

Abstract
Objective: To determine the molecular mechanism by which absent in melanoma 2 (AIM2) induces breast cancer cell apoptosis. Methods: Establish Tet-OffTM model system to induce AIM2 expression in great quantities, MCF-7 tTA-AIM2 cells were the experimental group; MCF-7 tTA-Luc cells were the control group. The expression and subcellular localization of AIM2 in breast cancer cell lines were determined via Western Blotting. AIM2 protein expression was determined after the addition of interferon-γ (102 U/ml). Flow cytometry was used to analyze the effects of AIM2 on the cell cycle. Apoptosis detection was performed by staining with Annexin V-FITC and propidium iodide. Apoptosis mechanism was detected via Western Blotting. The XTT assay was used to analyze the effects of AIM2 on cell growth. Result: This experiment established Tet-Off guidance system. This system results which promotes AIM2 gene transcription and increased AIM2 protein expression. Four days after induction, AIM2 expression was detected. AIM2 expression increased with the number of days post-induction. AIM2 is present in cytoplasm and nuclei. Interferon-γ (102 U/ml) induced AIM2 protein expression and significantly increased AIM2 expression. AIM2 expression had no significant effect on the cell cycle, With the increase of Cdk2 expression induced by days were gradually increased, and Cdk4, Cyclin E expression was no significantly difference. AIM2 expression can significantly promote the apoptosis of breast cancer cells. Increased AIM2 expression can inhibit the expression of the anti-apoptotic protein Bcl-xL, increase the expression of the apoptosis proteins Bad and Bax, and activate caspases, resulting in cleavage of the DNA repair protein PARP. The XTT assay showed that AIM2 expression slows the rate of cell growth. Conclusion: In this breast cancer Tet-OffTM system, AIM2 was expressed in the cytoplasm and nucleus, stimulated the mitochondria to promote apoptosis, and influenced cell survival and proliferation.
Keywords: Absent in melanoma 2, breast cancer cells, interferon-γ, cell cycle, cell apoptosis
Introduction
Breast cancer tends to affect young patients. Therefore, early treatment is one of the focuses in present study [1]. The role of interferon in cancer treatment has been important. Interferon can inhibit cancer cell growth by regulating the body’s immune system, and the anticancer capacity of interferon is primarily associated with interferon-induced proteins [2]. Human absent in melanoma 2 (AIM2) contains a 200-amino acid repeat domain (HIN-200). HIN-200 can inhibit the cell cycle and tumor growth [3].
This study investigated the suppressor role of AIM2 in breast cancer cells and the corresponding molecular mechanism. The Tet-OffTM model system was used to dramatically increase AIM2 expression. The distribution of AIM2 in breast cancer cells after induction was analyzed. The effects of AIM2 on the cell cycle, apoptosis, cell proliferation and other biological characteristics were also analyzed. We also investigated the AIM2 signaling pathway to further understand the mechanism of AIM2 in breast cancer.
Materials and methods
Antibodies and cell culture
Antibodies
The antibodies in this study included: cleaved caspase 7 (Asp198, anti-mouse), cleaved caspase 9 (Asp315, anti-mouse), Bcl-xL (H62, anti-mouse), beta-actin (C4, anti-mouse), PARP (F2, anti-mouse), HRP-conjugated goat anti-mouse IgG, Cdk2 (D12, anti-rabbit), Cdk4 (H22, anti-rabbit), cyclin E (c19, anti-rabbit), Bad (K17, anti-rabbit), Bax (B-9, anti-mouse), HRP-conjugated goat anti-rabbit IgG, and alpha-Tubulin (anti-mouse). All antibodies were purchased from Fuzhou Maixin Biotechnology Co., LTD, China.
