of such a complex results in its binding to and inhibition of calcineurin[170]. In the process of T-cell activation calcineurin, which is a calmodulin-activated serine phosphatase, associates with and dephosphorylates inactive nuclear factor of activated T cells (NFAT). This leads to NFAT translocation to the nucleus and, in association with other transcription factors such as AP-1, initiation of downstream events involved in T-cell activation[171-173]. Within the members of the NFAT family, NFAT1, NFAT2, and NFAT4 participate in T lymphocytes cytokines transcriptional activation such as IL-2, IL-4, and CD40L[174]. So, in T cells NFAT proteins not only regulate activation but also are involved in the control of thymocyte development, T-cell differentiation and self-tolerance. The functional versatility of NFAT proteins is explained by their complex mechanism of regulation and their ability to integrate calcium signalling with other signalling pathways. The drug-immunophilin complex forms an inhibitory association with calcium-calmodulin-activated calcineurin, preventing its binding and activation of NFAT. CsA also inhibits mRNA transcription of IFN-γ by inhibiting NFAT translocation into the nucleus (Figure 6).
Figure 6.
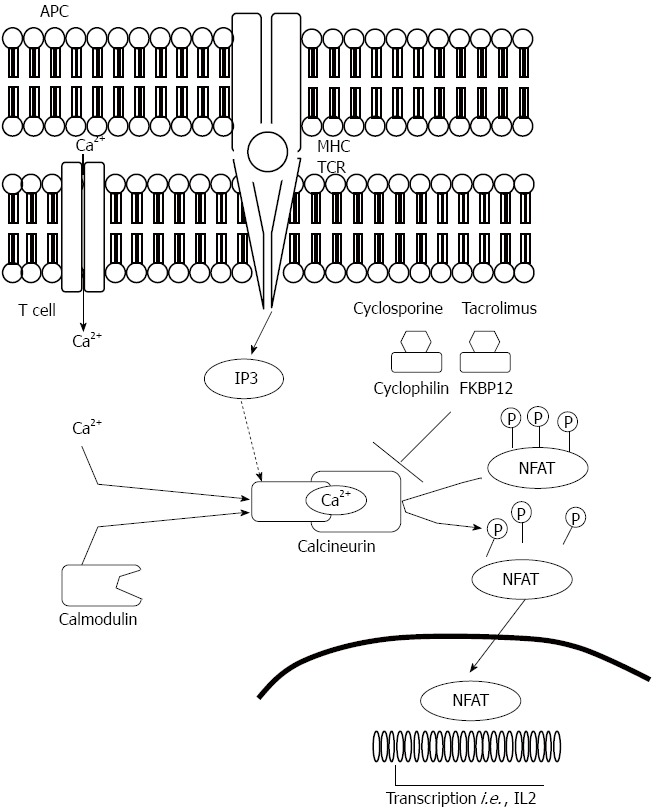
Mode of action of calcineurin inhibitors. APC: Antigen presenting cell; MHC: Major histocompatibility complex; NFAT: Nuclear factor of activated T cells; TCR: T cell receptor; IP3: Inositol triphosphate.
CsA and Tac, induce apoptosis of CD4+ T-lymphocytes[175]. CsA reduces the number of the anti-apoptotic Bcl-2-positive T cells[175]. The apoptotic activity of CsA is possibly mediated by the inhibition of cytokine release and the subsequent activation of ICE-like proteins (ICE is an acronym for IL-1β converting enzyme of caspase 1), known to play a chief role triggering the apoptotic cascade[176].
Apart from the above-mentioned effects on T lymphocytes, CsA inhibits antigen presenting cells activity and production of the B-lymphocyte activating factors[177]; attenuates adhesion interaction and trans-endothelial migration and infiltration of neutrophils by decreasing endothelial expression of cell adhesion molecules (E-selectin, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1) and inhibits the anti-apoptotic NF-κB, a central transcription factor mediating inflammatory injury[178].
Influence of genetics on pharmacological behavior
Pharmacokinetics: CsA and Tac are among the most commonly used immunosuppressants in patients with organ transplantation or autoimmune diseases. However, both have a narrow therapeutic window and large inter-individual variability, resulting in therapeutic drug monitoring (TDM), necessary for adjusting the dose in order to reduce the toxicity and improve the efficacy.
To date, most transplant centers utilize whole-blood measurements of CsA trough levels as a means of TDM. However, it was demonstrated that the correlation of “therapeutic” trough levels with the actual drug exposure[179,180] or with clinical outcomes[181] was relatively poor. The determination of total AUC is the most accurate measure of drug exposure, and its values possibly correlate to some degree with the rate of successful outcomes. However, due to the cost and inconvenience of multiple blood measurements required for AUC determination, this method is impractical. A decade ago, prospective studies were underway examining the utility of a single measurement of 2-h (C2) CsA level, which showed to be associated with renal allograft rejection (blood concentrations of CsA during the early post dose period had been shown to correlate well with inhibition of calcineurin and IL-2 but still was, logistically difficult and plagued by a high intra-individual variability)[182,183].
Unlike the case of CsA, the trough levels of Tac correlate reasonably well with AUC and are the most common measure of Tac treatment monitoring[184]. Although TDM is widely recommended in clinical practice and has been conducted for approximately 30 years, this strategy for calcineurin inhibitors therapy is controversial according to recent reports[185]. In the past decade, the understanding of the pharmacogenomics of calcineurin inhibitors in transplantation has improved. Polymorphisms of genes coding for enzymes and transport proteins involved in the metabolism of these compounds have been thoroughly studied. CYP3A4 oxidizes CsA at multiple positions and is known to convert CsA into three major primary metabolites (AM1, AM9 and AM4N). CYP3A5 preferentially attacks at amino acid 9 and metabolizes CsA to only one primary metabolite (AM9). For Tac, the intrinsic clearance for CYP3A5 is approximately 2-fold higher than for CYP3A4. CYP3A5 catalyses the formation of four primary metabolites (M1, M2, M3 and M6). It is well established that the CYP3A5 A6986G (*3) SNP influences the pharmacokinetics of Tac in renal recipients[186]. Almost all studies have reported that recipients with the CYP3A5*3/*3 genotype (non-expressers) exhibit higher dose-adjusted Tac exposure (C0/dose, C2/dose or AUC/dose), and a lower dose requirement compared with the CYP3A5*1/*1 or*1/*3 carriers (expressers). With respect to CsA and the CYP3A5*3SNP, the results from clinical studies have not been able to reach a conclusion. ABCB1 (MDR1), encoding the transport protein P-glycoprotein, which pumps calcineurin inhibitors out of intestinal enterocytes, has had several of its SNPs investigated in renal transplant patients. The influence of these SNPs on the pharmacokinetics of CsA and Tac remains uncertain, as CYP3A4 also demonstrates inter-individual variation in a metabolic capacity and functional SNPs are few in the CYP3A4 gene and most studies found no association with pharmacokinetics of CsA, and controversial associations with Tac[186,187].
Pharmacodynamics: Enzymatic and immunological strategies are the two types of methods which can be used to assess the pharmacodynamics of these compounds. The former directly determines calcineurin activity, while the latter measures immune responsiveness at several levels[187,188]. Although these strategies are still in a very early stage, and have not been validated in clinical practice, several clinical studies have reported associations of NFAT-regulated genes with biopsy-proven acute rejection and recurrent infections in renal recipients, making the expression of such genes a promising biomarker of pharmacodynamics[189,190]. In fact, functional polymorphisms in PPIA (coding for cyclophilin), FKBP1A (coding for FKBP12), PPP3CA/PPP3CB/PPP3R1 (coding for calcineurin), NFATC1/NFATC2/NFAT5 (coding for NFAT) and IL-2 (coding for IL-2) have been explored in other diseases[191-195].
Many important metabolic enzymes and transporters could also be modulated by orphan nuclear receptors, a large family of transcription factors regulating tissue gene expression, such as the pregnane X receptor (PXR), constitutive androstane receptor, the glucocorticoid receptor and more.
In recent years, epigenetics have been incorporated to the field of pharmacology referring to drug responses accounted for by epigenetic changes (DNA methylation, modification of histones in chromatin and RNA-mediated regulation of gene expression, as, miRNAs) instead of alterations in the DNA sequence. In contrast to SNPs, epigenetic characteristics can be altered by age, influenced by drugs and can interact with environments. Current evidence has revealed that the expression of CYP3A4 and CYP3A5 could be affected by a DNA methyltransferase inhibitor and miRNA-27b[196,197]. Besides the direct action on enzymes, miRNAs also regulate the expression of nuclear receptors, such as PXR[198].
Biologics
Mechanism of action - Anti TNF-α: TNF plays a central role as a pro-inflammatory cytokine that initiates the defense response to local injury. When present at low concentrations, it is believed to have beneficial effects, such as the augmentation of host defense mechanisms against infections. At high concentrations, TNF can lead to excess inflammation and organ injury. Both immune (macrophages, T cells, granulocytes, etc.) and non-immune cells (fibroblasts, neurons, smooth muscle cells) can produce TNF. It is initially produced as a cell surface-bound precursor (tmTNF), which can be enzymatically cleaved by TNF-α converting enzyme to form a soluble cytokine (sTNF). Both sTNF and tmTNF are biologically active and interact with either of 2 distinct receptors to exert their action: the p55 TNF receptor 1 (TNFR1) and the p75 TNFR2 that are present on a wide range of cell types. sTNF preferably binds to TNFR1 and generates the pro-inflammatory properties of TNF: Activation of nuclear factor kappa-B1 (NF-κB1), which will result in the transcription of several inflammatory genes and the expression of other pro-inflammatory cytokines including IL-1 and IL-6 and enhancement of leukocyte migration by inducing expression of adhesion molecules by endothelial cells and leukocytes; caspase-8- and caspase-3-dependent apoptosis. tmTNF preferably binds to TNFR2 and initiates the immune-regulatory properties of TNF. This effect is attributed to the possibility of tmTNF serving as a receptor instead of a ligand for TNFR2 and inducing reverse signaling through this membrane-anchored ligand and triggering cell activation, cytokine suppression or apoptosis of the tmTNF bearing cell[199].
The four TNF antagonists available in the treatment of IBD can be divided into two categories based on their structure: the full-length monoclonal antibodies (mAbs) and those with only an antibody fragment. Infliximab, adalimumab and golimumab are the full-length IgG1 antibodies and their Fc region is capable of complement fixation and Fc-receptor mediated biologic activities. Certolizumab pegol lacks this Fc region and is therefore not able to perform these effector functions. All of these agents have TNF as target and are capable of binding sTNF and tmTNF with high affinity. However, differences in structure between antagonists cause different kinetic-binding parameters that can result in variable clinical efficacy. For example, etanercept, an anti-TNF approved for rheumatological diseases, is able to bind both forms of TNF but is not effective in CD, so other mechanisms must be responsible for the action of these agents[200]. These compounds exert a down-regulation of inflammatory cells in the inflamed bowel mucosa that is believed to be induced by apoptosis in tmTNF carrying cells. There is also in vitro evidence that infliximab induces cell lysis through complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity, both Fc-dependent[201]. One other possible mechanism of action that has been shuffled is the induction of regulatory macrophages, also an Fc-dependent mechanism. These macrophages have immunosuppressive capacities, play a crucial role in wound healing and have been shown to be up-regulated in patients responding to infliximab therapy[202] (Figure 7).
Figure 7.
