

Abstract
Glycosyl triflates, which serve as important intermediates in glycosylation reactions, were generated and accumulated by the low-temperature electrochemical oxidation of thioglycosides such as thioglucosides, thiogalactosides, and thiomannosides in the presence of tetrabutylammonium triflate (Bu4NOTf) as a supporting electrolyte. Thus-obtained solutions of glycosyl triflates (glycosyl triflate pools) were characterized by low-temperature NMR measurements. The thermal stability of glycosyl triflates and their reactions with glycosyl acceptors were also examined.
Introduction
Among the many glycosyl cation equivalents, glycosyl triflates have been recognized as important intermediates in modern chemical glycosylation methodologies.1 Kronzer and Schuerch developed the silver triflate-assisted methanolysis of glycosyl chloride and proposed the existence of glycosyl triflate intermediates.2 Although related intermediates such as glycosyl toluenesulfonates were isolated and characterized in their subsequent studies,3 glycosyl triflates have been successfully characterized only recently using low-temperature NMR mesurements.1a,4 Since then several methods for iterative glycosylation have been developed based on the pre-generation of glycosyl triflates or related intermediates before the addition of glycosyl acceptors.5 These glycosyl triflates usually have been generated from thioglycosides, which are the glycosyl donors of choice, because of their stability under atmospheric conditions and a variety reaction conditions. However, this very stability on occasion leads to difficulties in the activation process. Indeed, strong thiophilic reagents are required for the generation of glycosyl triflates from thioglycosides resulting in the formation of activator-based byproducts, which can influence the stability and reactivity of glycosyl triflates. Therefore, a new straightforward method for the generation of glycosyl triflates is highly desired.
The electrochemical reactions serve as powerful methods for activation of organic compounds without using highly reactive chemical reagents.6 We have developed the “cation pool” method that involves the irreversible electrochemical oxidative generation and accumulation of highly reactive carbocations in the absence of nucleophiles.7 Successful generation and accumulation of alkoxy carbenium ions from α-silyl ethers encouraged us to generate and accumulate the glycosyl cations as “glycosyl cation pools” (Scheme 1). Unfortunately, the Bu4NBF4/CH2Cl2 system that is the standard supporting electrolyte/solvent system of the “cation pool” method gave fluoro glycosides from thioglycosides.8, 9 By using tetrabutylammonium perchlorate (Bu4NClO4) as a supporting electrolyte, however, we could accumulate glycosyl cation equivalents that gave glycosylation products after the addition of nucleophiles (Scheme 2).8 Although the formation of glycosyl perchlorates was most likely, it proved difficult to determine the electrochemically generated species by NMR. We envisioned that the glycosyl triflates, which serve as important intermediates in glycosylation reactions, could be generated and accumulated by the electrochemical method using tetrabutylammonium triflate (Bu4NOTf) as a supporting electrolyte. This idea has now been reduced to practice and we report herein the successful generation and accumulation of glycosyl triflates, and their characterization by the low-temperature NMR analysis. The thermal stability of glycosyl triflates and their reactions with glycosyl acceptors are also described.
Scheme 1.

Scheme 2.

Results and Discussion
Oxidation Potentials of Thioglycosides
We initiated our study by determining the oxidation potentials of thioglycosides by rotating disk electrode voltammetry using Bu4NOTf as a supporting electrolyte in CH2Cl2 (Figure 1). All these thioglycosides 1–3 have sufficiently low oxidation potentials for preparative electrochemical oxidation in the Bu4NOTf/CH2Cl2 system. It should be noted that the 4,6-O-benzylidene protecting group, which has been indispensable for the β-selective mannosylation, was oxidized and cleaved during the electrochemical oxidation. However, the use of the 4,6-O-p-chlorobenzylidene protecting group was effective for the selective oxidation of thiomannosides.
Figure 1.

Oxidation potentials of thioglucosides.
Coupling of Electrochemically Generated Glycosyl Triflates with Methanol
With data for oxidation potentials in hand, we examined the constant current electrochemical oxidation of thioglycosides 1–3 in the absence of an acceptor in CH2Cl2 at −78 °C, with Bu4NOTf (6.0 equiv) intended to serve as both the supporting electrolyte and the source of a triflate anion. The use of a smaller amount of Bu4NOTf resulted in higher cell voltage (>50 V). An H-type divided cell equipped with a carbon felt anode, a platinum plate cathode, and a glass filter separator were used, and the electrolysis was conducted under the conditions of constant current (4.0 mA, 1.5–2.0 F/mol of electricity). In the next step, the thus-generated intermediate was allowed to react with methanol (Figure 2). Under these conditions thioglycoside 1 afforded the corresponding methyl glucoside 4 in 84% yield (α/β = 6/94). Di(p-tolyl)disulfide was obtained as a byproduct (55%), which is inactive to thioglycosides.
Figure 2.

Electrochemical oxidation of thioglucosides followed by the reactions with methanol. a The reaction was carried out with 3.0 equiv. of Bu4NOTf.
Chemical generation of the glycosyl triflate from 1 was also examined. Treatment of 1 with trifluoromethanesulfonic acid in the presence of diphenylsulfoxide (DPSO) and tri-t- butylpyrimidine (TTPB) in CH2Cl2 at −78 °C for 15 min followed by the reaction of thus generated species with methanol at the same temperature for 1 h gave 4 in 87% yield (α/β = 36/64).10 It is important to note that α/β selectivity of the chemical method is lower than that of the electrochemical method, although the yield is comparable. Another advantage of the electrochemical method is no use of TTPB, which is rather difficult to remove by simple operation, whereas the supporting electrolyte (Bu4NOTf) can be easily removed by filtration through a short silica-gel column. Separation of di(p-tolyl)disulfide formed by the electrochemical method is also easy. Therefore, higher α/β selectivity and purification ease are benefits of the present electrochemical protocol from a synthetic point of view.