Generation of tetracycline-inducible AIM2 cell lines
MCF-7 Tet-Off cells were purchased from Fuzhou Maixin Biotechnology co., LTD and were maintained in DMEM/F-12 supplemented with 10% (v/v) fetal bovine serum and 250 mg/mL G418 (Invitrogen, Carlsbad, CA). For the selection of double transfectants and the induction of gene expression, the NH2-terminal FLAG tag peptide-fused AIM2 was cloned into the pBI-EGFP Tet plasmid (Clontech). The resulting vector was named pBI-EGFP-Tag-AIM2. The pBI-EGFP-Luc (Clontech) plasmid was used as a control. MCF-7 cells were transfected with pBI-EGFP-Tag-AIM2 or pBI-EGFP-Luc together with pcDNA6/c-myc/His (Invitrogen) using SN liposomes. After 2 days, the transfected cells were subjected to selection with 300 mg/mL blasticidin and the tetracycline derivative doxycycline (2 mg/mL; Sigma, St. Louis, MO). Blasticidin-resistant cells were screened for the induction of green fluorescent protein (GFP) by fluorescence microscopy or for AIM2 expression by Western blotting upon removal of the doxycycline. The induction of luciferase activity in pBI-EGFP-Luc transfectants was confirmed using a luciferase assay kit (Promega, Madison, WI). Two independent AIM2 expression stable lines (MCF-7 tTA-AIM2) were selected for subsequent experiments. The luciferase expression stable line (MCF-7 tTA-Luc) was also generated as a negative control. After transferring AIM2 to the Tet-Off system of MCF-7 cells, the cells were rinsed with PBS buffer to remove the doxycycline and induce AIM2 expression in great quantities. Interferon-γ (102 U/ml) was added to 1 ml of cell immediately after induction to prove that interferon could induce the experimental cells to express AIM2.
Protein isolation and quantification
In total, 4 × 104 cells were rinsed with 4°C PBS three times. The cells were blown; loaded into 1.5 mL centrifuge tubes; and 1 mL of protein was added to the cell lysis extraction buffer. The cells were incubated on ice for 30 mins. After high-speed centrifugation (1300 RPM, 10 mins), the supernatant was collected. The protein extract was analyzed with a spectrophotometer to measure the O.D. 595 nm absorbance value. Based on the protein concentration, a standard curve was plotted.
Western blot analysis
After protein quantification, the protein samples were mixed with protein buffer (0.6 mol/L DTT, 10% SDS, 1 mol/L Tris-HCL (pH 6.8), 30% glycerol, 1% bromophenol blue), boiled for 4 minutes, and incubated at -20°C for 1 h. The protein samples were loaded into an SDS-polyacrylamide gel and separated at 100 V. Then, the protein on the gel was blotted onto nitrocellulose membrane and blocked with 5% skim milk solution at room temperature for 30 to 60 minutes. Then, the target protein antibody was applied and incubated for one hour at room temperature. The gel was rinsed three times with TBST and HRP-conjugated secondary antibody was applied and incubated to react at room temperature for one hour. TBST washed away non-specific complexes. The ECL chemiluminescence substrate was reacted with the secondary antibody enzyme, and the results were recorded using highly photosensitive film.
Cell cycle analysis
The culture medium was removed from a sample of 4 × 105 cells. The cells were rinsed with PBS once before adding Trypsin/EDTA solution, after which they were incubated in a 37°C 5% CO2 incubator until they were detached from the surface of the plate. The cells were scattered using PBS before being collected by low-speed centrifugation (2000 rpm, 2 mins, 4°C). The cells were then mixed with 70% alcohol and stored in -20°C refrigerator overnight. On the next day, the cells were centrifuged and the alcohol solution was removed. The cells were resuspended in PBS, after which 0.1% Triton-100, RNase A (4 mg/mL), and propidium iodide (2 mg/mL) were added. The cells were incubated at room temperature away from light for 30 mins, and a 35-micron nylon mesh was then used to filter the cell suspension. The cells were analyzed using a flow cytometry instrument, and cell cycle analysis software was used to analyze the cell cycle distribution in the cell groups grown with different treatment conditions.