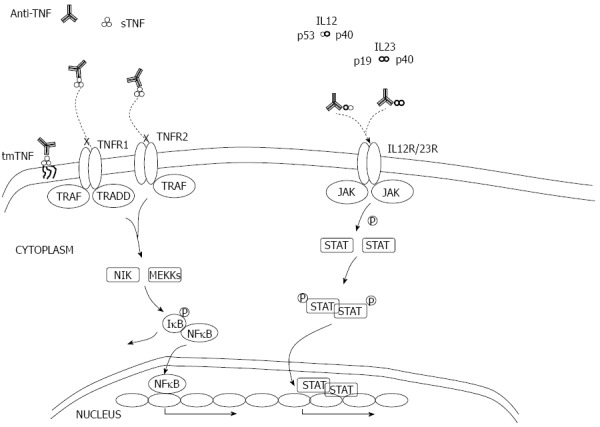
Mode of action of anti- tumor necrosis factor α and anti-IL-12/23. TNF: Tumor necrosis factor; tmTNF: Transmembrane TNF; sTNF: Soluble TNF; TRAF/TRADD: TLRs adaptors; JAK: Janus kinase; STAT: Signal transducer and activator of transcription.
Anti IL12/23: IL-12 is the key inducer of Th1 cells while recent studies conducted on human cells suggest that a cocktail of cytokines, such as IL-23 and IL-1β, are critical for Th17 differentiation. Human Th17 cells are thought to produce several pro-inflammatory cytokines, including IL-17A and F, TNFα, IL-22, IL-26 and IFNγ[203]. Similar to IL-12, IL-23 can contribute to functional responses of several effector cell subtypes other than CD4+ T cells, including CD8+ T cells, NK, NKT, γδ T cells, and innate lymphoid cells[204-207]. There is increasing evidence of plasticity amongst certain Th subtypes, depending upon the cytokine microenviroment[208,209].
Ustekinumab is a human IgG1 kappa (κ) mAb that binds to the IL-12p40 subunit. This subunit of IL-12 was also found to associate with a p19 subunit to form IL-23[210]. Ustekinumab prevents human IL-12 and IL-23 from binding to the IL-12Rβ1 receptor chain of IL-12 (IL-12Rβ1/β2) and IL-23 (IL-12Rβ1/IL-23R) receptor complexes on the surface of NK and T cells but cannot bind to endogenous IL-12 or IL-23 that is already bound to receptor complexes. Thus, ustekinumab is unlikely to mediate Fc effector functions.
Mechanism of action - Anti-adhesion molecules: L-selectins are expressed on leukocytes, and P- and E-selectins are found on the endothelium. Although strong, these selectin bonds are short-lived and, consequently, the T cells roll over the endothelium from one selectin bond to the next[211]. This results in slowing of the lymphocytes and allows for transient interactions which enable the cell to encounter the cytokine-rich microenvironment that triggers subsequent firm adhesion and consequent migration through the blood vessel wall[212,213]. Secondary adhesion molecules, all members of the integrin family, function to stop the rolling lymphocytes and allow migration. Integrins are leucocyte cell-surface adhesion molecules that mediate both cell-cell and cell-extracellular matrix interactions[214] (Figure 8). The expression of integrins is activated by chemokines, which are released by T cells[215]. Integrins involved in the T-cell migration are as follows: leucocyte function-associated antigen 1 (LFA-1 or α2β2) and the two α4-integrins (α4β1 and α4β7). For the migration of leucocytes, these integrins bind to specific ligands at the endothelium called addressins or adhesion molecules. The α2β2 integrin, expressed on neutrophils, interacts with intercellular adhesion molecule-1 (ICAM-1) that is expressed on leucocytes, dendritic cells, fibroblasts, epithelial cells and endothelial cells[216,217]. The α4β1 integrin is expressed on most leucocytes, but not on neutrophils and binds to vascular cell adhesion molecule-1 (VCAM-1) and to components of the extracellular matrix such as fibronectin and thrombospondin[218]. The third family is the α4β7 integrin, which is expressed on the lymphocytes that colonise the gut and gut-associated lymphoid tissues and interacts with the MAdCAM-1 and this interaction activates the migration of lymphocytes to Peyer’s patches[219,220]. Last, the αEβ7 integrin is another member of the β7 integrin family that it is expressed only in mucosal intraepithelial T lymphocytes and that binds selectively to E-cadherin on epithelial cells[221]. The expression of aEβ7 is elevated in UC and CD in the active phase of the disease[222,223], and the interaction of aEβ7/E-cadherin has been proposed to participate in the retention of T cells in the mucosal tissue[224]. Pro-inflammatory cytokines such as IL-1 and TNF[225-227] up-regulate the expression of ICAM-1 and MAdCAM-1. Treatment with IFX decreases the expression of ICAM-1[228]. Increased ICAM-1 expression in CD is present not only in the mucosa but also in the submucosa and muscular layers[229], which could be implied in the transmural nature of CD. Most anti-adhesion molecule therapies target the integrin family. The first drug developed was natalizumab, a mAb against α4 that is not gut-specific. However, its use in patients with CD has been limited by the development in some patients of progressive multifocal leukoencephalopathy (PML), an opportunistic brain infection that is caused by reactivation of latent JC polyomavirus. Newer monoclonal antibodies targeting the α4β7 integrin (vedolizumab, a humanized immunoglobulin G1 monoclonal antibody to α4β7 integrin, selectively blocks gut lymphocyte trafficking without interfering with trafficking to the central nervous system)[230,231] and β7 subunit of the heterodimeric integrins α4β7 and αEβ7 (etrolizumab)[232] are now in late clinical development.
Figure 8.
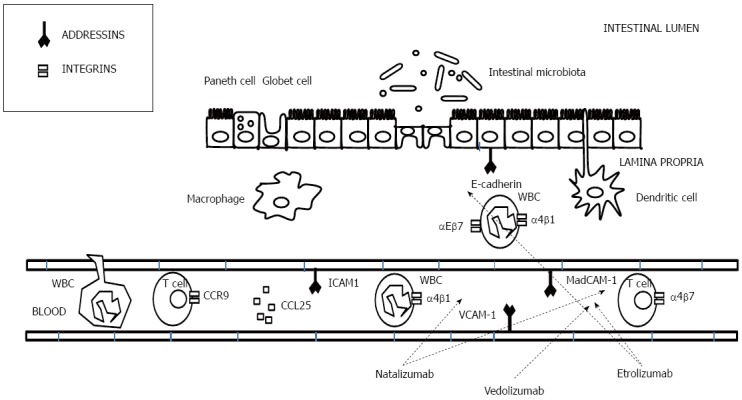
Mechanism of action of anti-adhesion drugs.
Evaluation of clinical response: Lack of long experience managing these compounds, variability in response and the high economic cost which limits its use, makes it difficult to find studies which describe predictors of response within this therapeutic group. Generally, response can be classified in non-genetic and genetic predictors.
Anti-TNF-α
Histological changes: The effects of anti-TNF agents are mediated by multiple mechanisms including direct neutralization of soluble TNF and interaction with membrane-bound TNF. Anti-TNF agents act by reduction of pro-inflammatory cytokine levels, elimination or clearance of active inflammatory cells from inflamed tissue which can conceptually be achieved by a number of mechanisms including apoptosis induction, antibody and complement mediated cytotoxicity and inhibition of cell migration into the intestinal tissue. Effects of anti-TNF agents may vary according to their physical contact with TNF, which may also be influenced by structural differences in the non-TNF binding domain affecting the ability of each drug to interact with the immune system.
The binding avidity of infliximab, adalimumab and etanercept to sTNF and mTNF appears to be similar[233]. But the bond between the anti-TNF agent and TNF may be reversible and as such, the anti-TNF molecule may actually serve as a TNF reservoir. In support of this possibility, the concentrations of immunoreactive TNF were shown to rise in the circulation following infliximab[234] and adalimumab[235] administration in rheumatoid arthritis, probably due to drug-TNF complexes. In vitro studies demonstrated that the rate of dissociation of etanercept from TNF is higher than that of infliximab and that the released TNF was bioavailable[236].
Infliximab has been shown to inhibit the production of GM-CSF and IFNγ[237] in vitro[238]. Infliximab, adalimumab and certolizumab inhibit the production of IL-1β from LPS-activated macrophages in CD. Down regulation of mucosal chemokine molecules following treatment with Infliximab was also shown in vivo for macrophage inflammatory protein (MIP)-1α, RANTES [Chemokine (C-C motif) ligand 5 (also CCL5)] and monocyte chemotactic protein (MCP)-1[239]. Another effect, less explored in CD and whose implications deserve further studies, is the change in tissue cellular populations following anti-TNF treatment. In one study, the population of FoxP3 positive cells was found to be reduced in the mucosa of active CD pediatric patients and infliximab treatment resulted in an increase on the mucosal number of these cells[240].
Ten Hove et al[241] observed apoptosis induction in mucosal CD3 positive T cells of CD patients treated with infliximab 24 h after drug administration. An additional study assessed mucosal biopsies from CD patients four weeks after treatment with standard infliximab induction doses and detected a significant increase in TUNEL-positive mucosal T cells[242]. Recently, another study used SPECT applied to infliximab-treated CD patients and detected positive up take in responders[243]. A similar profile of apoptosis induction was demonstrated in vitro for adalimumab[244]. However, no apoptosis induction was observed in vitro with certolizumab.
Another mechanism which is relevant for clearance of inflammatory cells is cytotoxicity. There is no study which definitively defines the relevance of these mechanisms in vivo. However, using Jurkat cells stably expressing uncleavable mTNF, Mitoma et al[246] demonstrated that infliximab and adalimumab exerted complement-dependent toxicity (CDC) equally, etanercept exerted CDC to a lesser extent and all three agents were capable of inducing antibody-dependent cell-mediated cytotoxicity (ADCC)[245]. Activation of p38 MAPK was demonstrated both in vitro and in vivo following exposure to infliximab. Differences between anti-TNF agents exist with respect to functional implications of reverse signaling. Kirchner et al[247] demonstrated that infliximab, but not etanercept inhibited LPS-induced cytokine secretion but both inhibited endothelial cell apoptotic factor. Similarly, infliximab and adalimumab inhibited secretion of IL-10 and IL-12 induced by LPS from monocytes, whereas etanercept did not[241]. In a different study, incubation of intestinal T cells derived from CD patients with infliximab and etanercept resulted in activation of the p38 MAPK pathway by infliximab only, although both reduced STAT3 activation[248].
Serological changes: Linked to the GCs chapter, anti-TNF-α therapies reduce MMP-7 levels but not to the levels of control patients as in GC therapy. α2M increase following treatment with infliximab. Anti-TNF-α therapy seems to induce α2M in serum, possibly to control disease activity. Anti-TNF-α therapy does not significantly alter serum TNF-α concentrations[249], and TNF-α can itself up-regulate MMP-7 expression[250]. This may explain why levels of MMP-7 remained elevated after anti-TNF-α treatment, compared with controls. Although TIMP-1 induction has been linked to the response to infliximab in CD in adults[251], unexpectedly weak TIMP-1 expression has been observed in pediatric IBD[252].
CD4+ T cells changes: infliximab appears to block up-regulation of CD73 on CD4+ T cells in response to TNF, but does not induce apoptosis in cells expressing high levels of CD73 ab initio. Doherty et al[243] speculated that the resolution of the inflammatory response induced by infliximab in the lamina propria results in downstream reduction of the inflammatory mediators of CD73 expression.