Thiogalactoside 2 also gave the corresponding β-methyl galactoside 5 with moderate selectively. On the other hand, the reaction of thiomannoside 3a afforded a ca. 1:1 mixture of the α- and β-methyl mannoside 6a. It is interesting to note that both yield and β-selectivity of the mannosylation reaction were improved by the introduction of p-chloro-4,6-O-benzilidene protecting group. Moreover, the use of a smaller amount of Bu4NOTf (3.0 equiv) led to better β selectivity in the case of the mannosides. These selectivities fit the pattern previously established for glycosylation reactions conducted with chemically generated glycosyl triflates.4 If the α-glycosyl triflate is viewed as a reservoir for a transient β-selective contact ion pair and an associated α-selective solvent separated ion pair, the decrease in β-selectivity seen with the benzylidene-protected mannosyl donor with increased concentration of Bu4NOTf must be due to the corresponding increase in solvent polarity which supports a greater concentration of the solvent separated ion pair.4k Alternatively, the possibility that increased triflate concentration promotes in situ anomerization and the intermediacy of a low concentration of a more reactive α-selective β-glycosyl triflate and associated contact ion pair can not be excluded.11 The generally increased β-selectivity seen with the benzylidene protected mannosyl triflate derived from 3b over that seen with the corresponding perbenzyl system 3a is due to the locking of the C5-C6 bond in its most electron-withdrawing trans-gauche conformation, which limits the concentration of the solvent separated ion pair.12
Electrochemical Generation of a Glucosyl Triflate Pool
In order to confirm the generation of glycosyl triflates, the anodic solution was characterized by low-temperature NMR spectroscopy. For example, the anodic solution obtained by the low temperature electrochemical oxidation of thioglucoside 1 in CD2Cl2 at −78 °C exhibited a single set of signals in its 1H NMR spectrum (Figure 3). Together with the absence of the signals from the starting thioglucoside 1, this indicates complete conversion and the formation of a single new species. A signal at δ 6.10 (doublet, 3JH1H2 = 2.9 Hz) was assigned to the anomeric proton. No singnal around 9.5 ppm due to the proton adjacent to the cationic carbon was not observed.7b Therefore, the observed species is covalent rather than ionic. The coupling constant points to the glucosyl triflate 7 being the α-anomer.13 A single set of signals were also observed by 13C NMR spectroscopy, with the resonance at δ 106.9 assigned to the anomeric carbon, as confirmed by the presence of a cross peak with the 1H signal at δ 6.10 in the 13C-1H HMQC spectrum. Based on these observations, it is reasonable to consider that an α-glucosyl triflate 7 was generated and accumulated under the conditions of the low temperature electrochemical oxidation.
Figure 3.

1H NMR spectrum of glucosyl triflates 7.
The thermal stability of glucosyl triflate 7 was investigated by the method we reported previously (Figure 4).7b A solution of glucosyl triflate 7, which was produced by the electrochemical oxidation of thioglycoside 1 at –78 °C, was allowed to warm to the second temperature. After equilibration for 30 min, the resulting solution was recooled to –78 °C and then allowed to react with methanol for 30 min. From Figure 4 yields of methyl glucoside 4 changed around 10% at temperatures between −78 °C to −50 °C. Thus, we assume that glucosyl triflate 7 is stable at temperatures lower than –50 °C.
Figure 4.

Thermal stability of glucosyl triflates 7.
Electrochemical Generation of a Galactosyl Triflate Pool
Successful characterization of glucosyl triflate 7 encouraged us to confirm the generation of other glycosyl triflates by low-temperature NMR spectroscopy. The anodic solution obtained by the low temperature electrochemical oxidation of thiogalactoside 2 in CD2Cl2 at −78 °C exhibited a single set of signals in its 1H NMR spectrum (Figure 5). Together with the absence of the signals from the starting thiogalactoside 2, this indicates complete conversion and the formation of a single new species. A signal at δ 6.14 (doublet, 3JH1H2 = 3.4 Hz) was assigned to the anomeric proton. The chemical shift indicated that the observed species is covalent rather than ionic. The coupling constant points to the galactosyl triflate 8 being the α-anomer. A single set of signals were also observed by 13C NMR spectroscopy, with the resonance at δ 108.2 assigned to the anomeric carbon, as confirmed by the presence of a cross peak with the 1H signal at δ 6.14 in the 13C-1H HMQC spectrum. Based on these observations, it is reasonable to consider that an α-galactosyl triflate 8 was also generated and accumulated under the conditions of the low temperature electrochemical oxidation.
Figure 5.

1H NMR spectrum of galactosyl triflates 8.
The thermal stability of galactosyl triflate 8 was also investigated by the same method as glucosyl triflate 7 (Figure 6). A solution of galactosyl triflate 8, which was produced by the electrochemical oxidation of thiogalactoside 2 at –78 °C, was allowed to warm to a second temperature. After equilibration for 30 min, the resulting solution was recooled to –78 °C and then allowed to react with methanol for 30 min. It is evident that galactosyl triflate 8 is stable at temperatures lower than –60 °C. This result indicates that the thermal stability of galactosyl triflate 8 is almost the same as that of glucosyl triflate 7.
Figure 6.

Thermal stability of galactosyl triflates 8.