Cell fractionation assay
In total, 4 × 104 cells were cleaned with 4°C PBS twice, and 500 µL pyrolysis buffer (20 mmol/L MgCl2, 0.5% NP-40, 100 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L PMSF, 1 mmol/L aprotinin) was added at 4°C for 20 mins. The cells were scraped with a scraper and incubated at 4°C on ice for 10 mins. The mixture was placed into the homogenizer, stirred 60 times, and centrifuged (4000 rpm, 5 mins, 4°C). The supernatant was placed in 1.5 mL centrifuge pipe and centrifuged (1300 rpm, 20 mins, 4°C). The supernatant fluid contained the cytoplasmic protein fraction. The nucleus particles were added to 1 mL pyrolysis buffer and were washed three times (4000 rpm, 5 mins, 4°C). The particles were suspended in 300 μL NETN buffer (0.5% NP-40, 1 mmol/L EDTA, 20 mmol/L Tis (pH 8.0), 150 mmol/L NaCl) with a sonicator (power 3-4, 20 seconds). To break the nuclei, the suspension was centrifuged (13000 rpm, 20 mins, 4°C), and the resulting supernatant fluid contained the nuclear proteins. The quantitated protein was mixed with sample buffer, boiled for 4 mins and placed on ice prior to protein electrophoresis separation.
Cytochrome c release assay
In total, 4 × 104 cells were distributed evenly in a 10 cm petri dish until the chosen number of induction days had elapsed. The cells were washed with 4°C PBS twice, and 500 µL buffer A (20 mmol/L Hepes (pH 7.4), 250 mmol/L sucrose, 10 mmol/L KCL, 1 mmol/L MgCl2, 1.5 mmol/L EDTA, 1.5 mmol/L EGTA, 1 mmol/L DTT) was added. A straw was used to homogeneously mix the cells 50 times. The cells were centrifuged (2000 rpm, 10 mins, 4°C) to separate the suspending liquid from the grains. After the suspending liquid was centrifuged (13000 rpm, 30 mins, 4°C), the supernatant fluid contained the cytoplasmic granules. The supernatant was removed, and 100 µL buffer B (50 mmol/L Hepes (pH 7.4), 1% NP-40, 10% glycerol, 1 mmol/L EDTA, 2 mmol/L DTT) was added to the remaining liquid. The buffer was centrifuged (13000 rpm, 30 mins, 4°C), and the supernatant contained mitochondria particles. Protein electrophoresis separation was performed,with the cytoplasmic granules and mitochondria granules, respectively.
Apoptosis detection: staining with Annexin V-FITC and propidium iodide
In total, 4 × 104 cells were cultured in petri dish until the desired number of induction days had elapsed. The nutrient solution was removed. The cells were washed with PBS. Trypsin/EDTA solution was applied until the cells detached, and 4°C PBS was used to scatter the cells prior to centrifugation (2000 rpm, 2 mins). The cell particles were mixed evenly with 70% alcohol and incubated in a -20°C environment. The next day, the alcohol was removed after centrifugation, and the Annexin V-FITC apoptosis detection kit was filled with the Annexin V-FITC, propidium iodide and membrane protein V combined buffer, respectively. The kit was incubated at room temperature for 30 mins, and a 35 µm nylon mesh was used to filter the solution. The cells were analyzed by flow cytometry. Finally, the WinMDI 2.9 software program was used to analyze the cell apoptosis distribution within the cell groups under different conditions.
XTT assay
MCF-7 tTA-AIM2 and MCF-7 tTA-Luc cells were induced for 2, 4, and 6 days, respectively. In a 96-well plate, 6 × 103 cells were distributed evenly. On the next day, the culture medium was changed. At the time of detection, a culture medium containing XTT reagent (DMEN, mixture of 1 mg/mL XTT markers, 0.383 mg/mL PMS (N-methyl dibenzopyrazine methyl sulfate) was used. After 3 hours reaction, the O.D. 420-590 nm absorbance values were determined by an ELISA reader.
Results
AIM2 colonization and Tet-Off system model
To study AIM2 expression in breast cancer cells and the mechanism of action, we placed AIM2 cDNA into a pCMV-Tag2C plasmid-pCMV-Tag-AIM2 (Figure 1A). With the Tet-OffTM system, we established pBI-EGFP Tet plasmids (Figure 1A) in which TRE, pminiCMV and the AIM2 gene were fused with the SV40 poly-adenylation signal. This Tet-OffTM induction model system allowed for complete doxycycline washes and the tetracycline transcription activation (tTA) to be combined with TRE. This system promoted AIM2 gene transcription and enabled the expression of the AIM2 protein in large amounts (Figure 1B).