Imaging changes: Recent progress concerning molecular imaging studies in animals and human patients implicates that this approach can be used to improve detection of mucosal lesions in wide-field imaging and for in vivo characterization of the mucosa with the ultimate goal of assessing the likelihood of response to targeted therapy with biological agents. In a recently published study, using confocal laser endomicroscopy with a fluorescent antibody in vivo demonstrated its potential to predict therapeutic response to subsequent biological treatment in human patients. As anti-TNF Abs suppress immune responses in CD by binding to membrane-bound TNF (mTNF) expressing mucosal cells, in vivo detection of such cells via fluorescent anti-TNF Abs was used to predict therapeutic efficacy in CD patients via molecular imaging[253,254].
Genetics and genomics: Genetic non-response to Infliximab appears to be stable in time implicating a genetic influence [patients homozygous for a TNF-α polymorphism (LTA NcoI-TNFc-aa13L-aa26 1-1-1-1 haplotype)][255]. Previous data have confirmed the association of the TNFα-1031C allele with CD and polymorphisms at the TNFα locus have been suggested as a tool to predict response to Infliximab[256]. Halavaty et al[257] reported that polymorphisms in FasL/Fas system and caspase-9 influenced the response to infliximab in luminal and fistulizing Crohn’s disease. In this study, Fas ligand-843 TT genotype was presented exhibiting the strongest association with the lack of response, while concomitant 6-MP/AZA therapy, however, was able to overcome the effect of unfavourable genotypes in luminal disease. Another study identified a 100% accurate predictive gene signature for (non)response to IFX in CD colitis (TNFAIP6, S100A8, IL11, G0S2, and S100A9), whereas no such a predictive gene set could be identified for CD ileitis[258]. In another interesting study, several parameters were investigated to determine early response to infliximab in patients with UC[255]. Homozygous carriers of IBD risk-increasing IL23R variants were more likely to respond to infliximab than were homozygous carriers of IBD risk decreasing IL23R variants. Similar conclusions were reached by Rismo et al[259] who found that high levels of Th17-defining cytokine IL-17A and Th1-defining cytokine IFN-γ can potentially predict a favorable outcome of infliximab therapy in patients with UC, whereas Th2 and T-reg related cytokines do not seem to be useful as predictive markers in relation to therapeutic outcome.
Anti IL-12/23 and anti-adhesion therapies
Although recently, IL-20, IL-21 and p40 have been proposed as potential biomarkers of treatment response to ustekinumab in psoriasis, no data have been published related to IBD until now[260].
CONCLUSION
In summary, great advances have been made in the last years for a better comprehension of the pathogenesis of IBD that undoubtedly will lead us to develop more accurate therapies and therapeutic strategies. Alterations in recognition and intracellular processing of bacterial components with involvement of ER stress and apoptotic cell death may have a meaningful role in the immunopathogenesis of the disease. The weakening of the mucosal defenses promoting excessive interactions between commensal microbiota and the mucosal immune system leads to a loss of tolerance and over-activation of dendritic cells starting the production of pro-inflammatory cytokines and a promotion of differentiation of effector T cells and CD8* lymphocytes. Also active IBD is dependent on the recruitment of mononuclear cells and leukocyte populations from the blood stream into the bowel wall. This recruitment is dependent on a series of steps coordinated by selective adhesion molecules on the surface of immune cells and mucosal addressins on endothelial cells.
SRecent years knowledge in the areas of Immunology and Genetics has allowed to start using some biomarkers to measure the responsiveness of CD and UC diseases to certain therapies: mucosal HSP60 and Hsp10 levels in UC treated with 5-ASA; serum MMP-7 in response to GC therapy; role of 6-TGN and 6-MMP in AZA’s treated patients; inverse correlation between red blood cells MTXGlu1-5 and MTX efficacy in CD; through levels of Tac and 2-h CsA blood concentrations and lately, the likehood of response to targeted therapies with biological agents using immune cells imaging.
The goal of physicians treating patients with IBD, is to have to their disposal a molecular (serum, DNA, tissue-based) profile of patients which would allow them to choose the most appropriate management of the disease: prognosis of the disease outcome, most adequate therapy for an individual patient to reach success, most adequate therapy from a safety perspective, prediction on the intensity of follow-up, etc. At the moment, only TPMT testing prior to start of thiopurine analogues has shown clinical applicability, although it does not replace blood monitoring during treatment and some authors even defend the idea that the latest can substitute the former. Other genetic associations for the different therapeutic classes in IBD have not yet shown consistent or robust results but in the close future it is anticipated that this genetic marker’s determination will be implemented in an integrated molecular diagnostic and prognostic approach to manage IBD patients.
ACKNOWLEDGMENTS
The authors would like to thank the unvaluable help of Katrin Seemayer (Grünenthal Information Management Services) for her contribution to the scientific information, criticism of the manuscript and subsequent corrections.
Footnotes
Conflict-of-interest statement: Emilio G Quetglas, Simone Wigge and Sebastian Wachten are currently Grünenthal Employees. Zlatan Mujagic and Daniel Keszthelyi declare no conflict of interest. Ad Masclee has received research funding from Grünenthal, Ferring Pharmaceuticals, Falk Medical and DSM. Walter Reinisch Walter Reinisch has served as a speaker, consultant, and/or advisory board member for Abbott Laboratories, AbbVie, Aesca, Amgen, Astellas, Astra Zeneca, Biogen IDEC, Bristol-Myers Squibb, Cellerix, Chemocentryx, Celgene, Centocor, Danone Austria, Elan, Ferring, Galapagos, Genentech, Grünenthal, Johnson and Johnson, Kyowa Hakko Kirin Pharma, Lipid Therapeutics, Millenium, Mitsubishi Tanabe Pharma Corporation, MSD, Novartis, Ocera, Otsuka, PDL, Pharmacosmos, Pfizer, Procter and Gamble, Prometheus, Robarts Clinical Trial, Schering-Plough, Setpointmedical, Shire, Takeda, Therakos, Tigenix, UCB, Vifor, Yakult, Zyngenia, Austria, and 4SC.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 7, 2015
First decision: May 18, 2015
Article in press: September 14, 2015
P- Reviewer: Ledder OD S- Editor: Yu J L- Editor: A E- Editor: Zhang DN
References
- 1.Iskandar HN, Ciorba MA. Biomarkers in inflammatory bowel disease: current practices and recent advances. Transl Res. 2012;159:313–325. doi: 10.1016/j.trsl.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 3.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 4.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 5.Shaw MH, Kamada N, Warner N, Kim YG, Nuñez G. The ever-expanding function of NOD2: autophagy, viral recognition, and T cell activation. Trends Immunol. 2011;32:73–79. doi: 10.1016/j.it.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramasundara M, Leach ST, Lemberg DA, Day AS. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol. 2009;24:202–208. doi: 10.1111/j.1440-1746.2008.05772.x. [DOI] [PubMed] [Google Scholar]
- 9.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 10.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura K, Kanai T, Hayashi A, Mikami Y, Sujino T, Mizuno S, Handa T, Matsuoka K, Hisamatsu T, Sato T, et al. Dysregulated balance of retinoid-related orphan receptor γt-dependent innate lymphoid cells is involved in the pathogenesis of chronic DSS-induced colitis. Biochem Biophys Res Commun. 2012;427:694–700. doi: 10.1016/j.bbrc.2012.09.091. [DOI] [PubMed] [Google Scholar]
- 13.Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, He B, Cassis L, Bigas A, Cols M, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science. 2013;342:447–453. doi: 10.1126/science.1237910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niess JH. Role of gut-resident dendritic cells in inflammatory bowel disease. Expert Rev Clin Immunol. 2009;5:451–461. doi: 10.1586/eci.09.20. [DOI] [PubMed] [Google Scholar]
- 15.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 16.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 17.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–131. [PMC free article] [PubMed] [Google Scholar]
- 18.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Bürgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rovedatti L, Kudo T, Biancheri P, Sarra M, Knowles CH, Rampton DS, Corazza GR, Monteleone G, Di Sabatino A, Macdonald TT. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58:1629–1636. doi: 10.1136/gut.2009.182170. [DOI] [PubMed] [Google Scholar]
- 21.Ziegler SF, Buckner JH. FOXP3 and the regulation of Treg/Th17 differentiation. Microbes Infect. 2009;11:594–598. doi: 10.1016/j.micinf.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vainer B, Nielsen OH, Hendel J, Horn T, Kirman I. Colonic expression and synthesis of interleukin 13 and interleukin 15 in inflammatory bowel disease. Cytokine. 2000;12:1531–1536. doi: 10.1006/cyto.2000.0744. [DOI] [PubMed] [Google Scholar]
- 23.Dong C, Nurieva RI. Regulation of immune and autoimmune responses by ICOS. J Autoimmun. 2003;21:255–260. doi: 10.1016/s0896-8411(03)00119-7. [DOI] [PubMed] [Google Scholar]
- 24.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 25.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, Kitazume MT, Nakazawa A, Sugita A, Koganei K, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57:1682–1689. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 27.Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, Fujiyama Y, Andoh A. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol. 2010;160:386–393. doi: 10.1111/j.1365-2249.2010.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahlén L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monteleone G, Boirivant M, Pallone F, MacDonald TT. TGF-beta1 and Smad7 in the regulation of IBD. Mucosal Immunol. 2008;1 Suppl 1:S50–S53. doi: 10.1038/mi.2008.55. [DOI] [PubMed] [Google Scholar]
- 30.Boirivant M, Pallone F, Di Giacinto C, Fina D, Monteleone I, Marinaro M, Caruso R, Colantoni A, Palmieri G, Sanchez M, et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology. 2006;131:1786–1798. doi: 10.1053/j.gastro.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 31.Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, Becker C, Macdonald TT, Pallone F, Neurath MF, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. 2009;136:1308–116, 1308-116. doi: 10.1053/j.gastro.2008.12.053. [DOI] [PubMed] [Google Scholar]