Electrochemical Generation of a Mannosyl Triflate Pool
Mannosyl triflate 9, generated from the corresponding thiomannoside 3b, was also characterized by NMR spectroscopy (Figure 7). A signal at δ 6.04 (singlet) was assigned to the anomeric proton. Again, the chemical shift indicated that the observed species is covalent rather than ionic. A single set of signals were also observed by 13C NMR spectroscopy, with the resonance at δ 105.3 assigned to the anomeric carbon, as confirmed by the presence of a cross peak with the 1H signal at δ 6.04 in the 13C-1H HMQC spectrum. Additionally, the 1H-NMR spectrum of triflate 9 generated by the electrochemical method matched that obtained from thioglycoside 3b by the more conventional activation with diphenyl sulfoxide and trifluoromethanesulfonic anhydride (see supporting information).10 In the glucose and galactose series, we assume that the β-selective glycosylations with methanol stem from the α-configuration of the intermediate glycosyl triflates. The reason for the lower β-selectivity observed in the reaction of mannosyl triflate 9, which also has the α-configuration, remains to be clarified.
Figure 7.

1H NMR spectrum of mannosyl triflate 9.
The thermal stability of mannosyl triflate 9 was also investigated (Figure 8). A solution of mannosyl triflate 9, which was produced by the electrochemical oxidation of thiomannoside 3b at –78 °C, was allowed to warm to a second temperature. After equilibration for 30 min, the resulting solution was recooled to – 78 °C and then allowed to react with methanol for 30 min. Although it is difficult to explain that yields were same at −40 °C to −78 °C, mannosyl triflate 9 gave the methyl mannoside in moderate yield even at −40 °C.14 This result indicates the higher thermal stability of mannosyl triflate 9. The diminished donation of electrons into the σ* orbital of the C-OTf bond by a carbon-oxygen bond relative to a carbon-hydrogen bond seems to be responsible for the enhanced stability of the mannosyl triflate.
Figure 8.

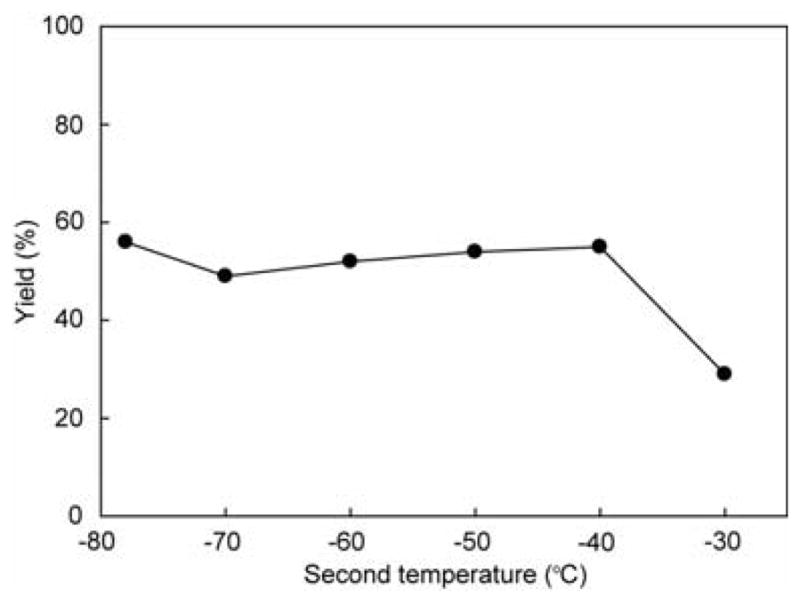
Thermal stability of mannosyl triflates 9.
Reactions of Glycosyl Triflates with Carbohydrate Acceptors
To examine reactivity of electrochemically generated glycosyl triflate pools, couplings to carbohydrate acceptors were performed (Figure 9). Reactions of triflates 7, 8, and 9 with methyl 2,3,4-tri-O-benzyl-α-D-glucoside gave the disaccharides 11, 12, and 13a, respectively, in moderate yields and moderate to high β-selectivity. It is interesting to note that the reaction of 9 with methyl 2,3-O-isopropylidene-α-L-rhamnopyranoside as a glycosyl acceptor gave only the β-isomer of the disaccharide 13b. The bulkiness of the acceptor may be responsible for the excellent stereoselectivity. In this method, it is advantageous that thioglycosides having a free hydroxyl group can be used as acceptors. For example, the electrochemical oxidation of thioglycosides 1 and 2 followed by reaction with a thioglycoside acceptor afforded disaccharides 14 and 15, respectively, which can be used as donors in further glycosylation reactions. The ability to work with thioglycoside bearing acceptors in this manner is significant. First, it indicates that, unlike the chemical systems where an additional thiophile such as a trialkyl phosphite must be added in such instances,13a the byproduct of the electrochemical activation process i.e. diaryldisulfide is not itself an oxidant for thioglycosides. Second, it opens up the possibility of sequential glycosylation sequences without the need for intermediate functional group manipulations.1b
Figure 9.

Preparation of disaccharides. a Reactions were carried out with 1.5 equiv. of Bu4NOTf.
Conclusions
In conclusion, we have developed a new and simple method for access to glycosyl triflates intermediates using electrochemical oxidation. Solutions of triflates generated by this method are free from by-products derived due to activator, which are inevitably present when chemical methods are employed. This feature should be advantageous not only from the viewpoint of mechanistic studies but also in the synthesis of polysaccharides. Further mechanistic studies and applications to iterative glycosylation are in progress.
Experimental Section
General Procedures
1H and 13C NMR spectra were recorded on Varian MERCURYplus-400 (1H 400 MHz, 13C 100 MHz). Low-temperature 1H, 13C NMR, and 13C-1H HMQC spectra were recorded on JEOL ECA-600P (1H 600 MHz, 13C 150 MHz). EI and CI mass spectra were recorded on JEOL JMS-SX102A spectrometer and FAB mass spectra were recorded on JEOL JMS-HX110A spectrometer. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. Dichloromethane was washed with water, distilled from P2O5, redistilled from dried K2CO3 to remove a trace amount of acid, and stored over molecular sieves 4A. Rotating-disk electrode voltammetry was carried out using BAS 600BS analyzer and BAS RDE-2 rotating disk electrode. Measurements were carried out in 0.1 M Bu4NOTf/CH2Cl2 using a glassy carbon disk working electrode, a platinum wire counter electrode, and an SCE reference electrode with sweep rate of 10 mV/s at 3000 r.p.m.