Figure 1.
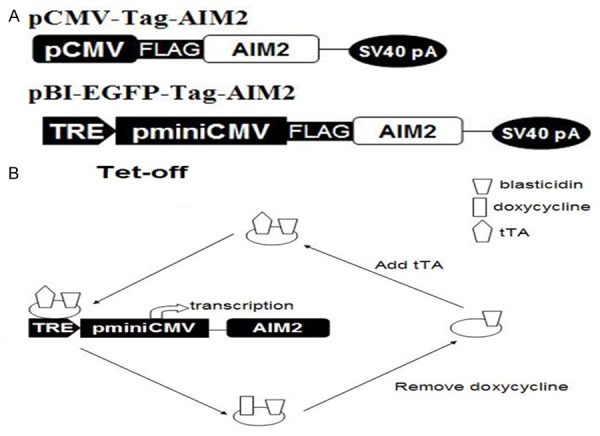
AIM2 gene transfer plasmid contructs and the Tet-Off model system. Note: A. pCMV-Tag-AIM2 and pBI-EGFP-Tag-AIM2 plasmids. B. Tet-Off induction model system.
Western blotting to test AIM2 expression in breast cancer cell lines
After induction (2, 4, 6, and 8 days), the cell protein extracts were submitted to Western Blotting to analyze AIM2 protein expression. The results demonstrated that at four days after induction, AIM2 expression can be detected. AIM2 expression gradually increased with the number of days post-induction (Figure 2A). However, the control group did not express AIM2 (Figure 2B).
Figure 2.
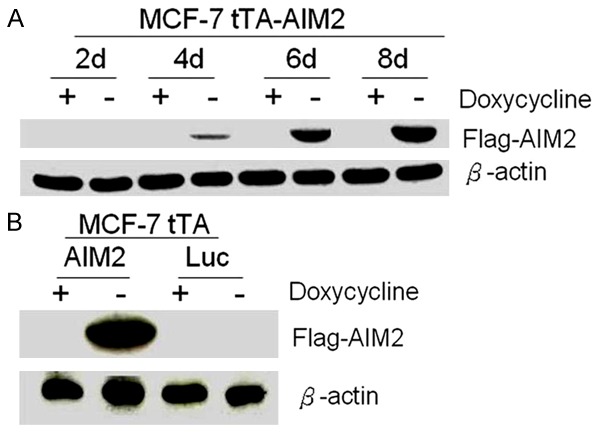
The experimental group steadily increased AIM2 protein expression. Note: A. AIM2 expression in the experimental group 2, 4, 6, and 8 days after induction B. Control cells did not express AIM2 protein 8 days after induction. (β-actin control, -indicates removal of doxycycline, + indicates addition of doxycycline).
AIM2 distribution in MCF-7 cells
To further understand the AIM2 distribution in breast cancer cells, nuclear protein extraction was performed on the MCF-7 cells eight days after induction. Western Blotting was performed on the extracts. AIM2 was found in the cytoplasm (C) and nucleus (N) (Figure 3).
Figure 3.
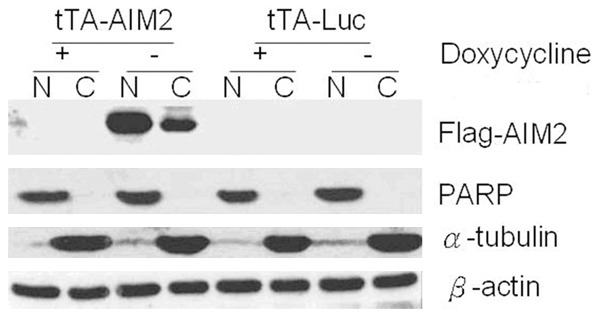
AIM2 expression in the nucleus and cytoplasm. Note: Cell fractionation was performed on the experimental and control cells eight days after induction to analyze AIM2 distribution within the cell. PARP was the nucleus protein marker, and α-tubulin was the cytoplasmic protein marker.
Induction of AIM2 protein expression by interferon
To prove that interferon could induce the experimental cells to express AIM2, interferon-γ (102 U/ml) was added immediately after induction. AIM2 expression was detected 2 days after induction. Without interferon-γ, no AIM2 protein expression was detected until four days after induction. Four days after induction, interferon-γ was used to treat the groups of cells, and AIM2 expression was obviously increased (Figure 4).