- 32.Podolsky DK. Selective adhesion-molecule therapy and inflammatory bowel disease--a tale of Janus? N Engl J Med. 2005;353:1965–1968. doi: 10.1056/NEJMe058212. [DOI] [PubMed] [Google Scholar]
- 33.Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014;20:6–21. doi: 10.3748/wjg.v20.i1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fina D, Sarra M, Fantini MC, Rizzo A, Caruso R, Caprioli F, Stolfi C, Cardolini I, Dottori M, Boirivant M, et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 35.Mowat C, Cole A, Windsor A, Ahmad T, Arnott I, Driscoll R, Mitton S, Orchard T, Rutter M, Younge L, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2011;60:571–607. doi: 10.1136/gut.2010.224154. [DOI] [PubMed] [Google Scholar]
- 36.Sutherland L, Macdonald JK. Oral 5-aminosalicylic acid for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2006;(2):CD000544. doi: 10.1002/14651858.CD000544.pub2. [DOI] [PubMed] [Google Scholar]
- 37.Bergman R, Parkes M. Systematic review: the use of mesalazine in inflammatory bowel disease. Aliment Pharmacol Ther. 2006;23:841–855. doi: 10.1111/j.1365-2036.2006.02846.x. [DOI] [PubMed] [Google Scholar]
- 38.Truelove SC, Watkinson G, Draper G. Comparison of corticosteroid and sulphasalazine therapy in ulcerative colitis. Br Med J. 1962;2:1708–1711. doi: 10.1136/bmj.2.5321.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Modigliani R, Mary JY, Simon JF, Cortot A, Soule JC, Gendre JP, Rene E. Clinical, biological, and endoscopic picture of attacks of Crohn’s disease. Evolution on prednisolone. Groupe d’Etude Thérapeutique des Affections Inflammatoires Digestives. Gastroenterology. 1990;98:811–818. doi: 10.1016/0016-5085(90)90002-i. [DOI] [PubMed] [Google Scholar]
- 40.Kane SV, Schoenfeld P, Sandborn WJ, Tremaine W, Hofer T, Feagan BG. The effectiveness of budesonide therapy for Crohn’s disease. Aliment Pharmacol Ther. 2002;16:1509–1517. doi: 10.1046/j.1365-2036.2002.01289.x. [DOI] [PubMed] [Google Scholar]
- 41.Ardizzone S, Maconi G, Russo A, Imbesi V, Colombo E, Bianchi Porro G. Randomised controlled trial of azathioprine and 5-aminosalicylic acid for treatment of steroid dependent ulcerative colitis. Gut. 2006;55:47–53. doi: 10.1136/gut.2005.068809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prefontaine E, Macdonald JK, Sutherland LR. Azathioprine or 6-mercaptopurine for induction of remission in Crohn’s disease. Cochrane Database Syst Rev. 2009;(4):CD000545. doi: 10.1002/14651858.CD000545.pub2. [DOI] [PubMed] [Google Scholar]
- 43.Prefontaine E, Sutherland LR, Macdonald JK, Cepoiu M. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2009;(1):CD000067. doi: 10.1002/14651858.CD000067.pub2. [DOI] [PubMed] [Google Scholar]
- 44.Alfadhli AA, McDonald JW, Feagan BG. Methotrexate for induction of remission in refractory Crohn’s disease. Cochrane Database Syst Rev. 2005;(1):CD003459. doi: 10.1002/14651858.CD003459.pub2. [DOI] [PubMed] [Google Scholar]
- 45.Patel V, Macdonald JK, McDonald JW, Chande N. Methotrexate for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev. 2009;(4):CD006884. doi: 10.1002/14651858.CD006884.pub2. [DOI] [PubMed] [Google Scholar]
- 46.Feagan BG, Rochon J, Fedorak RN, Irvine EJ, Wild G, Sutherland L, Steinhart AH, Greenberg GR, Gillies R, Hopkins M. Methotrexate for the treatment of Crohn’s disease. The North American Crohn’s Study Group Investigators. N Engl J Med. 1995;332:292–297. doi: 10.1056/NEJM199502023320503. [DOI] [PubMed] [Google Scholar]
- 47.El-Matary W, Huynh H, Vandermeer B. Diagnostic characteristics of given video capsule endoscopy in diagnosis of celiac disease: a meta-analysis. J Laparoendosc Adv Surg Tech A. 2009;19:815–820. doi: 10.1089/lap.2008.0380. [DOI] [PubMed] [Google Scholar]
- 48.Wahed M, Louis-Auguste JR, Baxter LM, Limdi JK, McCartney SA, Lindsay JO, Bloom SL. Efficacy of methotrexate in Crohn’s disease and ulcerative colitis patients unresponsive or intolerant to azathioprine /mercaptopurine. Aliment Pharmacol Ther. 2009;30:614–620. doi: 10.1111/j.1365-2036.2009.04073.x. [DOI] [PubMed] [Google Scholar]
- 49.Lichtiger S, Present DH, Kornbluth A, Gelernt I, Bauer J, Galler G, Michelassi F, Hanauer S. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. N Engl J Med. 1994;330:1841–1845. doi: 10.1056/NEJM199406303302601. [DOI] [PubMed] [Google Scholar]
- 50.Shibolet O, Regushevskaya E, Brezis M, Soares-Weiser K. Cyclosporine A for induction of remission in severe ulcerative colitis. Cochrane Database Syst Rev. 2005;(1):CD004277. doi: 10.1002/14651858.CD004277.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ogata H, Matsui T, Nakamura M, Iida M, Takazoe M, Suzuki Y, Hibi T. A randomised dose finding study of oral tacrolimus (FK506) therapy in refractory ulcerative colitis. Gut. 2006;55:1255–1262. doi: 10.1136/gut.2005.081794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–1549. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- 53.Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–1610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- 54.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 55.Egan LJ, Mays DC, Huntoon CJ, Bell MP, Pike MG, Sandborn WJ, Lipsky JJ, McKean DJ. Inhibition of interleukin-1-stimulated NF-kappaB RelA/p65 phosphorylation by mesalamine is accompanied by decreased transcriptional activity. J Biol Chem. 1999;274:26448–26453. doi: 10.1074/jbc.274.37.26448. [DOI] [PubMed] [Google Scholar]
- 56.Lawson MM, Thomas AG, Akobeng AK. Tumour necrosis factor alpha blocking agents for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2006;(3):CD005112. doi: 10.1002/14651858.CD005112.pub2. [DOI] [PubMed] [Google Scholar]
- 57.Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, Wahli W, Desvergne B, Naccari GC, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stenson WF, Lobos E. Sulfasalazine inhibits the synthesis of chemotactic lipids by neutrophils. J Clin Invest. 1982;69:494–497. doi: 10.1172/JCI110474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mahida YR, Lamming CE, Gallagher A, Hawthorne AB, Hawkey CJ. 5-Aminosalicylic acid is a potent inhibitor of interleukin 1 beta production in organ culture of colonic biopsy specimens from patients with inflammatory bowel disease. Gut. 1991;32:50–54. doi: 10.1136/gut.32.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaiser GC, Yan F, Polk DB. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor kappaB activation in mouse colonocytes. Gastroenterology. 1999;116:602–609. doi: 10.1016/s0016-5085(99)70182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahnfelt-Rønne I, Nielsen OH, Christensen A, Langholz E, Binder V, Riis P. Clinical evidence supporting the radical scavenger mechanism of 5-aminosalicylic acid. Gastroenterology. 1990;98:1162–1169. doi: 10.1016/0016-5085(90)90329-y. [DOI] [PubMed] [Google Scholar]
- 62.Macario AJ, Lange M, Ahring BK, Conway de Macario E. Stress genes and proteins in the archaea. Microbiol Mol Biol Rev. 1999;63:923–967, table of contents. doi: 10.1128/mmbr.63.4.923-967.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prohászka Z, Füst G. Immunological aspects of heat-shock proteins-the optimum stress of life. Mol Immunol. 2004;41:29–44. doi: 10.1016/j.molimm.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 64.Cappello F, Conway de Macario E, Di Felice V, Zummo G, Macario AJ. Chlamydia trachomatis infection and anti-Hsp60 immunity: the two sides of the coin. PLoS Pathog. 2009;5:e1000552. doi: 10.1371/journal.ppat.1000552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rodolico V, Tomasello G, Zerilli M, Martorana A, Pitruzzella A, Gammazza AM, David S, Zummo G, Damiani P, Accomando S, et al. Hsp60 and Hsp10 increase in colon mucosa of Crohn’s disease and ulcerative colitis. Cell Stress Chaperones. 2010;15:877–884. doi: 10.1007/s12192-010-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tomasello G, Rodolico V, Zerilli M, Martorana A, Bucchieri F, Pitruzzella A, Marino Gammazza A, David S, Rappa F, Zummo G, et al. Changes in immunohistochemical levels and subcellular localization after therapy and correlation and colocalization with CD68 suggest a pathogenetic role of Hsp60 in ulcerative colitis. Appl Immunohistochem Mol Morphol. 2011;19:552–561. doi: 10.1097/PAI.0b013e3182118e5f. [DOI] [PubMed] [Google Scholar]
- 67.Tomasello G, Sciumé C, Rappa F, Rodolico V, Zerilli M, Martorana A, Cicero G, De Luca R, Damiani P, Accardo FM, et al. Hsp10, Hsp70, and Hsp90 immunohistochemical levels change in ulcerative colitis after therapy. Eur J Histochem. 2011;55:e38. doi: 10.4081/ejh.2011.e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yun TJ, Harning EK, Giza K, Rabah D, Li P, Arndt JW, Luchetti D, Biamonte MA, Shi J, Lundgren K, et al. EC144, a synthetic inhibitor of heat shock protein 90, blocks innate and adaptive immune responses in models of inflammation and autoimmunity. J Immunol. 2011;186:563–575. doi: 10.4049/jimmunol.1000222. [DOI] [PubMed] [Google Scholar]
- 69.Dignass A, Lindsay JO, Sturm A, Windsor A, Colombel JF, Allez M, D’Haens G, D’Hoore A, Mantzaris G, Novacek G, et al. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 2: current management. J Crohns Colitis. 2012;6:991–1030. doi: 10.1016/j.crohns.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 70.Dignass A, Van Assche G, Lindsay JO, Lémann M, Söderholm J, Colombel JF, Danese S, D’Hoore A, Gassull M, Gomollón F, et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: Current management. J Crohns Colitis. 2010;4:28–62. doi: 10.1016/j.crohns.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 71.Vermeire S, Van Assche G, Rutgeerts P. Role of genetics in prediction of disease course and response to therapy. World J Gastroenterol. 2010;16:2609–2615. doi: 10.3748/wjg.v16.i21.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beato M, Herrlich P, Schütz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 73.Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–198. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 74.Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol. 2002;71:9–15. [PubMed] [Google Scholar]
- 75.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nordeen SK, Suh BJ, Kühnel B, Hutchison CA. Structural determinants of a glucocorticoid receptor recognition element. Mol Endocrinol. 1990;4:1866–1873. doi: 10.1210/mend-4-12-1866. [DOI] [PubMed] [Google Scholar]
- 77.Catley M. Dissociated steroids. ScientificWorldJournal. 2007;7:421–430. doi: 10.1100/tsw.2007.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, Nordhues U, Sorg C, Sunderkötter C, Roth J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2007;109:1265–1274. doi: 10.1182/blood-2006-02-001115. [DOI] [PubMed] [Google Scholar]