Low-Temperature NMR Analysis of Electrochemically Generated Glycosyl Triflates
The anodic oxidation was carried out in an H-type divided cell (4G glass filter) equipped with a carbon felt anode (Nippon Carbon JF-20-P7, ca. 160 mg, dried at 250 °C/1 mmHg for 2.5 h before use) and a platinum plate cathode (10 mm x 10 mm). In the anodic chamber were placed 1 (64.6 mg, 0.10 mmol) and 0.1 M Bu4NOTf in CD2Cl2 (5.0 ml). In the cathodic chamber were placed trifluoromethanesulfonic acid (22 μl, 0.25 mmol) and 0.1 M Bu4NOTf in CD2Cl2 (5.0 mL). The constant current electrolysis (4.0 mA) was carried out at −78 °C with magnetic stirring. After 1.25 F/mol of electricity was consumed, an aliquot of the anodic solution was transferred to a 5 mm ϕ NMR tube with a septum cap under Ar atmosphere at −78 °C. The NMR measurement was carried out at −80 °C. Chemical shifts were reported using signals of CH2Cl2 at 5.32 ppm (1H NMR) and CD2Cl2 at 53.8 ppm (13C NMR) as standards.
Triflyl 2,3,4,6-tetra-O-benzyl-α-D-glucopyranoside (7)
Selected data for 7 (6.5 – 3.5 ppm for 1H NMR and 110 – 60 ppm for 13C NMR). 1H NMR (CD2Cl2, 600 MHz) δ 6.10 (d, J = 2.8 Hz, 1 H, anomeric H), 4.90 (d, J = 10.3 Hz, 1 H), 4.80 (d, J = 9.6 Hz, 1 H), 4.79 (d, J = 9.7 Hz, 1 H), 4.72 (d, J = 11.6 Hz, 1 H), 4.69 (d, J = 12.4 Hz, 1 H), 4.49 (d, J = 11.0 Hz, 1 H), 4.42 (d, J = 10.3 Hz, 1 H), 4.36 (d, J = 11.0 Hz, 1 H), 3.90 – 3.86 (m, 2 H), 3.81 (d, J = 9.6 Hz, 1 H), 3.77 (dm, J = 8.9 Hz, 1 H), 3.71 (dd, J = 9.6, 2.8 Hz, 1 H), 3.62 (d, J = 9.6 Hz, 1 H). 13C NMR (CD2Cl2, 150 MHz) δ 106.9 (C1), 79.7, 76.4, 75.5, 75.0, 74.4, 73.8, 73.0, 72.5, 66.1.
Triflyl 2,3,4,6-tetra-O-benzyl-α-D-galactopyranoside (8)
The anodic oxidation of 2 (64.6 mg, 0.10 mmol) afforded 8. Selected data for 8. (6.5 – 3.4 ppm for 1H NMR and 110 – 60 ppm for 13C NMR). 1H NMR (CD2Cl2, 600 MHz) δ 6.14 (d, J = 3.4 Hz, 1 H, anomeric H), 4.86 (d, J = 10.3 Hz, 1 H), 4.78 – 4.72 (m, 4 H), 4.47 (d, J = 11.7 Hz, 1 H), 4.44 (d, J = 10.3 Hz, 1 H), 4.38 (d, J = 11.7 Hz, 1 H), 4.15 – 4.11 (m, 3 H), 3.90 (dd, J = 10.3, 2.0 Hz, 1 H), 3.49 (d, J = 6.8 Hz, 2 H). 13C NMR (CD2Cl2, 150 MHz) δ 108.2 (C1), 77.1, 74.8, 73.4, 72.94, 72.89, 72.7, 72.1, 71.8, 66.6.
Triflyl 2,3-di-O-benzyl-4,6-O-p-chlorobenzylidene-α-D-mannopyranoside (9)
The anodic oxidation of 3b (125 mg, 0.21 mmol) afforded 9. Selected data for 9 (6.5 – 3.4 ppm for 1H NMR and 110 – 60 ppm for 13C NMR). 1H NMR (CD2Cl2, 600 MHz) δ 6.04 (s, 1 H, anomeric H), 5.59 (s, 1 H), 4.74 (d, J = 11.7 Hz, 1 H), 4.73 (d, J = 11.0 Hz, 1 H), 4.64 (d, J = 11.0 Hz, 1 H), 4.60 (d, J = 11.7 Hz, 1 H), 4.25 – 4.21 (m, 2 H), 4.00 – 3.96 (m, 2 H), 3.92 (td, J = 10.3, 4.8 Hz, 1 H), 3.82 (sm, 4 H). 13C NMR (CD2Cl2, 150 MHz) δ 105.3 (C1), 100.2, 76.2, 74.14, 74.11, 73.9, 72.6, 66.9, 66.7.