Figure 4.
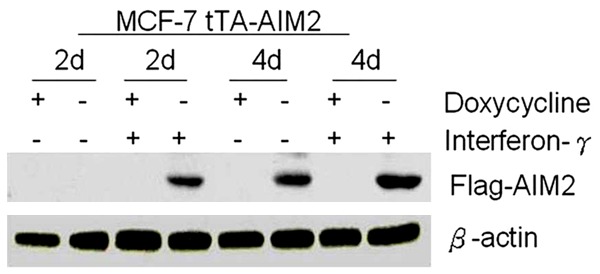
Interferon-γ induced the expression of AIM2 (β-actin control, -removal of doxycycline, + addition of doxycycline).
Influence of AIM2 expression on cell cycle
The experimental cells (2, 4, and 6 days after induction of AIM2 expression) were use flow type instrument to analyzed the cell cycle phases. The results are shown in Figure 5A, 5B. AIM2 induction had no significant effect on the cell cycle. However, for control cells, 4 and 6 days after removing doxycycline, the S phase and G2 phase increased significantly (two independent sample t-test). Then, Western Blotting was used to analyze the cell cycle regulatory proteins Cdk2, Cdk4, and Cyclin E (Figure 5C). For experimental cells, the level of Cdk2 gradually increased with the number of days post-induction, the expression of Cyclin E and Cdk4 were unchanged (two independent sample t-test).
Figure 5.
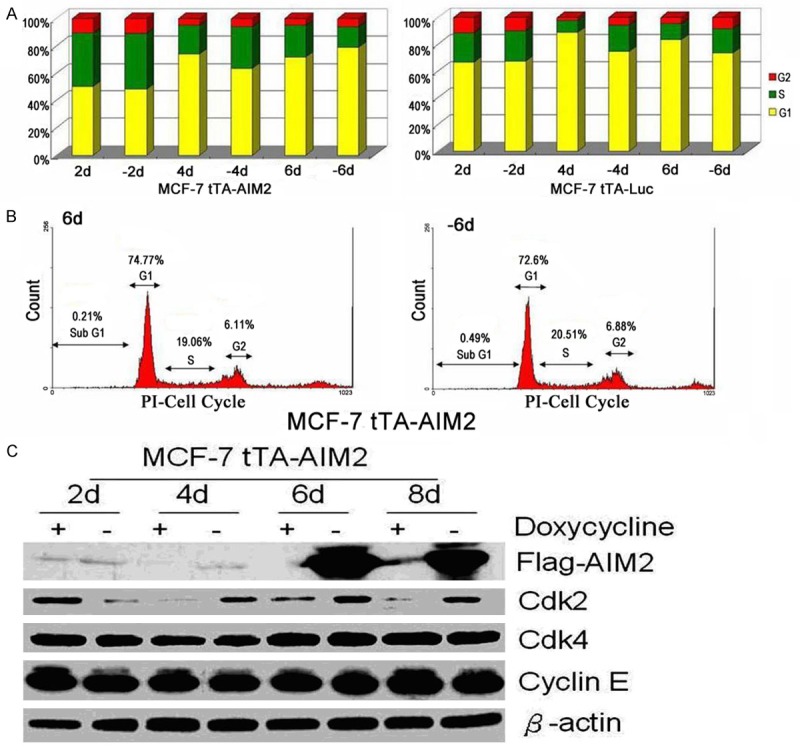
High AIM2 expression did not affect cell cycle regulation. Note: A. 2, 4, and 6 days after the experimental and control cells were induced, the cell cycle was analyzed with flow cytometry. B. The cell cycle distribution in the experimental cells six days after induction. C. Western Blotting was used to analyze the expression of cell cycle regulatory proteins (Cdk2, Cdk4, Cyclin E) in experimental cells 2, 4, 6, and 8 days after induction.