- 79.Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. 2004;101:227–232. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 81.Stahn C, Löwenberg M, Hommes DW, Buttgereit F. Molecular mechanisms of glucocorticoid action and selective glucocorticoid receptor agonists. Mol Cell Endocrinol. 2007;275:71–78. doi: 10.1016/j.mce.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 82.Löwenberg M, Stahn C, Hommes DW, Buttgereit F. Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids. 2008;73:1025–1029. doi: 10.1016/j.steroids.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, Drouin J, Karin M. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 84.Ray A, Prefontaine KE. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. 1994;91:752–756. doi: 10.1073/pnas.91.2.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stöcklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 86.Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol. 2001;79:376–384. doi: 10.1046/j.1440-1711.2001.01025.x. [DOI] [PubMed] [Google Scholar]
- 87.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 88.Miner JN, Yamamoto KR. The basic region of AP-1 specifies glucocorticoid receptor activity at a composite response element. Genes Dev. 1992;6:2491–2501. doi: 10.1101/gad.6.12b.2491. [DOI] [PubMed] [Google Scholar]
- 89.Cascorbi I, Gerloff T, Johne A, Meisel C, Hoffmeyer S, Schwab M, Schaeffeler E, Eichelbaum M, Brinkmann U, Roots I. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–174. doi: 10.1067/mcp.2001.114164. [DOI] [PubMed] [Google Scholar]
- 90.Schaeffeler E, Eichelbaum M, Brinkmann U, Penger A, Asante-Poku S, Zanger UM, Schwab M. Frequency of C3435T polymorphism of MDR1 gene in African people. Lancet. 2001;358:383–384. doi: 10.1016/S0140-6736(01)05579-9. [DOI] [PubMed] [Google Scholar]
- 91.Kam JC, Szefler SJ, Surs W, Sher ER, Leung DY. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J Immunol. 1993;151:3460–3466. [PubMed] [Google Scholar]
- 92.Adcock IM, Lane SJ, Brown CR, Lee TH, Barnes PJ. Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. J Exp Med. 1995;182:1951–1958. doi: 10.1084/jem.182.6.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dumont A, Hehner SP, Schmitz ML, Gustafsson JA, Lidén J, Okret S, van der Saag PT, Wissink S, van der Burg B, Herrlich P, et al. Cross-talk between steroids and NF-kappa B: what language? Trends Biochem Sci. 1998;23:233–235. doi: 10.1016/s0968-0004(98)01212-2. [DOI] [PubMed] [Google Scholar]
- 95.Bantel H, Schmitz ML, Raible A, Gregor M, Schulze-Osthoff K. Critical role of NF-kappaB and stress-activated protein kinases in steroid unresponsiveness. FASEB J. 2002;16:1832–1834. doi: 10.1096/fj.02-0223fje. [DOI] [PubMed] [Google Scholar]
- 96.Farrell RJ, Murphy A, Long A, Donnelly S, Cherikuri A, O’Toole D, Mahmud N, Keeling PW, Weir DG, Kelleher D. High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology. 2000;118:279–288. doi: 10.1016/s0016-5085(00)70210-1. [DOI] [PubMed] [Google Scholar]
- 97.Brant SR, Panhuysen CI, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, Zhang L, Swanson E, Datta LW, Moran T, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73:1282–1292. doi: 10.1086/379927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ho GT, Nimmo ER, Tenesa A, Fennell J, Drummond H, Mowat C, Arnott ID, Satsangi J. Allelic variations of the multidrug resistance gene determine susceptibility and disease behavior in ulcerative colitis. Gastroenterology. 2005;128:288–296. doi: 10.1053/j.gastro.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 99.Griga T, Wilkens C, Wirkus N, Epplen J, Schmiegel W, Klein W. A polymorphism in the macrophage migration inhibitory factor gene is involved in the genetic predisposition of Crohn’s disease and associated with cumulative steroid doses. Hepatogastroenterology. 2007;54:784–786. [PubMed] [Google Scholar]
- 100.Cucchiara S, Latiano A, Palmieri O, Canani RB, D’Incà R, Guariso G, Vieni G, De Venuto D, Riegler G, De’Angelis GL, et al. Polymorphisms of tumor necrosis factor-alpha but not MDR1 influence response to medical therapy in pediatric-onset inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2007;44:171–179. doi: 10.1097/MPG.0b013e31802c41f3. [DOI] [PubMed] [Google Scholar]
- 101.Bonyadi MJ, Gerami SM, Somi MH, Khoshbaten M. Effect of the C3435T polymorphism of the multidrug resistance 1 gene on the severity of inflammatory bowel disease in Iranian Azeri Turks. Saudi J Gastroenterol. 2013;19:172–176. doi: 10.4103/1319-3767.114515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Curran S, Murray GI. Matrix metalloproteinases: molecular aspects of their roles in tumour invasion and metastasis. Eur J Cancer. 2000;36:1621–1630. doi: 10.1016/s0959-8049(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 103.Ravi A, Garg P, Sitaraman SV. Matrix metalloproteinases in inflammatory bowel disease: boon or a bane? Inflamm Bowel Dis. 2007;13:97–107. doi: 10.1002/ibd.20011. [DOI] [PubMed] [Google Scholar]
- 104.Baugh MD, Perry MJ, Hollander AP, Davies DR, Cross SS, Lobo AJ, Taylor CJ, Evans GS. Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology. 1999;117:814–822. doi: 10.1016/s0016-5085(99)70339-2. [DOI] [PubMed] [Google Scholar]
- 105.Matsuno K, Adachi Y, Yamamoto H, Goto A, Arimura Y, Endo T, Itoh F, Imai K. The expression of matrix metalloproteinase matrilysin indicates the degree of inflammation in ulcerative colitis. J Gastroenterol. 2003;38:348–354. doi: 10.1007/s005350300062. [DOI] [PubMed] [Google Scholar]
- 106.Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 107.Louis E, Ribbens C, Godon A, Franchimont D, De Groote D, Hardy N, Boniver J, Belaiche J, Malaise M. Increased production of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 by inflamed mucosa in inflammatory bowel disease. Clin Exp Immunol. 2000;120:241–246. doi: 10.1046/j.1365-2249.2000.01227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mäkitalo L, Sipponen T, Kärkkäinen P, Kolho KL, Saarialho-Kere U. Changes in matrix metalloproteinase (MMP) and tissue inhibitors of metalloproteinases (TIMP) expression profile in Crohn’s disease after immunosuppressive treatment correlate with histological score and calprotectin values. Int J Colorectal Dis. 2009;24:1157–1167. doi: 10.1007/s00384-009-0756-5. [DOI] [PubMed] [Google Scholar]
- 109.Hayden DM, Forsyth C, Keshavarzian A. The role of matrix metalloproteinases in intestinal epithelial wound healing during normal and inflammatory states. J Surg Res. 2011;168:315–324. doi: 10.1016/j.jss.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 110.Mäkitalo L, Rintamäki H, Tervahartiala T, Sorsa T, Kolho KL. Serum MMPs 7-9 and their inhibitors during glucocorticoid and anti-TNF-α therapy in pediatric inflammatory bowel disease. Scand J Gastroenterol. 2012;47:785–794. doi: 10.3109/00365521.2012.677954. [DOI] [PubMed] [Google Scholar]
- 111.Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, Kohgo Y. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–866. doi: 10.1016/s0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- 112.Fujishima S, Takeda H, Kawata S, Yamakawa M. The relationship between the expression of the glucocorticoid receptor in biopsied colonic mucosa and the glucocorticoid responsiveness of ulcerative colitis patients. Clin Immunol. 2009;133:208–217. doi: 10.1016/j.clim.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 113.De Iudicibus S, Lucafò M, Martelossi S, Pierobon C, Ventura A, Decorti G. MicroRNAs as tools to predict glucocorticoid response in inflammatory bowel diseases. World J Gastroenterol. 2013;19:7947–7954. doi: 10.3748/wjg.v19.i44.7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sionov RV. MicroRNAs and Glucocorticoid-Induced Apoptosis in Lymphoid Malignancies. ISRN Hematol. 2013;2013:348212. doi: 10.1155/2013/348212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.De Iudicibus S, Franca R, Martelossi S, Ventura A, Decorti G. Molecular mechanism of glucocorticoid resistance in inflammatory bowel disease. World J Gastroenterol. 2011;17:1095–1108. doi: 10.3748/wjg.v17.i9.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.De Iudicibus S, Stocco G, Martelossi S, Londero M, Ebner E, Pontillo A, Lionetti P, Barabino A, Bartoli F, Ventura A, et al. Genetic predictors of glucocorticoid response in pediatric patients with inflammatory bowel diseases. J Clin Gastroenterol. 2011;45:e1–e7. doi: 10.1097/MCG.0b013e3181e8ae93. [DOI] [PubMed] [Google Scholar]
- 117.Montero-Meléndez T, Llor X, García-Planella E, Perretti M, Suárez A. Identification of novel predictor classifiers for inflammatory bowel disease by gene expression profiling. PLoS One. 2013;8:e76235. doi: 10.1371/journal.pone.0076235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, Lehr HA, Wirtz S, Becker C, Atreya R, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111:1133–1145. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Colombel JF, Ferrari N, Debuysere H, Marteau P, Gendre JP, Bonaz B, Soulé JC, Modigliani R, Touze Y, Catala P, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn’s disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–1030. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 120.Gisbert JP, Niño P, Rodrigo L, Cara C, Guijarro LG. Thiopurine methyltransferase (TPMT) activity and adverse effects of azathioprine in inflammatory bowel disease: long-term follow-up study of 394 patients. Am J Gastroenterol. 2006;101:2769–2776. doi: 10.1111/j.1572-0241.2006.00843.x. [DOI] [PubMed] [Google Scholar]
- 121.Rashidi MR, Beedham C, Smith JS, Davaran S. In vitro study of 6-mercaptopurine oxidation catalysed by aldehyde oxidase and xanthine oxidase. Drug Metab Pharmacokinet. 2007;22:299–306. doi: 10.2133/dmpk.22.299. [DOI] [PubMed] [Google Scholar]
- 122.Al Hadithy AF, de Boer NK, Derijks LJ, Escher JC, Mulder CJ, Brouwers JR. Thiopurines in inflammatory bowel disease: pharmacogenetics, therapeutic drug monitoring and clinical recommendations. Dig Liver Dis. 2005;37:282–297. doi: 10.1016/j.dld.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 123.Blaker PA, Arenas-Hernandez M, Smith MA, Shobowale-Bakre EA, Fairbanks L, Irving PM, Sanderson JD, Marinaki AM. Mechanism of allopurinol induced TPMT inhibition. Biochem Pharmacol. 2013;86:539–547. doi: 10.1016/j.bcp.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 124.Zimm S, Collins JM, O’Neill D, Chabner BA, Poplack DG. Inhibition of first-pass metabolism in cancer chemotherapy: interaction of 6-mercaptopurine and allopurinol. Clin Pharmacol Ther. 1983;34:810–817. doi: 10.1038/clpt.1983.254. [DOI] [PubMed] [Google Scholar]
- 125.Schwarz G. Molybdenum cofactor biosynthesis and deficiency. Cell Mol Life Sci. 2005;62:2792–2810. doi: 10.1007/s00018-005-5269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Peretz H, Naamati MS, Levartovsky D, Lagziel A, Shani E, Horn I, Shalev H, Landau D. Identification and characterization of the first mutation (Arg776Cys) in the C-terminal domain of the Human Molybdenum Cofactor Sulfurase (HMCS) associated with type II classical xanthinuria. Mol Genet Metab. 2007;91:23–29. doi: 10.1016/j.ymgme.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 127.Cuffari C, Hunt S, Bayless T. Utilisation of erythrocyte 6-thioguanine metabolite levels to optimise azathioprine therapy in patients with inflammatory bowel disease. Gut. 2001;48:642–646. doi: 10.1136/gut.48.5.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cuffari C, Théorêt Y, Latour S, Seidman G. 6-Mercaptopurine metabolism in Crohn’s disease: correlation with efficacy and toxicity. Gut. 1996;39:401–406. doi: 10.1136/gut.39.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dubinsky MC, Lamothe S, Yang HY, Targan SR, Sinnett D, Théorêt Y, Seidman EG. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–713. doi: 10.1016/s0016-5085(00)70140-5. [DOI] [PubMed] [Google Scholar]