Reaction of Electrochemically Generated Glycosyl Triflates. Typical procedure
The anodic oxidation was carried out in an H-type divided cell (4G glass filter) equipped with a carbon felt anode (Nippon Carbon JF-20-P7, ca. 160 mg, dried at 250 °C/1 mmHg for 2.5 h before use) and a platinum plate cathode (10 mm x 20 mm). In the anodic chamber was placed a solution of tolyl thioglucoside 1 (64.2 mg, 0.10 mmol) in 0.1 M Bu4NOTf/CH2Cl2 (5 mL). In the cathodic chamber was placed a solution of TfOH (22 μl, 0.25 mmol) in 0.1 M Bu4NOTf/CH2Cl2 (5 mL). The constant current electrolysis (4 mA) was carried out at −78 °C with magnetic stirring until 1.5 F/mol of electricity was consumed. Methyl 2,3,4-tri-O-benzyl-α-D-glucopyranoside (93 mg, 0.2 mmol, 0.2 M CH2Cl2 solution) was added to the solution in the anodic chamber. After additional stirring (−78 °C, 1 h), Et3N (70 μl, 0.5 mmol) was added and the mixture was warmed to room temperature. After filtering through a short column (2 x 3 cm) of silica gel to remove Bu4NOTf, and evaporation of the solvent under reduced pressure, the crude product was purified with preparative gel permeation chromatography (eluent: CHCl3) to afford a mixture of methyl glucosides 11α and 11β in 68% yield (66.3 mg, 0.067 mmol, 11α/11β = 12/88). Selected data of major product:
Methyl 2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl-(1→6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (11β).15
1H NMR (400 MHz) δ 7.34 – 7.14 (m, 35 H), 4.96 (d, J = 11.2 Hz, 1 H), 4.95 (d, J = 10.4 Hz, 1 H), 4.89 (d, J = 10.8 Hz, 1 H), 4.79 (d, J = 9.6 Hz, 1 H), 4.77 (d, J = 11.2 Hz, 2 H), 4.73 (d, J = 11.2 Hz, 1 H), 4.70 (d, J = 10.8 Hz, 1 H), 4.64 (d, J = 12.0 Hz, 1 H), 4.59 (d, J = 3.6 Hz, 1 H), 4.55 (d, J = 11.6 Hz, 2 H), 4.52 (d, J = 10.0 Hz, 1 H), 4.50 (d, J = 10.0 Hz, 1 H), 4.33 (d, J = 7.6 Hz, 1 H), 4.17 (dd, J = 10.8, 2.0 Hz, 1 H), 3.98 (t, J = 8.8 Hz, 1 H), 3.84 – 3.80 (m, 1 H), 3.71 (dd, J = 10.8, 2.0 Hz, 1 H), 3.66 (d, J = 10.8 Hz, 1 H), 3.65 (d, J = 10.8 Hz, 1 H), 3.61 (d, J = 8.8 Hz, 1 H), 3.57 (d, J = 9.2 Hz, 1 H), 3.53 – 3.49 (m, 3 H), 3.46 (d, J = 8.8 Hz, 1 H), 3.42 (ddd, J = 9.6, 4.4, 2.0 Hz, 1 H), 3.32 (s, 3 H). 13C NMR (100 MHz) δ 138.7, 138.4, 138.2, 138.2, 138.1, 138.0, 137.9, 128.3, 128.2, 128.2, 128.2, 128.0, 127.9, 127.8, 127.8, 127.8, 127.7, 127.7, 127.6, 127.6, 127.5, 127.5, 127.4, 127.4, 127.4, 103.7, 98.0, 84.8, 82.1, 82.0, 79.8, 78.0, 77.9, 75.7, 75.7, 75.0, 75.0, 74.9, 73.4, 73.3, 69.9, 69.0, 68.6, 55.2.
Methyl 2,3-di-O-benzyl-4,6-O-p-chlorobenzyliden-α-D-mannopyranosyl-(1→6)-2,3,4-tri-O-benzyl-α-D-gluco-pyranoside (13aα)
Glycosylation of 3b (176 mg, 0.30 mmol) with methyl 2,3,4-tri-O-benzyl-α-D-glucopyranoside (284 mg, 0.61 mmol) afforded 13aα (138 mg, 0.15 mmol) and 13aβ (12 mg, 0.013 mmol) in 54% yield (13aα/13bβ = 8/92). TLC (hexane / ethyl acetate 5:2): Rf 0.30 (13aα), 0.25 (13aβ). 1H NMR (CDCl3, 400 MHz) δ 7.39 – 7.18 (m, 29 H), 5.57 (s, 1 H), 4.99 (d, J = 10.8 Hz, 1 H), 4.88 – 4.85 (m, 2 H), 4.82 – 4.77 (m, 3 H), 4.74 – 4.61 (m, 4 H), 4.55 (d, J = 3.6 Hz, 1 H), 4.49 (d, J = 10.8 Hz, 1 H), 4.21 (t, J = 9.6 Hz,1 H), 4.14 (dd, J = 9.6, 3.6 Hz, 1 H), 3.97 (t, J = 9.2 Hz, 1 H), 3.88 (dd, J = 9.6, 2.8 Hz, 1 H), 3.84 – 3.75 (m, 4 H), 3.69 – 3.66 (m, 1 H), 3.60 (d, J = 11.2 Hz, 1 H), 3.45 (dd, J = 9.6, 3.6 Hz, 1 H), 3.38 – 3.32 (m, 1 H), 3.29 (s, 3 H). 13C NMR (CDCl3, 150 MHz) δ 138.5, 138.2, 138.0, 137.9, 136.1, 134.5, 128.4, 128.3, 128.21, 128.18, 127.88, 127.85, 127.81, 127.78, 127.66, 127.56, 127.4, 100.7, 99.6, 97.8, 82.0, 80.0, 79.1, 77.6, 76.3, 75.8, 75.7, 75.0, 73.5, 73.3, 72.9, 69.8, 68.8, 66.2, 64.3, 55.1. HRMS (FAB) m/z calcd for C55H57ClO11 [M-H]+: 927.3511, found: 927.3527.