High AIM2 expression can promote breast cancer cell apoptosis
Early apoptotic cells were stained with Annexin V-FITC, and dead cells were stained with propidium iodide 6 days after induction. A flow cytometry instrument was used to analyze cell apoptosis. As shown in Figure 6, following high AIM2 expression was induced (-6d), Apoptosis percentage (FITC fluorescence ) increased significantly (two independent sample t-test).
Figure 6.
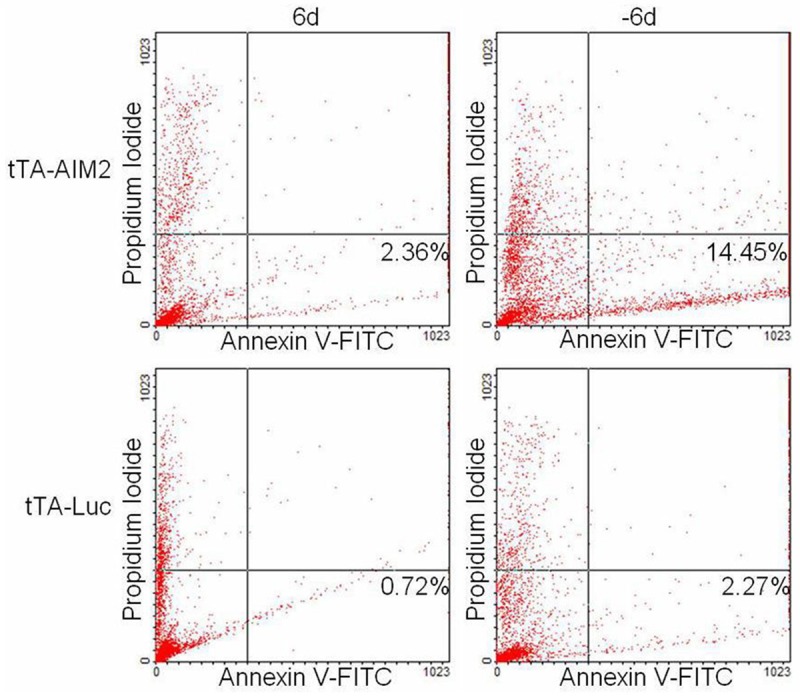
Annexin V-FITC was used to analyze cell apoptosis following high AIM2 expression.
Mechanism of AIM2 promoting breast cancer cell apoptosis
On the sixth induction day, take experimental and control cells to perform cytoplasm and mitochondria separation, and extraction protein. After AIM2 expression, cytochrome c released into the cytoplasm was detected (Figure 7A). In addition, with increasing induction days, high AIM2 levels can suppress anti-apoptosis Bcl-xL expression, increase pro-apoptosis Bad and Bax expression, which activate caspases and the DNA repair protein PARP cleavage (Figure 7B, 7C). In contrast, 6 days and 8 days after induction, Bcl-xL expression increased, Bad and Bax levels were reduced in control cells. PARP expression increased (Figure 7C). Demonstrated AIM2 expression promotes MCF-7 cell apoptosis through mitochondrial mechanism.
Figure 7.
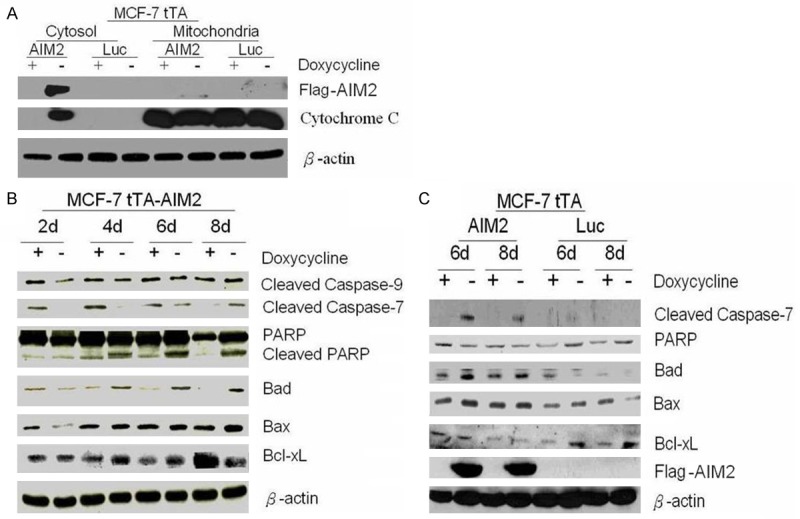
Induced AIM2 expression can promote MCF-7 cell apoptosis through a mitochondrial mechanism. Note: A. Western blot analysis was performed with a cytochrome c antibody. B. The expression of PARP, cleaved caspase 9, cleaved caspase 7, Bad, Bax, and Bcl-xL in experimental cells. C. The expression of PARP, cleaved caspase 9, cleaved caspase 7, Bad, Bax, and Bcl-xL in control cells.