- 130.Dubinsky MC, Feldman EJ, Abreu MT, Targan SR, Vasiliauskas EA. Thioguanine: a potential alternate thiopurine for IBD patients allergic to 6-mercaptopurine or azathioprine. Am J Gastroenterol. 2003;98:1058–1063. doi: 10.1111/j.1572-0241.2003.07413.x. [DOI] [PubMed] [Google Scholar]
- 131.Dubinsky MC, Yang H, Hassard PV, Seidman EG, Kam LY, Abreu MT, Targan SR, Vasiliauskas EA. 6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel disease. Gastroenterology. 2002;122:904–915. doi: 10.1053/gast.2002.32420. [DOI] [PubMed] [Google Scholar]
- 132.Herrlinger KR, Kreisel W, Schwab M, Schoelmerich J, Fleig WE, Ruhl A, Reinshagen M, Deibert P, Fellermann K, Greinwald R, et al. 6-thioguanine--efficacy and safety in chronic active Crohn’s disease. Aliment Pharmacol Ther. 2003;17:503–508. doi: 10.1046/j.1365-2036.2003.01440.x. [DOI] [PubMed] [Google Scholar]
- 133.Bonaz B, Boitard J, Marteau P, Lémann M, Coffin B, Flourié B, Belaiche J, Cadiot G, Metman EH, Cortot A, et al. Tioguanine in patients with Crohn’s disease intolerant or resistant to azathioprine/mercaptopurine. Aliment Pharmacol Ther. 2003;18:401–408. doi: 10.1046/j.1365-2036.2003.01683.x. [DOI] [PubMed] [Google Scholar]
- 134.Dubinsky MC, Vasiliauskas EA, Singh H, Abreu MT, Papadakis KA, Tran T, Martin P, Vierling JM, Geller SA, Targan SR, et al. 6-thioguanine can cause serious liver injury in inflammatory bowel disease patients. Gastroenterology. 2003;125:298–303. doi: 10.1016/s0016-5085(03)00938-7. [DOI] [PubMed] [Google Scholar]
- 135.Smith M, Blaker P, Patel C, Marinaki A, Arenas M, Escuredo E, Anderson S, Irving P, Sanderson J. The impact of introducing thioguanine nucleotide monitoring into an inflammatory bowel disease clinic. Int J Clin Pract. 2013;67:161–169. doi: 10.1111/ijcp.12039. [DOI] [PubMed] [Google Scholar]
- 136.Smith MA, Marinaki AM, Sanderson JD. Pharmacogenomics in the treatment of inflammatory bowel disease. Pharmacogenomics. 2010;11:421–437. doi: 10.2217/pgs.10.4. [DOI] [PubMed] [Google Scholar]
- 137.Sparrow MP, Hande SA, Friedman S, Cao D, Hanauer SB. Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine. Clin Gastroenterol Hepatol. 2007;5:209–214. doi: 10.1016/j.cgh.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 138.Clarke DA, Elion GB, Hitchings GH, Stock CC. Structure-activity relationships among purines related to 6-mercaptopurine. Cancer Res. 1958;18:445–456. [PubMed] [Google Scholar]
- 139.Atreya I, Neurath MF. Understanding the delayed onset of action of azathioprine in IBD: are we there yet? Gut. 2009;58:325–326. doi: 10.1136/gut.2008.163485. [DOI] [PubMed] [Google Scholar]
- 140.Jolivet J, Schilsky RL, Bailey BD, Drake JC, Chabner BA. Synthesis, retention, and biological activity of methotrexate polyglutamates in cultured human breast cancer cells. J Clin Invest. 1982;70:351–360. doi: 10.1172/JCI110624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jolivet J, Cowan KH, Curt GA, Clendeninn NJ, Chabner BA. The pharmacology and clinical use of methotrexate. N Engl J Med. 1983;309:1094–1104. doi: 10.1056/NEJM198311033091805. [DOI] [PubMed] [Google Scholar]
- 142.Jolivet J, Chabner BA. Intracellular pharmacokinetics of methotrexate polyglutamates in human breast cancer cells. Selective retention and less dissociable binding of 4-NH2-10-CH3-pteroylglutamate4 and 4-NH2-10-CH3-pteroylglutamate5 to dihydrofolate reductase. J Clin Invest. 1983;72:773–778. doi: 10.1172/JCI111048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Seideman P, Beck O, Eksborg S, Wennberg M. The pharmacokinetics of methotrexate and its 7-hydroxy metabolite in patients with rheumatoid arthritis. Br J Clin Pharmacol. 1993;35:409–412. doi: 10.1111/j.1365-2125.1993.tb04158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baggott JE, Morgan SL, Vaughn WH. Differences in methotrexate and 7-hydroxymethotrexate inhibition of folate-dependent enzymes of purine nucleotide biosynthesis. Biochem J. 1994;300(Pt 3):627–629. doi: 10.1042/bj3000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Genestier L, Paillot R, Quemeneur L, Izeradjene K, Revillard JP. Mechanisms of action of methotrexate. Immunopharmacology. 2000;47:247–257. doi: 10.1016/s0162-3109(00)00189-2. [DOI] [PubMed] [Google Scholar]
- 146.Shiroky JB, Neville C, Esdaile JM, Choquette D, Zummer M, Hazeltine M, Bykerk V, Kanji M, St-Pierre A, Robidoux L. Low-dose methotrexate with leucovorin (folinic acid) in the management of rheumatoid arthritis. Results of a multicenter randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 1993;36:795–803. doi: 10.1002/art.1780360609. [DOI] [PubMed] [Google Scholar]
- 147.Weinblatt ME, Maier AL, Coblyn JS. Low dose leucovorin does not interfere with the efficacy of methotrexate in rheumatoid arthritis: an 8 week randomized placebo controlled trial. J Rheumatol. 1993;20:950–952. [PubMed] [Google Scholar]
- 148.Joyce DA, Will RK, Hoffman DM, Laing B, Blackbourn SJ. Exacerbation of rheumatoid arthritis in patients treated with methotrexate after administration of folinic acid. Ann Rheum Dis. 1991;50:913–914. doi: 10.1136/ard.50.12.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Constantin A, Loubet-Lescoulié P, Lambert N, Yassine-Diab B, Abbal M, Mazières B, de Préval C, Cantagrel A. Antiinflammatory and immunoregulatory action of methotrexate in the treatment of rheumatoid arthritis: evidence of increased interleukin-4 and interleukin-10 gene expression demonstrated in vitro by competitive reverse transcriptase-polymerase chain reaction. Arthritis Rheum. 1998;41:48–57. doi: 10.1002/1529-0131(199801)41:1<48::AID-ART7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 150.Durez P, Appelboom T, Vray B, Pira C, Goldman M. Methotrexate inhibits LPS-induced tumor necrosis factor production in vivo. Eur Cytokine Netw. 1998;9:669–672. [PubMed] [Google Scholar]
- 151.Yamaki K, Uchida H, Harada Y, Li X, Yanagisawa R, Takano H, Hayashi H, Taneda S, Mori Y, Yoshino S. Effect of methotrexate on Th1 and Th2 immune responses in mice. J Pharm Pharmacol. 2003;55:1661–1666. doi: 10.1211/0022357022269. [DOI] [PubMed] [Google Scholar]
- 152.Hildner K, Finotto S, Becker C, Schlaak J, Schirmacher P, Galle PR, Märker-Hermann E, Neurath MF. Tumour necrosis factor (TNF) production by T cell receptor-primed T lymphocytes is a target for low dose methotrexate in rheumatoid arthritis. Clin Exp Immunol. 1999;118:137–146. doi: 10.1046/j.1365-2249.1999.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Becker C, Barbulescu K, Hildner K, Meyer zum Büschenfelde KH, Neurath MF. Activation and methotrexate-mediated suppression of the TNF alpha promoter in T cells and macrophages. Ann N Y Acad Sci. 1998;859:311–314. doi: 10.1111/j.1749-6632.1998.tb11153.x. [DOI] [PubMed] [Google Scholar]
- 154.Rudwaleit M, Yin Z, Siegert S, Grolms M, Radbruch A, Braun J, Sieper J. Response to methotrexate in early rheumatoid arthritis is associated with a decrease of T cell derived tumour necrosis factor alpha, increase of interleukin 10, and predicted by the initial concentration of interleukin 4. Ann Rheum Dis. 2000;59:311–314. doi: 10.1136/ard.59.4.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS. Inhibition of TNF-alpha expression by adenosine: role of A3 adenosine receptors. J Immunol. 1996;156:3435–3442. [PubMed] [Google Scholar]
- 156.Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene. 2003;22:1206–1218. doi: 10.1038/sj.onc.1206184. [DOI] [PubMed] [Google Scholar]
- 157.Seitz M, Zwicker M, Wider B. Enhanced in vitro induced production of interleukin 10 by peripheral blood mononuclear cells in rheumatoid arthritis is associated with clinical response to methotrexate treatment. J Rheumatol. 2001;28:496–501. [PubMed] [Google Scholar]
- 158.Laverdière C, Chiasson S, Costea I, Moghrabi A, Krajinovic M. Polymorphism G80A in the reduced folate carrier gene and its relationship to methotrexate plasma levels and outcome of childhood acute lymphoblastic leukemia. Blood. 2002;100:3832–3834. doi: 10.1182/blood.V100.10.3832. [DOI] [PubMed] [Google Scholar]
- 159.Chave KJ, Ryan TJ, Chmura SE, Galivan J. Identification of single nucleotide polymorphisms in the human gamma-glutamyl hydrolase gene and characterization of promoter polymorphisms. Gene. 2003;319:167–175. doi: 10.1016/s0378-1119(03)00807-2. [DOI] [PubMed] [Google Scholar]
- 160.Cheng Q, Wu B, Kager L, Panetta JC, Zheng J, Pui CH, Relling MV, Evans WE. A substrate specific functional polymorphism of human gamma-glutamyl hydrolase alters catalytic activity and methotrexate polyglutamate accumulation in acute lymphoblastic leukaemia cells. Pharmacogenetics. 2004;14:557–567. doi: 10.1097/01.fpc.0000114761.78957.7e. [DOI] [PubMed] [Google Scholar]
- 161.Ulrich CM, Yasui Y, Storb R, Schubert MM, Wagner JL, Bigler J, Ariail KS, Keener CL, Li S, Liu H, et al. Pharmacogenetics of methotrexate: toxicity among marrow transplantation patients varies with the methylenetetrahydrofolate reductase C677T polymorphism. Blood. 2001;98:231–234. doi: 10.1182/blood.v98.1.231. [DOI] [PubMed] [Google Scholar]
- 162.Mahmud N, Molloy A, McPartlin J, Corbally R, Whitehead AS, Scott JM, Weir DG. Increased prevalence of methylenetetrahydrofolate reductase C677T variant in patients with inflammatory bowel disease, and its clinical implications. Gut. 1999;45:389–394. doi: 10.1136/gut.45.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.van der Put NM, Gabreëls F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, van den Heuvel LP, Blom HJ. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62:1044–1051. doi: 10.1086/301825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Weisberg I, Tran P, Christensen B, Sibani S, Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab. 1998;64:169–172. doi: 10.1006/mgme.1998.2714. [DOI] [PubMed] [Google Scholar]
- 165.Urano W, Taniguchi A, Yamanaka H, Tanaka E, Nakajima H, Matsuda Y, Akama H, Kitamura Y, Kamatani N. Polymorphisms in the methylenetetrahydrofolate reductase gene were associated with both the efficacy and the toxicity of methotrexate used for the treatment of rheumatoid arthritis, as evidenced by single locus and haplotype analyses. Pharmacogenetics. 2002;12:183–190. doi: 10.1097/00008571-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 166.Rots MG, Pieters R, Peters GJ, Noordhuis P, Van Zantwijk CH, Henze G, Janka-Schaub GE, Veerman AJ, Jansen G. Methotrexate resistance in relapsed childhood acute lymphoblastic leukaemia. Br J Haematol. 2000;109:629–634. doi: 10.1046/j.1365-2141.2000.02071.x. [DOI] [PubMed] [Google Scholar]