Methyl 2,3-di-O-benzyl-4,6-O-p-chloro-benzyliden-β-D-manno-pyranosyl-(1→6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (13aβ)
1H NMR (CDCl3, 400 MHz) δ 7.43 – 7.13 (m, 29 H), 5.53 (s, 1 H), 5.15 (d, J = 10.8 Hz, 1 H), 4.90 (d, J = 12.0 Hz, 1 H), 4.84 – 4.75 (m, 5 H), 4.66 (d, J = 11.6 Hz, 2 H), 4.57 (d, J = 11.6 Hz, 2 H), 4.49 (d, J = 11.6 Hz, 1 H), 4.23 (dd, J = 10.4, 4.8 Hz, 1 H), 4.15 (t, J = 9.6 Hz, 1 H), 4.07 – 4.05 (m, 1 H), 4.01 (t, J = 9.6 Hz, 1 H), 3.88 (t, J = 10.0 Hz, 1 H), 3.84 – 3.68 (m, 2 H), 3.51 – 3.41 (m, 4 H), 3.32 (s, 3 H), 3.19 (td, J = 9.6, 4.8 Hz, 1 H). 13C NMR (CDCl3, 100 MHz) δ 138.6, 138.2, 138.1, 137.9, 136.0, 134.6, 128.6, 128.4, 128.33, 128.26, 128.22, 128.1, 128.02, 127.97, 127.92, 127.83, 127.79, 127.54, 127.48, 127.45, 127.39, 101.9, 100.6, 97.8, 82.2, 79.8, 78.6, 77.8, 75.7, 75.6, 74.7, 74.6, 73.3, 72.4, 69.6, 68.5, 68.3, 67.5, 55.1. HRMS (FAB) m/z calcd for C55H57ClO11 [M-H]+: 927.3511, found: 927.3527.
Methyl 2,3-di-O-benzyl-4,6-O-p-chlorobenzylidene-β-D-manno-pyranosyl-(1→4)-2,3-O-isopropylidene-α-L-rhamno-pyranoside (13b)
Glycosylation of 3b (217 mg, 0.37 mmol) with methyl 2,3-O-isopropylidene-α-L-rhaminoside (165 mg, 0.76 mmol) afforded 13b (173 mg, 0.25 mmol) as a sole product in 68% yield TLC (hexane / ethyl acetate 5:1): Rf 0.30. 1H NMR (CDCl3, 400 MHz) δ 7.46 – 7.41 (m, 4 H), 7.36 – 7.26 (m, 10 H), 5.58 (s, 1 H), 5.00 (s, 1 H), 4.92 (d, J = 12.0 Hz, 1 H), 4.87 (s, 1 H), 4.81 (d J = 12.4 Hz, 1 H), 4.63 (d, J = 12.8 Hz, 1 H), 4.59 (d, J = 12.4 Hz, 1 H), 4.26 (dd, J = 10.4, 4.8 Hz, 1 H), 4.19 (t, J = 9.6 Hz, 1 H), 4.14 – 4.09 (m, 2 H), 3.98 – 3.92 (m, 2 H), 3.71 – 3.61 (m, 3 H), 3.41 (s, 3 H), 3.37 – 3.27 (m, 1 H), 1.51 (s, 3 H), 1.34 (s, 6 H). 13C NMR (CDCl3, 100 MHz) δ 138.4, 138.1, 136.0, 134.5, 128.2, 128.0, 127.4, 127.4, 127.4, 127.3, 109.2, 100.6, 100.1, 97.8, 78.6, 78.4, 78.0, 77.8, 76.4, 76.1, 74.9, 72.1, 68.6, 67.6, 64.2, 54.9, 27.9, 26.5, 17.8. HRMS (FAB) m/z calcd for C37H43ClO10 [M-H]+: 681.2467, found: 681.2470.
Supplementary Material
Acknowledgments
This work was financially supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science. AAB thanks JSPS and NSF for financial supports for a study visit to Japan, and the NIH (GM62160) for the support of his work in Chicago. The authors thank Dr. Takeshi Yamada of Kyoto University for fruitful discussion. J.Y. and T. N. dedicate this paper to late Professor Yoshihiko Ito in honor of his outstanding contribution to synthetic chemistry.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data of compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.a) Crich D. J Carbohydr Chem. 2002;21:667. [Google Scholar]; b) Codée JDC, Litjens REJN, van den Bos LJ, Overkleeft HS, van der Marel GA. Chem Soc Rev. 2005;34:769. doi: 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]; c) Toshima K, Tatsuta K. Chem Rev. 1993;93:1503. [Google Scholar]
- 2.a) Kronzer FJ, Schuerch C. Carbohydr Res. 1973;27:379. [Google Scholar]; b) Lucas TJ, Schuerch C. Carbohydr Res. 1975;39:39. doi: 10.1016/s0008-6215(00)82649-x. [DOI] [PubMed] [Google Scholar]; c) Srivastava VK, Schuerch C. Carbohydr Res. 1980;79:C13. [Google Scholar]; d) Srivastava VK, Schuerch C. J Org Chem. 1981;46:1121. [Google Scholar]; e) Leroux J, Perlin AS. Carbohydr Res. 1976;47:C8. [Google Scholar]
- 3.Kronzer FJ, Schuerch C. Carbohydr Res. 1973;27:379. [Google Scholar]
- 4.a) Crich D, Patel M. Carbohydr Res. 2006;341:1467. doi: 10.1016/j.carres.2006.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Crich D, Banerjee A. J Am Chem Soc. 2006;128:8078. doi: 10.1021/ja061594u. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Crich D, Bowers AA. J Org Chem. 2006;71:3452. doi: 10.1021/jo0526688. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Crich D, Jayalath P, Hutton TK. J Org Chem. 2006;71:3064. doi: 10.1021/jo0526789. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Crich D, Jayalath P. Org Lett. 2005;7:2277. doi: 10.1021/ol050680g. [DOI] [PubMed] [Google Scholar]; f) Crich D, Banerjee A. Org Lett. 2005;7:1395. doi: 10.1021/ol050224s. [DOI] [PubMed] [Google Scholar]; g) Crich D, Vinod AU. J Org Chem. 2005;70:1291. doi: 10.1021/jo0482559. [DOI] [PubMed] [Google Scholar]; h) Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asymmetry. 