AIM2 affects cell proliferation and survival
This study used the XTT assay to analyze the influence of AIM2 on cell growth. As demonstrated in Figure 8, 6 days after MCF-7 tTA-AIM2 cell lines were induced (-6d), their growth rate was slower than that of the non-induced cell lines (6d). However, following doxycycline removal (tTA-Luc, -6d), Control group MCF-7 tTA-Luc cell growth was accelerated (two independent sample t-test).
Figure 8.
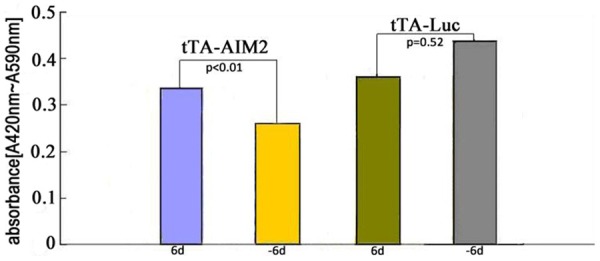
AIM2 affects cell proliferation and survival.
Discussion
This study explored the role of AIM2 in breast cancer cells. The n-terminal region of the AIM2 gene sequence was fused to the FLAG tag, which contains eight amino acids. This construct was termed the pCMV-tag-AIM2 plasmid. AIM2 protein expression was detected via the FLAG tag. Excessive AIM2 expression can inhibit cell proliferation. Thus, we used the Tet-OffTM genetic system to filter the transduced cells that could be induced (MCF-7 tTA-AIM2). AIM2 expression was induced by removing doxycycline. The experimental results demonstrate that AIM2 expression increased gradually during the post-induction period. However, doxycycline itself can inhibit the growth of cells and accelerate cell aging. Thus, to control for the effects of doxycycline, the MCF-7 tTA-Luc cell line served as a control group.
In breast cancer cells, AIM2 protein expression was found in both the nucleus and cytoplasm. Because the IFI-200 family proteins have a PAAD domain and can be combined with a variety of proteins (transcription factors) [4], AIM2 may depend on other proteins to enter the nucleus to regulate immune-and apoptosis-related processes. IFN-γ can stimulate AIM2 expression, and AIM2 expression can be detected 4 days after induction. Because AIM2 has an HIN-200 domain that can be induced by IFN and is known to inhibit cell growth and regulate cell differentiation and apoptosis [5], we investigated the physiological role of AIM2 in breast cancer cells.
Mammalian cell division via protein kinases-cell cycle protein dependent kinases (CDKS)-and cyclins to regulate the cell cycle [6]. The cell cycle is divided into G0, G1, S, G2, and M phases, and each cycle is controlled by specific CDKS-cyclin complexes. Specifically, Cdk4/6-Cylin D, Cdk2-Cylin E, Cdk2-Cylin A, and Cdk1-Cylin B for the G0, G1, S, and G2/M phases, respectively [7]. Previous studies have demonstrated that (in mice) the interferon-induced protein-p202 has the ability to delay cell growth and stuck cells in the G0/G1 phase [8]. This study used flow cytometry to analyze AIM2-induced effects on breast cancer cells. The experimental cells demonstrated no significant effects on the cell cycle after 2, 4, or 6 days. In contrast, in the control cells (after 4 and 6 induction days), the S phase and G2/M phases were increased significantly. Western blotting demonstrated that Cdk2 expression also increased gradually With the increase of induction days. The expression of Cdk4 and Cyclin E were unchanged, which indicates that AIM2 does not cause breast cancer cell cycle stagnation.