- 167.Liani E, Rothem L, Bunni MA, Smith CA, Jansen G, Assaraf YG. Loss of folylpoly-gamma-glutamate synthetase activity is a dominant mechanism of resistance to polyglutamylation-dependent novel antifolates in multiple human leukemia sublines. Int J Cancer. 2003;103:587–599. doi: 10.1002/ijc.10829. [DOI] [PubMed] [Google Scholar]
- 168.Brooks AJ, Begg EJ, Zhang M, Frampton CM, Barclay ML. Red blood cell methotrexate polyglutamate concentrations in inflammatory bowel disease. Ther Drug Monit. 2007;29:619–625. doi: 10.1097/FTD.0b013e31811f39bb. [DOI] [PubMed] [Google Scholar]
- 169.Bram RJ, Hung DT, Martin PK, Schreiber SL, Crabtree GR. Identification of the immunophilins capable of mediating inhibition of signal transduction by cyclosporin A and FK506: roles of calcineurin binding and cellular location. Mol Cell Biol. 1993;13:4760–4769. doi: 10.1128/mcb.13.8.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cardenas ME, Hemenway C, Muir RS, Ye R, Fiorentino D, Heitman J. Immunophilins interact with calcineurin in the absence of exogenous immunosuppressive ligands. EMBO J. 1994;13:5944–5957. doi: 10.1002/j.1460-2075.1994.tb06940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature. 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- 172.Shaw KT, Ho AM, Raghavan A, Kim J, Jain J, Park J, Sharma S, Rao A, Hogan PG. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci USA. 1995;92:11205–11209. doi: 10.1073/pnas.92.24.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- 174.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 175.Ying S, Khan LN, Meng Q, Barnes NC, Kay AB. Cyclosporin A, apoptosis of BAL T-cells and expression of Bcl-2 in asthmatics. Eur Respir J. 2003;22:207–212. doi: 10.1183/09031936.03.00098902. [DOI] [PubMed] [Google Scholar]
- 176.Kountouras J, Kouklakis G, Zavos C, Chatzopoulos D, Moschos J, Molyvas E, Zavos N. Apoptosis, inflammatory bowel disease and carcinogenesis: overview of international and Greek experiences. Can J Gastroenterol. 2003;17:249–258. doi: 10.1155/2003/527060. [DOI] [PubMed] [Google Scholar]
- 177.Sandborn WJ, Tremaine WJ. Cyclosporine treatment of inflammatory bowel disease. Mayo Clin Proc. 1992;67:981–990. doi: 10.1016/s0025-6196(12)60930-6. [DOI] [PubMed] [Google Scholar]
- 178.Rafiee P, Johnson CP, Li MS, Ogawa H, Heidemann J, Fisher PJ, Lamirand TH, Otterson MF, Wilson KT, Binion DG. Cyclosporine A enhances leukocyte binding by human intestinal microvascular endothelial cells through inhibition of p38 MAPK and iNOS. Paradoxical proinflammatory effect on the microvascular endothelium. J Biol Chem. 2002;277:35605–35615. doi: 10.1074/jbc.M205826200. [DOI] [PubMed] [Google Scholar]
- 179.Barone G, Chang CT, Choc MG, Klein JB, Marsh CL, Meligeni JA, Min DI, Pescovitz MD, Pollak R, Pruett TL, et al. The pharmacokinetics of a microemulsion formulation of cyclosporine in primary renal allograft recipients. The Neoral Study Group. Transplantation. 1996;61:875–880. doi: 10.1097/00007890-199603270-00005. [DOI] [PubMed] [Google Scholar]
- 180.Lindholm A, Kahan BD. Influence of cyclosporine pharmacokinetics, trough concentrations, and AUC monitoring on outcome after kidney transplantation. Clin Pharmacol Ther. 1993;54:205–218. doi: 10.1038/clpt.1993.132. [DOI] [PubMed] [Google Scholar]
- 181.Bowers LD, Canafax DM, Singh J, Seifedlin R, Simmons RL, Najarian JS. Studies of cyclosporine blood levels: analysis, clinical utility, pharmacokinetics, metabolites, and chronopharmacology. Transplant Proc. 1986;18:137–143. [PubMed] [Google Scholar]
- 182.Batiuk TD, Kung L, Halloran PF. Evidence that calcineurin is rate-limiting for primary human lymphocyte activation. J Clin Invest. 1997;100:1894–1901. doi: 10.1172/JCI119719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Halloran PF, Helms LM, Kung L, Noujaim J. The temporal profile of calcineurin inhibition by cyclosporine in vivo. Transplantation. 1999;68:1356–1361. doi: 10.1097/00007890-199911150-00023. [DOI] [PubMed] [Google Scholar]
- 184.Jusko WJ, Piekoszewski W, Klintmalm GB, Shaefer MS, Hebert MF, Piergies AA, Lee CC, Schechter P, Mekki QA. Pharmacokinetics of tacrolimus in liver transplant patients. Clin Pharmacol Ther. 1995;57:281–290. doi: 10.1016/0009-9236(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 185.Sommerer C, Giese T, Meuer S, Zeier M. Pharmacodynamic monitoring of calcineurin inhibitor therapy: is there a clinical benefit? Nephrol Dial Transplant. 2009;24:21–27. doi: 10.1093/ndt/gfn556. [DOI] [PubMed] [Google Scholar]
- 186.Staatz CE, Goodman LK, Tett SE. Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: Part I. Clin Pharmacokinet. 2010;49:141–175. doi: 10.2165/11317350-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 187.Hirota T, Ieiri I, Takane H, Maegawa S, Hosokawa M, Kobayashi K, Chiba K, Nanba E, Oshimura M, Sato T, et al. Allelic expression imbalance of the human CYP3A4 gene and individual phenotypic status. Hum Mol Genet. 2004;13:2959–2969. doi: 10.1093/hmg/ddh313. [DOI] [PubMed] [Google Scholar]
- 188.van Rossum HH, de Fijter JW, van Pelt J. Pharmacodynamic monitoring of calcineurin inhibition therapy: principles, performance, and perspectives. Ther Drug Monit. 2010;32:3–10. doi: 10.1097/FTD.0b013e3181c0eecb. [DOI] [PubMed] [Google Scholar]
- 189.Sommerer C, Zeier M, Meuer S, Giese T. Individualized monitoring of nuclear factor of activated T cells-regulated gene expression in FK506-treated kidney transplant recipients. Transplantation. 2010;89:1417–1423. doi: 10.1097/TP.0b013e3181dc13b6. [DOI] [PubMed] [Google Scholar]
- 190.Sommerer C, Giese T, Schmidt J, Meuer S, Zeier M. Ciclosporin A tapering monitored by NFAT-regulated gene expression: a new concept of individual immunosuppression. Transplantation. 2008;85:15–21. doi: 10.1097/01.tp.0000296824.58884.55. [DOI] [PubMed] [Google Scholar]
- 191.Ferraresso M, Turolo S, Ghio L, Tirelli AS, Belingheri M, Villa R, Groppali E, Edefonti A. Association between CYP3A5 polymorphisms and blood pressure in kidney transplant recipients receiving calcineurin inhibitors. Clin Exp Hypertens. 2011;33:359–365. doi: 10.3109/10641963.2011.561896. [DOI] [PubMed] [Google Scholar]
- 192.Iyer V, Hajjar RJ, Armoundas AA. Mechanisms of abnormal calcium homeostasis in mutations responsible for catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;100:e22–e31. doi: 10.1161/01.RES.0000258468.31815.42. [DOI] [PubMed] [Google Scholar]
- 193.Yücel A, Dilek K, Saba D, Ozçimen AA, Yurtkuran M, Oral HB. Interleukin-2 gene polymorphism in Turkish patients with Behçet’s disease and its association with ocular involvement. Int J Immunogenet. 2013;40:349–355. doi: 10.1111/iji.12039. [DOI] [PubMed] [Google Scholar]
- 194.Song N, Han S, Lee KM, Choi JY, Park SK, Jeon S, Lee Y, Ahn HS, Shin HY, Kang HJ, et al. Genetic variants in interleukin-2 and risk of lymphoma among children in Korea. Asian Pac J Cancer Prev. 2012;13:621–623. doi: 10.7314/apjcp.2012.13.2.621. [DOI] [PubMed] [Google Scholar]
- 195.Jafari N, Broer L, van Duijn CM, Janssens AC, Hintzen RQ. Perspectives on the use of multiple sclerosis risk genes for prediction. PLoS One. 2011;6:e26493. doi: 10.1371/journal.pone.0026493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dannenberg LO, Edenberg HJ. Epigenetics of gene expression in human hepatoma cells: expression profiling the response to inhibition of DNA methylation and histone deacetylation. BMC Genomics. 2006;7:181. doi: 10.1186/1471-2164-7-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pan YZ, Gao W, Yu AM. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab Dispos. 2009;37:2112–2117. doi: 10.1124/dmd.109.027680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Takagi S, Nakajima M, Mohri T, Yokoi T. Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J Biol Chem. 2008;283:9674–9680. doi: 10.1074/jbc.M709382200. [DOI] [PubMed] [Google Scholar]
- 199.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 200.Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, Tremaine WJ, Johnson T, Diehl NN, Zinsmeister AR. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121:1088–1094. doi: 10.1053/gast.2001.28674. [DOI] [PubMed] [Google Scholar]
- 201.Scallon BJ, Moore MA, Trinh H, Knight DM, Ghrayeb J. Chimeric anti-TNF-alpha monoclonal antibody cA2 binds recombinant transmembrane TNF-alpha and activates immune effector functions. Cytokine. 1995;7:251–259. doi: 10.1006/cyto.1995.0029. [DOI] [PubMed] [Google Scholar]
- 202.Vos AC, Wildenberg ME, Arijs I, Duijvestein M, Verhaar AP, de Hertogh G, Vermeire S, Rutgeerts P, van den Brink GR, Hommes DW. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm Bowel Dis. 2012;18:401–408. doi: 10.1002/ibd.21818. [DOI] [PubMed] [Google Scholar]
- 203.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 204.Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol. 2005;35:469–475. doi: 10.1002/eji.200425677. [DOI] [PubMed] [Google Scholar]
- 205.van de Wetering D, de Paus RA, van Dissel JT, van de Vosse E. IL-23 modulates CD56+/CD3- NK cell and CD56+/CD3+ NK-like T cell function differentially from IL-12. Int Immunol. 2009;21:145–153. doi: 10.1093/intimm/dxn132. [DOI] [PubMed] [Google Scholar]
- 206.Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, Nussenblatt RB, Caspi RR. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.O’Brien RL, Roark CL, Born WK. IL-17-producing gammadelta T cells. Eur J Immunol. 2009;39:662–666. doi: 10.1002/eji.200839120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 209.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 210.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 211.Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. 1991;65:859–873. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 212.Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 213.Campbell JJ, Qin S, Bacon KB, Mackay CR, Butcher EC. Biology of chemokine and classical chemoattractant receptors: differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J Cell Biol. 1996;134:255–266. doi: 10.1083/jcb.134.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 215.Thomas S, Baumgart DC. Targeting leukocyte migration and adhesion in Crohn’s disease and ulcerative colitis. Inflammopharmacology. 2012;20:1–18. doi: 10.1007/s10787-011-0104-6. [DOI] [PubMed] [Google Scholar]
- 216.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 217.Panés J, Granger DN. Leukocyte-endothelial cell interactions: molecular mechanisms and implications in gastrointestinal disease. Gastroenterology. 1998;114:1066–1090. doi: 10.1016/s0016-5085(98)70328-2. [DOI] [PubMed] [Google Scholar]