2005;16:105. [Google Scholar]; i) Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]; j) Crich D, Banerjee A, Yao Q. J Am Chem Soc. 2004;126:14930. doi: 10.1021/ja047194t. [DOI] [PubMed] [Google Scholar]; k) Crich D, Chandrasekera NS. Angew Chem Int Ed. 2004;43:5386. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]; l) Crich D, Yao Q. J Am Chem Soc. 2004;126:8232. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]; m) Crich D, Vinod AU, Picione J. J Org Chem. 2003;68:8453. doi: 10.1021/jo035003j. [DOI] [PubMed] [Google Scholar]; n) Crich D, de la Mora M, Vinod AU. J Org Chem. 2003;68:8142. doi: 10.1021/jo0349882. [DOI] [PubMed] [Google Scholar]; o) Crich D, Yao Q. Org Lett. 2003;5:2189. doi: 10.1021/ol034741r. [DOI] [PubMed] [Google Scholar]; p) Crich D, Picione J. Org Lett. 2003;5:781. doi: 10.1021/ol0340890. [DOI] [PubMed] [Google Scholar]; q) Dudkin VY, Crich D. Tetrahedron Lett. 2003;44:1787. [Google Scholar]; r) Crich D, Smith M. J Am Chem Soc. 2002;124:8867. doi: 10.1021/ja011406u. [DOI] [PubMed] [Google Scholar]; s) Crich D, Li H. J Org Chem. 2002;67:4640. doi: 10.1021/jo0108818. [DOI] [PubMed] [Google Scholar]; t) Crich D, Dudkin V. J Am Chem Soc. 2002;124:2263. doi: 10.1021/ja0123958. [DOI] [PubMed] [Google Scholar]; u) Crich D, de la Mora M, Cruz R. Tetrahedron. 2002;58:35. [Google Scholar]; v) Crich D, Smith M. J Am Chem Soc. 2001;123:9015. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]; w) Crich D, Dudkin V. J Am Chem Soc. 2001;123:6819. doi: 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]; x) Crich D, Li H, Yao Q, Wink DJ, Sommer RD, Rheingold AL. J Am Chem Soc. 2001;123:5826. doi: 10.1021/ja015985e. [DOI] [PubMed] [Google Scholar]
- 5.a) Nguyen HM, Poole JL, Gin DY. Angew Chem Int Ed. 2001;40:414. doi: 10.1002/1521-3773(20010119)40:2<414::AID-ANIE414>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]; b) Nguyen HM, Chen Y, Duron SG, Gin DY. J Am Chem Soc. 2001;123:8766. doi: 10.1021/ja015968p. [DOI] [PubMed] [Google Scholar]; c) Yamago S, Yamada T, Maruyama T, Yoshida J. Angew Chem Int Ed. 2004;43:2145. doi: 10.1002/anie.200353552. [DOI] [PubMed] [Google Scholar]; d) Huang X, Huang L, Wang H, Ye X. Angew Chem Int Ed. 2004;43:5221. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]; e) Yamada T, Kinjyo S, Yoshida J, Yamago S. Chem Lett. 2005;34:1556. [Google Scholar]; f) Yamada T, Takemura K, Yoshida J, Yamago S. Angew Chem Int Ed. 2006;45:7575. doi: 10.1002/anie.200602699. [DOI] [PubMed] [Google Scholar]
- 6.a) Sainsbury M, editor. Rodd’s Chemisty of Carbon Compounds, Vol. V, Organic Electrochemistry. Elsevier Science B. V; Amsterdam: 2002. [Google Scholar]; b) Lund H, Hammerich O, editors. Organic Electrochemistry. 4. Marcel Dekker; New York: 2001. [Google Scholar]; c) Torii S, editor. Novel Trends in Electroorganic Synthesis. Springer-Verlag; Tokyo: 1998. [Google Scholar]
- 7.a) Yoshida J, Suga S, Suzuki S, Kinomura N, Yamamoto A, Fujiwara K. J Am Chem Soc. 1999;121:9546. [Google Scholar]; b) Suga S, Suzuki S, Yamamoto A, Yoshida J. J Am Chem Soc. 2000;122:10244. [Google Scholar]; c) Suga S, Okajima M, Yoshida J. Tetrahedron Lett. 2001;42:2173. [Google Scholar]; d) Suga S, Okajima M, Fujiwara K, Yoshida J. J Am Chem Soc. 2001;123:7941. doi: 10.1021/ja015823i. [DOI] [PubMed] [Google Scholar]; e) Suga S, Suzuki S, Yoshida J. J Am Chem Soc. 2002;124:30. doi: 10.1021/ja0171759. [DOI] [PubMed] [Google Scholar]; f) Yoshida J, Suga S. Chem Eur J. 2002;8:2650. [Google Scholar]; g) Suga S, Watanabe M, Yoshida J. J Am Chem Soc. 2002;124:14824. doi: 10.1021/ja028663z. [DOI] [PubMed] [Google Scholar]; h) Suga S, Nagaki A, Yoshida J. Chem Commun. 