Regarding apoptosis, this experiment used Annexin V-FITC and flow cytometry to identify early apoptotic cells. The percentage of cells with FITC fluorescence increased with AIM2 induction, which suggests that high AIM2 expression can promote cell apoptosis.
The mechanism by which AIM2 affects cell apoptosis has been rarely studied. In this study, AIM2 expression led to the release of cytochrome c from the mitochondria into the cytoplasm. AIM2 also inhibited the expression of the anti-apoptotic protein Bcl-xL, increased the expression of pro-apoptotic proteins Bad and Bax, activate caspases, leading to cleavage of DNA repair proteins such as PARP and, finally, leading to cancer cell apoptosis. However, the control cells demonstrated different results, which also indicate that the changes in apoptosis-related protein expression were not caused by doxycycline. Interferon can modulate cell growth and differentiation. In the present study, interferon-inducible protein include p202, p204, IFI-16, IFIX and AIM2 proteins, which have physiological roles in cell growth inhibition [9,10]. The XTT assay demonstrated that the expression of AIM2 can reduce the rate of cell growth.
To summarize, in the Tet-offTM induction system with the AIM2 expression in breast cancer cells, AIM2 can be expressed in the cytoplasm and nucleus. This expression stimulates mitochondria, promotes cell apoptosis and influences cell proliferation and survival.
Acknowledgements
We thank Dr. Xiao-Ming Zou (The Second Affiliated Hospital of Harbin Medical University) for support in polishing the report.
Disclosure of conflict of interest
None.
References
- 1.Yang L, Sun T, Yuan Y, Wang N. Relationship between female breast cancer incidence and the socioeconomic status in Beijing. Zhonghua Zhong Liu Za Zhi. 2014;36:713–716. [PubMed] [Google Scholar]
- 2.Long XS, Wu Q, Song F, Huang J, Zhang LJ. RAS signal pathway participates in interferon-α mediated inhibition of proliferation of vascular smooth muscle cells in rats. Basic & Clinical Medicine. 2014;34:882–885. [Google Scholar]
- 3.Lee J, Li L, Gretz N, Gebert J, Dihlmann S. Absent in Melanoma 2 (AIM2) is an important mediator of interferon-dependent and -independent HLA-DRA and HLA-DRB gene expression in colorectal cancers. Oncogene. 2012;31:1242–1253. doi: 10.1038/onc.2011.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun C, Liu C, Dong J, Li D, Li W. Effects of the myeloid cell nuclear differentiation antigen on the proliferation,apoptosis and migration of osteosarcoma cells. Oncol Lett. 2014;7:815–819. doi: 10.3892/ol.2014.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Wang J, Wang J, Cao LS, Wang ZX, Wu JW. Structural mechanism of DNA recognition by the p202 HINa domain: insights into the inhibition of Aim2-mediated inflammatory signalling. Acta Crystallogr F Struct Biol Commun. 2014;70:21–29. doi: 10.1107/S2053230X1303135X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moritani M, Ishimi Y. Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J Biochem. 2013;154:363–372. doi: 10.1093/jb/mvt062. [DOI] [PubMed] [Google Scholar]
- 7.Dai L, Liu Y, Liu J, Wen X, Xu Z, Wang Z, Sun H, Tang S, Maguire AR, Quan J, Zhang H, Ye T. A novel cyclinE/cyclinA-CDK inhibitor targets p27 (Kip1) degradation, cell cycle progression and cell survival: implications in cancer therapy. Cancer Lett. 2013;333:103–112. doi: 10.1016/j.canlet.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Liu F, Guo H, Zhu Z, Jiao Y. Role of interferon-inducible protein 202 (p202) in the regulation of adipogenesis in mouse adipose-derived stem cells. Mol Cell Endocrinol. 2014;382:814–824. doi: 10.1016/j.mce.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Yu L, Liu P, Liu Z, Zhu W, Yan K, Chen Q, Han D. p204-mediated innate antiviral responses in mouse adipose cells and their effects on cell functions. Immunol Cell Biol. 2014;93:147–157. doi: 10.1038/icb.2014.83. [DOI] [PubMed] [Google Scholar]
- 10.Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 2012;5:173–183. doi: 10.1038/mi.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]