- 218.Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, Lobb RR. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 1990;60:577–584. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- 219.Tsuzuki Y, Miura S, Suematsu M, Kurose I, Shigematsu T, Kimura H, Higuchi H, Serizawa H, Yagita H, Okumura K, et al. alpha 4 integrin plays a critical role in early stages of T lymphocyte migration in Peyer’s patches of rats. Int Immunol. 1996;8:287–295. doi: 10.1093/intimm/8.3.287. [DOI] [PubMed] [Google Scholar]
- 220.Hu MC, Crowe DT, Weissman IL, Holzmann B. Cloning and expression of mouse integrin beta p(beta 7): a functional role in Peyer’s patch-specific lymphocyte homing. Proc Natl Acad Sci USA. 1992;89:8254–8258. doi: 10.1073/pnas.89.17.8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cepek KL, Parker CM, Madara JL, Brenner MB. Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J Immunol. 1993;150:3459–3470. [PubMed] [Google Scholar]
- 222.Elewaut D, Van Damme N, De Keyser F, Baeten D, De Paepe P, Van Vlierberghe H, Mielants H, Cuvelier C, Verbruggen G, Veys EM, et al. Altered expression of alpha E beta 7 integrin on intra-epithelial and lamina propria lymphocytes in patients with Crohn’s disease. Acta Gastroenterol Belg. 1998;61:288–294. [PubMed] [Google Scholar]
- 223.Oshitani N, Watanabe K, Maeda K, Fujiwara Y, Higuchi K, Matsumoto T, Arakawa T. Differential expression of homing receptor CD103 on lamina propria lymphocytes and association of CD103 with epithelial adhesion molecules in inflammatory bowel disease. Int J Mol Med. 2003;12:715–719. [PubMed] [Google Scholar]
- 224.Schön MP, Arya A, Murphy EA, Adams CM, Strauch UG, Agace WW, Marsal J, Donohue JP, Her H, Beier DR, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol. 1999;162:6641–6649. [PubMed] [Google Scholar]
- 225.Bernstein CN, Sargent M, Gallatin WM. Beta2 integrin/ICAM expression in Crohn’s disease. Clin Immunol Immunopathol. 1998;86:147–160. doi: 10.1006/clin.1997.4462. [DOI] [PubMed] [Google Scholar]
- 226.Meenan J, Spaans J, Grool TA, Pals ST, Tytgat GN, van Deventer SJ. Altered expression of alpha 4 beta 7, a gut homing integrin, by circulating and mucosal T cells in colonic mucosal inflammation. Gut. 1997;40:241–246. doi: 10.1136/gut.40.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Biancheri P, Di Sabatino A, Rovedatti L, Giuffrida P, Calarota SA, Vetrano S, Vidali F, Pasini A, Danese S, Corazza GR, et al. Effect of tumor necrosis factor-α blockade on mucosal addressin cell-adhesion molecule-1 in Crohn’s disease. Inflamm Bowel Dis. 2013;19:259–264. doi: 10.1097/MIB.0b013e31828100a4. [DOI] [PubMed] [Google Scholar]
- 228.Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, Geboes K, Rutgeerts PJ. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. 1999;116:22–28. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 229.Hogaboam CM, Snider DP, Collins SM. Activation of T lymphocytes by syngeneic murine intestinal smooth muscle cells. Gastroenterology. 1996;110:1456–1466. doi: 10.1053/gast.1996.v110.pm8613051. [DOI] [PubMed] [Google Scholar]
- 230.Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, Lukas M, Fedorak RN, Lee S, Bressler B, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369:711–721. doi: 10.1056/NEJMoa1215739. [DOI] [PubMed] [Google Scholar]
- 231.Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, Van Assche G, Axler J, Kim HJ, Danese S, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699–710. doi: 10.1056/NEJMoa1215734. [DOI] [PubMed] [Google Scholar]
- 232.Vermeire S, O’Byrne S, Keir M, Williams M, Lu TT, Mansfield JC, Lamb CA, Feagan BG, Panes J, Salas A, et al. Etrolizumab as induction therapy for ulcerative colitis: a randomised, controlled, phase 2 trial. Lancet. 2014;384:309–318. doi: 10.1016/S0140-6736(14)60661-9. [DOI] [PubMed] [Google Scholar]
- 233.Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, Salfeld J, Sasso EH. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131:308–316. doi: 10.1016/j.clim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 234.Charles P, Elliott MJ, Davis D, Potter A, Kalden JR, Antoni C, Breedveld FC, Smolen JS, Eberl G, deWoody K, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. 1999;163:1521–1528. [PubMed] [Google Scholar]
- 235.Barrera P, Joosten LA, den Broeder AA, van de Putte LB, van Riel PL, van den Berg WB. Effects of treatment with a fully human anti-tumour necrosis factor alpha monoclonal antibody on the local and systemic homeostasis of interleukin 1 and TNFalpha in patients with rheumatoid arthritis. Ann Rheum Dis. 2001;60:660–669. doi: 10.1136/ard.60.7.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, Wagner C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 237.Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D, Panaccione R, Wolf D, Pollack P. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: the CLASSIC-I trial. Gastroenterology. 2006;130:323–33; quiz 591. doi: 10.1053/j.gastro.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 238.Agnholt J, Kelsen J, Brandsborg B, Jakobsen NO, Dahlerup JF. Increased production of granulocyte-macrophage colony-stimulating factor in Crohn’s disease--a possible target for infliximab treatment. Eur J Gastroenterol Hepatol. 2004;16:649–655. doi: 10.1097/01.meg.0000108344.41221.8b. [DOI] [PubMed] [Google Scholar]
- 239.Van Deventer SJ. Tumour necrosis factor and Crohn’s disease. Gut. 1997;40:443–448. doi: 10.1136/gut.40.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ricciardelli I, Lindley KJ, Londei M, Quaratino S. Anti tumour necrosis-alpha therapy increases the number of FOXP3 regulatory T cells in children affected by Crohn’s disease. Immunology. 2008;125:178–183. doi: 10.1111/j.1365-2567.2008.02839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut. 2002;50:206–211. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Di Sabatino A, Ciccocioppo R, Cinque B, Millimaggi D, Morera R, Ricevuti L, Cifone MG, Corazza GR. Defective mucosal T cell death is sustainably reverted by infliximab in a caspase dependent pathway in Crohn’s disease. Gut. 2004;53:70–77. doi: 10.1136/gut.53.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Van den Brande JM, Koehler TC, Zelinkova Z, Bennink RJ, te Velde AA, ten Cate FJ, van Deventer SJ, Peppelenbosch MP, Hommes DW. Prediction of antitumour necrosis factor clinical efficacy by real-time visualisation of apoptosis in patients with Crohn’s disease. Gut. 2007;56:509–517. doi: 10.1136/gut.2006.105379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shen C, Maerten P, Geboes K, Van Assche G, Rutgeerts P, Ceuppens JL. Infliximab induces apoptosis of monocytes and T lymphocytes in a human-mouse chimeric model. Clin Immunol. 2005;115:250–259. doi: 10.1016/j.clim.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 245.Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, To K, Harashima S, Hatta N, Harada M. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008;58:1248–1257. doi: 10.1002/art.23447. [DOI] [PubMed] [Google Scholar]
- 246.Waetzig GH, Seegert D, Rosenstiel P, Nikolaus S, Schreiber S. p38 mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–5351. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 247.Kirchner S, Holler E, Haffner S, Andreesen R, Eissner G. Effect of different tumor necrosis factor (TNF) reactive agents on reverse signaling of membrane integrated TNF in monocytes. Cytokine. 2004;28:67–74. doi: 10.1016/j.cyto.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 248.Rosenstiel P, Agnholt J, Kelsen J, Medici V, Waetzig GH, Seegert D, Schreiber S. Differential modulation of p38 mitogen activated protein kinase and STAT3 signalling pathways by infliximab and etanercept in intestinal T cells from patients with Crohn’s disease. Gut. 2005;54:314–315; author reply 316-316. [PMC free article] [PubMed] [Google Scholar]
- 249.Martínez-Borra J, López-Larrea C, González S, Fuentes D, Dieguez A, Deschamps EM, Pérez-Pariente JM, López-Vázquez A, de Francisco R, Rodrigo L. High serum tumor necrosis factor-alpha levels are associated with lack of response to infliximab in fistulizing Crohn’s disease. Am J Gastroenterol. 2002;97:2350–2356. doi: 10.1111/j.1572-0241.2002.05990.x. [DOI] [PubMed] [Google Scholar]
- 250.Marti HP, McNeil L, Thomas G, Davies M, Lovett DH. Molecular characterization of a low-molecular-mass matrix metalloproteinase secreted by glomerular mesangial cells as PUMP-1. Biochem J. 1992;285(Pt 3):899–905. doi: 10.1042/bj2850899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Mäkitalo L, Kolho KL, Karikoski R, Anthoni H, Saarialho-Kere U. Expression profiles of matrix metalloproteinases and their inhibitors in colonic inflammation related to pediatric inflammatory bowel disease. Scand J Gastroenterol. 2010;45:862–871. doi: 10.3109/00365520903583863. [DOI] [PubMed] [Google Scholar]
- 252.Derer S, Waetzig GH, Seegert D, Nikolaus S, Schreiber S, Rosenstiel P. A possible link between TIMP-1 induction and response to infliximab. Gut. 2009;58:888; author reply 888–889. [PubMed] [Google Scholar]
- 253.Atreya R, Neurath MF. Novel imaging modalities for immune cell monitoring in the intestine. Curr Opin Gastroenterol. 2014;30:553–558. doi: 10.1097/MOG.0000000000000120. [DOI] [PubMed] [Google Scholar]
- 254.Atreya R, Zimmer M, Bartsch B, Waldner MJ, Atreya I, Neumann H, Hildner K, Hoffman A, Kiesslich R, Rink AD, et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14⁺ macrophages. Gastroenterology. 2011;141:2026–2038. doi: 10.1053/j.gastro.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 255.Siegel CA, Melmed GY. Predicting response to Anti-TNF Agents for the treatment of crohn’s disease. Therap Adv Gastroenterol. 2009;2:245–251. doi: 10.1177/1756283X09336364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Arnott ID, Watts D, Satsangi J. Azathioprine and anti-TNF alpha therapies in Crohn’s disease: a review of pharmacology, clinical efficacy and safety. Pharmacol Res. 2003;47:1–10. doi: 10.1016/s1043-6618(02)00264-5. [DOI] [PubMed] [Google Scholar]
- 257.Hlavaty T, Pierik M, Henckaerts L, Ferrante M, Joossens S, van Schuerbeek N, Noman M, Rutgeerts P, Vermeire S. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn’s disease. Aliment Pharmacol Ther. 2005;22:613–626. doi: 10.1111/j.1365-2036.2005.02635.x. [DOI] [PubMed] [Google Scholar]
- 258.Arijs I, Quintens R, Van Lommel L, Van Steen K, De Hertogh G, Lemaire K, Schraenen A, Perrier C, Van Assche G, Vermeire S, et al. Predictive value of epithelial gene expression profiles for response to infliximab in Crohn’s disease. Inflamm Bowel Dis. 2010;16:2090–2098. doi: 10.1002/ibd.21301. [DOI] [PubMed] [Google Scholar]
- 259.Rismo R, Olsen T, Cui G, Christiansen I, Florholmen J, Goll R. Mucosal cytokine gene expression profiles as biomarkers of response to infliximab in ulcerative colitis. Scand J Gastroenterol. 2012;47:538–547. doi: 10.3109/00365521.2012.667146. [DOI] [PubMed] [Google Scholar]
- 260.Gedebjerg A, Johansen C, Kragballe K, Iversen L. IL-20, IL-21 and p40: potential biomarkers of treatment response for ustekinumab. Acta Derm Venereol. 2013;93:150–155. doi: 10.2340/00015555-1440. [DOI] [PubMed] [Google Scholar]