2003;354 doi: 10.1039/b211433j. [DOI] [PubMed] [Google Scholar]; i) Suga S, Nagaki A, Tsutsui Y, Yoshida J. Org Lett. 2003;5:945. doi: 10.1021/ol0341243. [DOI] [PubMed] [Google Scholar]; j) Suga S, Kageyama Y, Babu G, Itami K, Yoshida J. Org Lett. 2004;6:2709. doi: 10.1021/ol049049q. [DOI] [PubMed] [Google Scholar]; k) Suga S, Suzuki S, Maruyama T, Yoshida J. Bull Chem Soc Jpn. 2004;77:1545. [Google Scholar]; l) Suga S, Nishida T, Yamada D, Nagaki A, Yoshida J. J Am Chem Soc. 2004;126:14338. doi: 10.1021/ja0455704. [DOI] [PubMed] [Google Scholar]; m) Nagaki A, Kawamura K, Suga S, Ando T, Sawamoto M, Yoshida J. J Am Chem Soc. 2004;126:14702. doi: 10.1021/ja044879k. [DOI] [PubMed] [Google Scholar]; n) Okajima M, Suga S, Itami K, Yoshida J. J Am Chem Soc. 2005;127:6930. doi: 10.1021/ja050414y. [DOI] [PubMed] [Google Scholar]; o) Maruyama T, Suga S, Yoshida J. J Am Chem Soc. 2005;127:7324. doi: 10.1021/ja0511218. [DOI] [PubMed] [Google Scholar]; p) Nagaki A, Togai M, Suga S, Aoki N, Mae K, Yoshida J. J Am Chem Soc. 2005;127:11666. doi: 10.1021/ja0527424. [DOI] [PubMed] [Google Scholar]; q) Suga S, Matsumoto K, Ueoka K, Yoshida J. J Am Chem Soc. 2006;128:7710. doi: 10.1021/ja0625778. [DOI] [PubMed] [Google Scholar]; r) Maruyama T, Suga S, Yoshida J. Tetrahedron. 2006;62:6519. [Google Scholar]; s) Maruyama T, Mizuno Y, Shimizu I, Suga S, Yoshida J. J Am Chem Soc. 2007;129:1902. doi: 10.1021/ja068589a. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki S, Matsumoto K, Kawamura K, Suga S, Yoshida J. Org Lett. 2004;6:3755. doi: 10.1021/ol048524h. [DOI] [PubMed] [Google Scholar]
- 9.Electrochemical glycosylation: Noyori R, Kurimoto I. J Org Chem. 1986;51:4322.Amatore C, Jutand A, Mallet JM, Meyer G, Sinay P. J Chem Soc, Chem Commun. 1990:718.Balavoine G, Gref A, Fischer J-c, Lubineau A. Tetrahedron Lett. 1990;31:5761.Mallet JM, Meyer G, Yvelin F, Jutand A, Amatore C, Sinay P. Carbohydr Res. 1993;244:237. doi: 10.1016/0008-6215(83)85004-6.Amatore C, Jutand A, Meyer G, Bourhis P, Machetto F, Sinay P, Tabeur C, Zhang YM. J Appl Electrochem. 1994;24:725.Balavoine G, Berteina S, Gref A, Fischer J-c, Lubineau A. J Carbohydr Chem. 1995;14:1217.Balavoine G, Berteina S, Gref A, Fischer J-c, Lubineau A. J Carbohydr Chem. 1995;14:1237.Yamago S, Kokubo K, Yoshida J. Chem Lett. 1997:111.Nokami J, Osafune M, Ito Y, Miyake F, Sumida S, Torii S. Chem Lett. 1999:1053.Yamago S, Kokubo K, Hara O, Masuda S, Yoshida J. J Org Chem. 2002;67:8584. doi: 10.1021/jo0261350.France RR, Rees NV, Wadhawan JD, Fairbanks AJ, Compton RG. Org Biomol Chem. 2004;2:2188. doi: 10.1039/b316720h.France RR, Compton RG, Davis BG, Fairbanks AJ, Rees NV, Wadhawan JD. Org Biomol Chem. 2004;2:2188. doi: 10.1039/b316720h.Mitsudo K, Kawaguchi T, Miyahara S, Matsuda W, Kuroboshi M, Tanaka H. Org Lett. 2005;7:4649. doi: 10.1021/ol051776d.
- 10.a) Codée JDC, Litjens REJN, den Heeten R, Overkleeft HS, Van Boom JH, van der Marel GA. Org Lett. 2003;5:1519. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]; b) van den Bos LJ, Codée JDC, Van Boom JH, Overkleeft HS, van der Marel GA. Org Biomol Chem. 2003;1:4160. doi: 10.1039/b309823k. [DOI] [PubMed] [Google Scholar]; c) Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1947. [Google Scholar]; d) Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;40:1057. [Google Scholar]; e) van den Bos LJ, Codée JDC, van der Toorn JC, Boltje TJ, van Boom JH, Overkleeft HS, van der Marel GA. Org Lett. 2004;6:2165. doi: 10.1021/ol049380+. [DOI] [PubMed] [Google Scholar]; f) Litjens REJN, van den Bos LJ, Codée JDC, van den Berg RJBHN, Overkleeft HS, van der Marel GA. Eur J Org Chem. 2005;918 [Google Scholar]
- 11.Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056. [Google Scholar]
- 12.a) Jensen HH, Nordstrom M, Bols M. J Am Chem Soc. 2004;126:9205. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]; b) Litjens REJN, van den Bos LJ, Codée JDC, Overkleeft HS, van der Marel GA. Carbohydr Res. 2007;342:419. doi: 10.1016/j.carres.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Crich D, Sun S. J Am Chem Soc. 1997;119:11217. [Google Scholar]
- 14.Although we elongated the reaction time of mannosyl triflate 9 with methanol, yields were not improved.
- 15.Jona H, Mandai H, Chavasiri W, Takeuchi K, Mukaiyama T. Bull Chem Soc Jpn. 2002;75:291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.